

Abstract
There is experimental and clinical evidence that some exanthematous allergic drug hypersensitivity reactions are mediated by drug-specific T cells. We hypothesized that the capacity of certain drugs to directly stimulate the innate immune system may contribute to generate drug-specific T cells. Here we analyzed whether abacavir, an HIV-1 reverse transcriptase inhibitor often inducing severe delayed-type drug hypersensitivity, can trigger innate immune activation that may contribute to its allergic potential. We show that abacavir fails to generate direct innate immune activation in human monocytes but potently triggers IL-1β release upon pro-inflammatory priming with phorbol ester or Toll-like receptor stimulation. IL-1β processing and secretion were sensitive to Caspase-1 inhibition, NLRP3 knockdown, and K+ efflux inhibition and were not observed with other non-allergenic nucleoside reverse transcriptase inhibitors, identifying abacavir as a specific inflammasome activator. It further correlated with dose-dependent mitochondrial reactive oxygen species production and cytotoxicity, indicating that inflammasome activation resulted from mitochondrial damage. However, both NLRP3 depletion and inhibition of K+ efflux mitigated abacavir-induced mitochondrial reactive oxygen species production and cytotoxicity, suggesting that these processes were secondary to NLRP3 activation. Instead, depletion of cardiolipin synthase 1 abolished abacavir-induced IL-1β secretion, suggesting that mitochondrial cardiolipin release may trigger abacavir-induced inflammasome activation. Our data identify abacavir as a novel inflammasome-stimulating drug allergen. They implicate a potential contribution of innate immune activation to medication-induced delayed-type hypersensitivity, which may stimulate new concepts for treatment and prevention of drug allergies.
Keywords: allergy, caspase 1 (CASP1), inflammasome, innate immunity, interleukin 1 (IL-1), NLRP3, abacavir, drug hypersensitivity
Introduction
Immune-mediated, allergic drug hypersensitivity represents a significant proportion of all adverse drug reactions (1). The clinical spectrum of delayed-type hypersensitivity ranges from uncomplicated maculopapular exanthemas to systemic reactions with profound liver involvement (drug reaction with eosinophilia and systemic symptoms (DRESS)) and severe bullous skin eruptions such as Stevens-Johnson syndrome and toxic epidermal necrolysis (1, 2).
Although there is evidence that some of these delayed-type exanthematous drug reactions are induced by drug-specific T cells, two questions still remain unanswered. First, how are small drug molecules processed to become antigens capable of triggering T cell receptors? Second, which pathways provide the additional “danger signals” during such a drug-T cell interaction that are necessary for efficient T cell activation?
A prime example is the abacavir hypersensitivity syndrome (ABC-HS)2 induced by the nucleoside analog reverse transcriptase inhibitor (NRTI) abacavir (ABC) (2). ABC is used in HIV-1 virus/AIDS combination therapy and elicits a severe cytotoxic CD8+ T cell-mediated allergic response that develops in about 5–8% of patients within 6 weeks after first treatment (3). Typical symptoms include fever, malaise, nausea, and diarrhea as well as mild to moderate exanthema as a late feature in ∼70% of affected patients (4). The development of ABC-HS is closely associated with positivity for a specific HLA class I allele, HLA-B*57:01 (5, 6), which has been accounted for by direct binding of ABC to the HLA-B*57:01-binding groove that alters the repertoire of presented self-peptides (7, 8). This “altered self” presentation is thought to trigger activation of ABC-cross-reactive CD8+ T memory cells originally directed against prevalent or persistent viral antigens, which might cause an immune response resembling organ graft rejection (9). Indeed, Lucas et al. (9) recently reported isolation of ABC-reactive memory T cells from ABC-naive patients. However, in 74% of all cases, ABC-HS occurs more than 5 days after treatment initiation (9), which suggests that de novo generation of drug-reactive T cells is also likely to play a role (1, 2).
According to the “danger model,” the mounting of an adaptive immune response critically requires the presence of a “danger signal” that indicates a potential hazard for the organism in addition to proper sensing of foreignness (10). Thus, principally de novo production of drug-specific T cells should ensue only if foreign peptide presentation by antigen-presenting cells is simultaneously accompanied by innate immune activation. The capacity to stimulate the innate immune system either may be an intrinsic property of the drug or its metabolites, for instance by direct activation of an innate immune receptor, or could be due to drug-induced release of endogenous damage-associated molecular patterns (DAMPs), for example from necrotic cells that secondarily trigger innate immune activation. Alternatively, an external cue such as a simultaneous infection may replace, prime, or amplify the drug-induced innate immune signal so that the necessary threshold for sensitization is exceeded (11).
Candidate mechanisms of drug-induced innate immune activation include stimulation of innate immune receptors of the Toll-like receptor (TLR) family and activation of the “inflammasome” sensory system, which have both been shown to be relevant for innate immune activation and disease development in allergic contact dermatitis, another T-cell-mediated hypersensitivity disorder (12, 13). The inflammasome is a multicomponent protease complex commonly composed of a NOD-like receptor protein (NLRP; usually NLRP3), the adaptor protein ASC, and the pro-inflammatory protease Caspase-1, which is required for processing and secretion of mature IL-1β from its inactive pro-form (14, 15). Unlike TLR activation, which triggers immediate release of critical cytokines such as TNF and IL-8 by transcriptional activation via the pro-inflammatory IKK2/NFκB signaling pathway (16), inflammasome activation is a two-step process that requires adequate priming, usually by TLR activation (signal I), which induces transcription of pro-IL-1β and expression of inflammasome components such as NLRP3 by transcriptional and post-translational mechanisms in addition to NLR activation by a distinct stimulus (signal II) (15). Thus, inflammasome activation can be considered as conditional innate immune activation as it critically depends on a preceding or coincident priming stimulus such as a bacterial or viral infection, without which the allergen fails to reach a sufficient level of innate immune activation for generation of allergen-specific T cells.
Although the factors governing inflammasome priming are well characterized, the actual triggers for inflammasome activation are controversial. For NLRP3, two major routes of activation have been proposed: 1) lysosomal damage associated with cytoplasmic cathepsin B release (17), and 2) mitochondrial damage associated with increased production of mitochondrial reactive oxygen species (mROS) as well as release of mitochondrial DNA and the mitochondria-localized lipid cardiolipin, which have all been implicated in NLRP3 inflammasome activation (18–21). Moreover, K+ efflux and necrosis induction emerged as mechanisms contributing to NLRP3 activation (22–24), but it is currently unclear whether these two events are causal or secondary to organelle damage and represent genuinely independent mechanisms promoting inflammasome activation.
Results
Recent evidence for a crucial role of innate immune activation in allergic contact dermatitis prompted us to ask whether drugs eliciting T cell-mediated delayed-type hypersensitivity such as ABC might also directly or conditionally induce an innate immune response. To this end, we stimulated human THP1 cells, which express a wide variety of innate immune receptors and represent an established model for inflammasome activation (25) with increasing ABC concentrations. Unlike the TLR4 agonist LPS, ABC failed to induce production of IL-8 (Fig. 1A), a strictly IKK2/NFκB-restricted cytokine (26), nor did it induce secretion of pro-IL-1β in the absence of additional stimuli (Fig. 1B). However, when THP1 cells were pre-stimulated with the phorbol ester TPA, an established priming stimulus for inflammasome activation in THP1 cells that induces both pro-IL-1β expression (25, 27) and their maturation into macrophage-like cells (28), ABC dose-dependently triggered IL-1β release (Fig. 1B). IL-1β secretion by ABC was statistically significant at concentrations ≥0.5 mg/ml, at which it reached levels comparable with stimulation with ATP, an established danger signal triggering NLRP3 inflammasome activation (29) (Fig. 1B). Similar results were obtained using the TLR4 agonist LPS or the TLR1/2 ligand Pam3CSK4 as alternative priming stimuli, although ABC was slightly less potent at triggering IL-1β release than ATP in this setting and overall levels of secreted IL-1β were about 10-fold lower than with TPA (Fig. 1B and data not shown). Importantly, ABC also increased IL-1β secretion from LPS-primed primary human blood monocytes (Fig. 1C), suggesting that ABC-mediated IL-1β release was not restricted to the THP1 cell line.
FIGURE 1.

ABC fails to trigger direct innate immune activation in human monocytes but dose-dependently induces IL-1β release upon TPA or TLR priming. A and B, THP1 cells were directly stimulated with the indicated ABC concentrations, LPS (100 ng/ml), or medium (ctrl) for 16 h (A), or stimulated for 16 h with medium (ctrl), ABC or ATP (5 mm) after priming with medium, 100 nm TPA (+TPA), or 100 ng/ml LPS (+LPS) (B), respectively. A, IL-8 ELISA; B, IL-1β ELISA of culture supernatants. C, IL-1β ELISAs of supernatants from medium- or LPS-primed (5 h, 100 ng/ml) primary human monocytes stimulated with 0.5 mg/ml ABC, 5 mm ATP, or medium (ctrl) for 16 h. D, IL-1β ELISAs of supernatants (SN) from THP1 that were pre-incubated with medium or the indicated TLR8 agonists CL075 (5 μg/ml) or VTX-2337 (10 μm) for 16 h and after 8 h of incubation in TLR8 agonist-free medium stimulated with 0.5 mg/ml ABC or medium for an additional 16 h, respectively. The immunoblots shown are representative of n = 3 experiments; bar diagrams represent mean cytokine concentrations ± S.D. from n ≥ 3 experiments. Statistical differences to the respective experimental controls (unstimulated cells in A and primed controls in B–D) are indicated by asterisks (**, p < 0.01, ***, p < 0.001, n.s. (non-significant) p > 0.05, one-way ANOVA (A) or two-way ANOVA (B–D) with Dunnett's (A and B) or Sidak's (C and D) multiplicity correction).
Considering that HIV-1 infection has recently been reported to trigger TLR8 activation and TLR8-dependent pro-IL-1β expression in human macrophages and monocytes (30, 31), we wondered whether TLR8 stimulation might also be able to prime for ABC-induced IL-1β secretion. Indeed, pre-stimulation of THP1 cells with two different TLR8 agonists, CL075 or VTX-2337, could also license for ABC-mediated IL-1β release from THP-1 cells, and resulted in processing of pro-IL-1β to partially cleaved IL-1β p28 and its biologically active p17 product (Fig. 1D). These data suggest that virus-mediated TLR8 activation might serve as a priming signal for ABC-induced inflammasome activation.
To analyze whether ABC-induced IL-1β secretion was due to inflammasome activation, we employed Z-YVAD-fmk, a Caspase-1 inhibitor that binds to cleaved Caspase-1, thereby inhibiting further proteolytic cleavage of IL-1β (32). Co-treatment of TPA-primed THP1 cells with Z-YVAD-fmk abrogated ABC-induced IL-1β release and its cleavage to IL-1β p28 and p17 (Fig. 2A).
FIGURE 2.

ABC-induced IL-1β release is mediated by the NLRP3 inflammasome. A, IL-1β ELISA (top) or IL-1β immunoblot (bottom) of culture SN from TPA-primed THP1 stimulated with the indicated stimuli in the presence of 10 μm of the Caspase-1 inhibitor Z-YVAD-fmk (YVAD) or an equivalent amount of diluent. B, comparison of ABC-induced IL-1β release and Caspase-1 (Casp1) cleavage from TPA-primed wild-type THP1 (THP1-WT) and functionally NLRP3-deficient THP1 (THP1-defNLRP3) stably expressing a shRNA for NLRP3. Top, IL-1β ELISA; bottom, immunoblots of culture SN for the indicated proteins. C, IL-8 ELISAs of culture supernatants (top) or immunoblots of total lysates (TL) (bottom) from non-primed or TPA-primed THP1-WT cells and functionally NLRP3-deficent THP1-defNLRP3, showing similar IL-8 and pro-IL-1β induction and efficient knockdown of NLRP3 protein in the THP1-defNLRP3 cell line. An immunoblot for tubulin is shown as loading control. D, IL-1β ELISA (top) and immunoblots of SN illustrating ABC-induced release of the indicated proteins from TPA-primed THP1 in the presence or absence of 5 μm of the small-compound NLRP3 inhibitor MCC950. E, IL-1β and Caspase-1 p10 immunoblot of culture SN of TPA-primed THP1 stimulated with 0.5 mg/ml of the specified NRTIs or medium (ctrl) for 16 h. Immunoblots are representative of n = 3 experiments; bar diagrams show mean cytokine concentrations ± S.D. from n ≥ 3 experiments (***, p < 0.001, n.s. (non-significant) p > 0.05; two-way ANOVA with Sidak's multiplicity testing).
Because various NLR proteins as well as the PYHIN (pyrin and HIN domain-containing protein) family members absent in melanoma 2 (AIM2) and IFNγ-inducible protein 16 (IFI16) can serve as relevant receptors for inflammasome activation (15), we next asked which inflammasome receptor may mediate the response to ABC. We first tested the requirement of NLRP3, the main inflammasome-activating NLR mediating IL-1β release upon contact allergen stimulation (13, 33). Comparison of IL-1β cleavage and release from TPA-primed wild-type THP1 versus THP1 cells functionally deficient for NLRP3 due to stable NLRP3 shRNA expression (THP1-defNLRP3) (34) revealed an absolute requirement of NLRP3 for ABC- and ATP-induced IL-1β p17 release and Caspase-1 cleavage (Fig. 2B). This was not due to defective priming or an off-target effect of the shRNA stably expressed in the THP1-defNLRP3 cell line as TPA pre-stimulation induced pro-IL-1β and IL-8 expression to comparable levels as in their wild-type counterparts (Fig. 2C) and the small-molecule NLRP3 inhibitor MCC950 (35) similarly decreased IL-1β release and Caspase-1 cleavage in the parental THP1 cell line (Fig. 2D). Thus, ABC-induced IL-1β cleavage and release critically require Caspase-1 and NLRP3 activation.
To explore whether ABC-induced inflammasome activation might be a class effect of NRTIs, we next tested the capacity of two non-allergenic NRTIs to induce inflammasome activation. Neither stavudine (d4T) nor zidovudine/azidothymidine (AZT) was able to trigger IL-1β cleavage and release or Caspase-1 activation at similar concentrations (Fig. 2E), excluding a class effect of NRTIs on inflammasome activation.
NLRP3 inflammasome activation is believed to result from either lysosomal or mitochondrial damage (36). A hallmark of the former is its sensitivity to actin depolymerization as lysosomal incorporation commonly ensues by phagocytosis, which requires an intact actin cytoskeleton. Accordingly, treatment with the actin-depolymerizing drug cytochalasin D has previously been shown to inhibit inflammasome-dependent IL-1β release by lysosomal signal II activators such as silica or alum (17, 37). However, unlike alum, ABC-induced Caspase-1 cleavage and IL-1β p17 release were insensitive to cytochalasin D treatment (Fig. 3A), suggesting that phagocytosis/lysosomal damage were unlikely to contribute to ABC-induced inflammasome activation. To analyze whether ABC-induced inflammasome activation thus may result from mitochondrial damage, we measured mROS accumulation, which commonly indicates mitochondrial damage. FACS-based quantification of the mitochondria-localized ROS sensor MitoSOX, which is converted from a colorless starting substance into a red fluorescent product upon mROS-dependent oxidation, revealed that ABC treatment resulted in dose-dependent mROS production in TPA-primed THP1, as evident by a gradual increase of both the percentage of MitoSOX positivity (Fig. 3B) and MitoSOX median fluorescence intensity, which reached statistical significance at a concentration of 0.5 mg/ml (data not shown). At this concentration, we also observed a substantial release of IL-1β p17 and Caspase-1 p10 in Western blots of culture supernatants (Fig. 3C) and a clear time-dependent increase of MitoSOX positivity by live cell fluorescence imaging (supplemental Movie 1). ABC-induced mROS production coincided with Caspase-1 activation, as evident by co-localization of the MitoSOX signal with cleaved Caspase-1 detected by co-staining with a fluorochrome-coupled Caspase-1 inhibitor (FAM-YVAD) (Fig. 3D). These data suggest that ABC-induced inflammasome activation is associated with mitochondrial damage.
FIGURE 3.

ABC-induced IL-1β release and Caspase-1 activation are independent of phagocytosis but associated with dose-dependent mROS production. A–C, TPA-primed THP1 cells were stimulated with medium (ctrl), ABC at 0.5 mg/ml or the indicated concentrations, alum (120 μg/ml), or 5 mm ATP for 16 h, and cells and culture supernatants were harvested separately for the different assays. A, impact of cytochalasin D (2 μm) or diluent (ctrl) co-incubation on ABC- or alum-induced IL-1β- and Caspase-1 (Casp1) processing and release. Top, IL-1β ELISA showing mean IL-1β secretion ± S.D. from n = 4 experiments (**, p < 0.01, n.s. (non-significant) p > 0.05; two-way ANOVA with Dunnett's multiplicity testing). Bottom, representative (n = 3) immunoblots of culture SN for cleaved and uncleaved IL-1β and active Caspase-1 p10, respectively. B, flow cytometric quantification of mROS-induced red fluorescence upon incubation of the differently stimulated cells with the fluorogenic mROS sensor MitoSOX. Histograms show representative overlays of logarithmic MitoSOX (PE-A) fluorescence from live-gated unstained control cells (ctrl unstained, gray dotted line), MitoSOX-loaded control stimulations (ctrl MitoSOX, black thin line), and MitoSOX incubations of the indicated ABC stimulations (bold red line) from n = 3 experiments, respectively. Numbers denote the percentage of MitoSOX-positive cells among the ABC-treated live population. C, representative (n = 3) Western blots of protein-precipitated culture SN, showing dose-dependent cleavage of IL-1β and Caspase-1 in response to increasing ABC concentrations or 5 mm ATP. D, combined DIC and fluorescence microscopy images showing co-localization (yellow) of sites of mROS production (red) and Caspase-1 cleavage (green) in ABC-stimulated cells. TPA-primed THP1 were treated with 0.5 mg/ml ABC for 4 h, and live cells were co-stained with the fluorogenic mROS detector MitoSOX and a green fluorochrome-coupled Caspase-1 inhibitor (FAM-YVAD) to allow for co-detection of mROS and cleaved Caspase-1 in situ. Pos. 1, position 1; Pos. 2, position 2.
Recently, necrosis induction has been proposed as a key mechanism for release of pro-IL-1β and its cleavage products by several NLRP3 inflammasome activators (22). Consistently, we observed a gradual increase of cytotoxicity upon exposure of TPA-primed THP1 cells with increasing ABC doses (Fig. 4A) that was not observed with other NRTIs (Fig. 4B). ABC-induced cytotoxicity closely correlated with its capacity to trigger IL-1β release and Caspase-1 cleavage (Fig. 3C) and appeared to occur via necrosis, as evident by high-amplitude swelling and ballooning followed by membrane permeabilization first apparent about 6–8 h after ABC exposure when mROS production was also evident (Fig. 4C, supplemental Movie 1). Moreover, it was accompanied by a time- and dose-dependent release of tubulin (Fig. 4, D and E), a protein normally found in the cytoplasm. The kinetics of tubulin release closely correlated with the appearance of cleaved IL-1β in the culture supernatant (Fig. 4D), suggesting that cytotoxicity/necrosis may contribute to ABC-induced IL-1β release. Of note, we did not observe a delayed kinetics of ABC-induced tubulin release and IL-1β cleavage in comparison with ATP (Fig. 4D), suggesting that ABC-induced inflammasome activation is unlikely to involve cytotoxicity-induced DAMP release.
FIGURE 4.
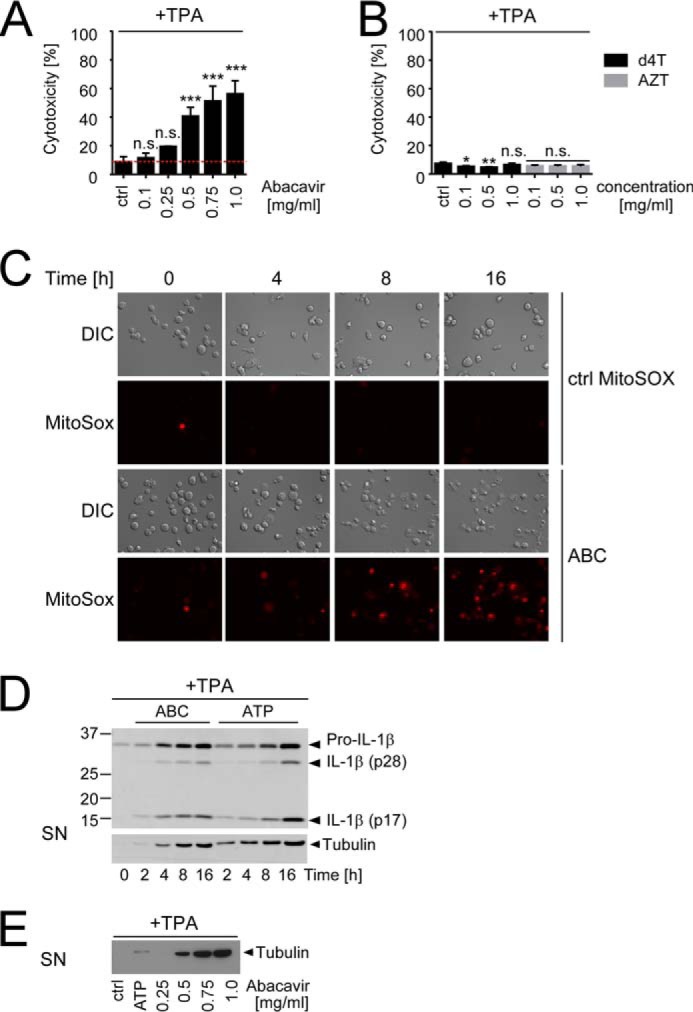
ABC-induced inflammasome activation coincides with cytotoxicity and mROS accumulation. A–E, TPA-primed THP1 cells were stimulated with medium (ctrl), 5 mm ATP, 0.5 mg/ml ABC, or the indicated concentrations of the specified NRTIs for 16 h or the given times and then analyzed for cellular cytotoxicity by LDH release assay (A and B), examined for visible signs of cytotoxicity and mROS-dependent MitoSOX fluorescence induction (red) by combined DIC and fluorescence time-lapse microscopy (C), or investigated for time- and dose-dependent IL-1β cleavage (D) or tubulin release (D and E) by immunoblots of culture SN, respectively. Bar diagrams show mean percentage of cytotoxicity ± S.D. of n ≥ 3 independent experiments related to a lysed control (set to 100%) with the red dotted line in A indicating background cytotoxicity upon TPA priming alone (*, p < 0.05, **, p < 0.01, ***, p < 0.001, n.s. (non-significant) p > 0.05; one-way ANOVA (A) or two-way ANOVA (B) with Dunnett's multiplicity testing). All other data are representative of n = 3 independent experiments.
To examine whether ABC-induced cytotoxicity was a cause or a consequence of NLRP3 activation, we repeated our cytotoxicity assays and tubulin Western blots using THP1-defNLRP3 cells or the NLRP3 inhibitor MCC950. Both approaches strongly reduced ABC-induced tubulin release (Fig. 5, A and B) and blunted ABC-induced cytotoxicity (Fig. 5C and data not shown). Thus, ABC-induced cytotoxicity obviously depends on NLRP3.
FIGURE 5.

ABC-induced cytotoxicity requires NLRP3. A and C, tubulin immunoblot of protein-precipitated culture SN (A) or LDH release assays (C), showing tubulin release or cytotoxicity of TPA-primed wild-type THP-1 cells (THP1-WT) and NLRP3-deficient THP1 cells (THP1-defNLRP3) upon treatment with medium (ctrl), ATP (5 mm), 0.5 mg/ml ABC, or the indicated ABC doses, respectively. B, immunoblots of SN, showing ABC-induced tubulin release from TPA-primed THP1 in the absence or presence of 5 μm of the small-compound NLRP3 inhibitor MCC950. Data are representative (A and B) or averaged (C) from n = 3 experiments with the diagram showing mean percentage of cytotoxicity ± S.D. in relation to a lysed control set to 100% (**, p < 0.01, ***, p < 0.001, two-way ANOVA, Sidak's multiplicity correction).
Another feature shared by many inflammasome activators is the induction of K+ efflux from the cytoplasm (24). Elevation of extracellular K+ was previously shown to suppress IL-1β release by multiple stimuli (23, 24) and has recently been reported to inhibit inflammasome-associated necrosis (22). Consistently, elevation of extracellular K+ levels dose-dependently reduced ABC-induced cytotoxicity (Fig. 6A) and prevented ABC-induced Caspase-1 activation and secretion of mature IL-1β p17 and tubulin (Fig. 6B). Moreover, elevation of extracellular KCl reduced ABC-induced mROS production (Fig. 6C). Thus, decrease of intracellular K+ obviously not only participates in the induction of ABC-induced cytotoxicity and IL-1β release but also contributes to mROS production.
FIGURE 6.

Increase of the extracellular K+ concentration inhibits ABC-induced cytotoxicity, IL-1β release, and mROS production. TPA-primed THP1 cells were stimulated for 16 h with 0.5 mg/ml ABC in standard medium containing low K+ (5 mm, ctrl) or medium containing the indicated KCl concentrations. A, LDH release assays showing decreased cytotoxicity upon ABC stimulation in high-KCl medium. B, IL-1β ELISA (top) and Western blots for IL-1β and tubulin (bottom) from SN of TPA-primed THP1 cells stimulated with ABC in normal culture medium (ctrl) or in medium containing the indicated KCl concentrations. Casp1, Caspase-1. C, effect of increased KCl medium concentrations on ABC-induced mROS production in TPA-primed THP1 cells as determined by flow cytometric quantification of PE-A fluorescence of the fluorogenic mROS sensor MitoSOX. Bar diagrams in A and B show means ± S.D. of n = 3 or n = 4 independent experiments, respectively. Immunoblots and histograms are representative of n = 4 independent experiments. Histograms display overlays of logarithmic MitoSOX (PE-A) fluorescence from the respective live-gated populations of unstained cells (ctrl unstained, gray dotted line) and MitoSOX-stained control cells (ctrl MitoSOX, black thin line) maintained at the indicated KCl concentration, and the respective ABC-stimulated condition (red thick line). The percentages of MitoSOX-positivity for the respective ABC-stimulated samples are indicated. Statistically significant differences are marked by asterisks (**, p < 0.01, ***, p < 0.001, n.s. (non-significant) p > 0.05, two-way-ANOVA with Dunnett's (A) or Sidak's (B) multiplicity correction).
mROS production has previously been proposed to be directly responsible for NLRP3 activation by mitochondria-damaging signal II stimuli (19). However, a causal role of mROS in NLRP3 activation has recently been challenged by the observation that IL-1β release by some mROS-inducing signal II stimuli such as linezolid was not affected by pharmacological ROS inhibition (21). Because we also failed to observe an inhibitory effect of ROS scavengers on ABC-induced IL-1β secretion (data not shown), we considered the possibility that ABC-induced mROS production might be a consequence of NLRP3 activation. Remarkably, both decreased NLRP3 expression and NLRP3 inhibition suppressed ABC-induced mROS production (Fig. 7A and data not shown), which was also evident by statistical analysis of mean MitoSOX median fluorescence intensity from multiple independent experiments (Fig. 7B and data not shown). Thus, ABC-induced mROS production obviously occurs secondary to NLRP3 activation.
FIGURE 7.

ABC-induced inflammasome activation is initiated upstream of mROS production and requires cardiolipin. A and B, flow cytometric analysis of the percentage of MitoSOX positivity (A) or MitoSOX median fluorescence intensity (MFI MitoSOX) (B), showing mROS production of TPA-primed wild-type THP-1 cells (THP1-WT) and NLRP3-deficient THP1 cells (THP1-defNLRP3) upon treatment with medium (ctrl), or 0.5 mg/ml ABC. C, left, analysis of medium- (ctrl), ABC-, or ATP-induced IL-1β release from TPA-primed THP1 after transfection with a scrambled siRNA (si scrambled) or a pool of two siRNAs against cardiolipin synthase 1 (si CRLS1). Right, evaluation of siRNA-mediated CRLS1 mRNA knockdown and its impact on TPA-mediated IL-1β mRNA induction as quantified by qRT-PCR. Bar diagrams show mean values ± S.D. of n ≥ 3 experiments with qRT-PCR data in C presented as -fold GAPDH-normalized expression of the indicated mRNAs in relation to the respective TPA-treated si scrambled control arbitrarily set to 1. Histograms in A are representative of n = 3 experiments with overlays showing logarithmic MitoSOX (PE-A) fluorescence intensity obtained for live-gated populations of the indicated ABC-stimulated cell line (bold red line), an unstained THP1-WT control (ctrl unstained, gray dotted line), and the respective unstimulated MitoSOX-stained control (ctrl MitoSOX, black thin line), respectively. Indicated percentages denote MitoSOX positivity among the ABC-exposed live population (*, p < 0.05, **, p < 0.01, ***, p < 0.001, one-sample t test (C, right) or two-way ANOVA (B and C, left), Sidak's multiplicity correction).
Because cytoplasmic release of the mitochondrial lipid cardiolipin has recently been demonstrated to contribute to inflammasome activation by mitochondria-damaging agents (21), we depleted a rate-limiting enzyme of cardiolipin synthesis, cardiolipin synthase 1 (CRLS1), by siRNA. CLRS1 knockdown significantly decreased ABC-induced IL-1β release from TPA-primed THP1 but had no effect on ATP-dependent IL-1β secretion that is mROS-dependent (18) (Fig. 7C, left). It further did not impair TPA-induced IL-1β mRNA expression (Fig. 7C, right), suggesting that the decreased ABC-mediated-IL-1β release under conditions of reduced cardiolipin synthesis was not due to flawed inflammasome priming but relied on defective signal II activation. Together these data imply an important role of cardiolipin in ABC-induced NLRP3 activation.
Discussion
Here we provide the first evidence that ABC, a substance frequently inducing T cell-mediated delayed-type drug hypersensitivity, can potently trigger inflammasome activation in human monocytic cells. Importantly, inflammasome activation was specific for ABC because other non-allergenic NRTIs failed to promote inflammasome activation. This fits previous observations that the NRTIs employed, d4T and AZT, had inhibitory rather than stimulating effects on inflammasome activation (38) and provokes the speculation that the capacity of ABC to induce inflammasome activation may contribute to its allergenicity. In accordance, Weston and Uetrecht (39) previously reported a positive correlation of inflammasome activation and idiosyncratic drug responses comparing IL-1β release from TPA-primed THP1 upon stimulation with two pairs of similar chemically reactive drugs, telaprevir/boceprevir and dimethyl fumarate/ethacrynic acid, of which only one compound per pair was capable of triggering skin reactivity. Although the reported levels of drug-induced IL-1β release in this study were comparably low and the authors failed to provide direct evidence for an involvement of the NLRP3 inflammasome therein, Weston's results support the suspicion that NLRP3 inflammasome activation may not be a unique feature of ABC but could be a more general trait of drug allergens. This underscores our hypothesis that NLRP3 inflammasome activation may contribute to the development of drug hypersensitivity syndromes such as ABC-HS. Unfortunately, the strict HLA specificity of ABC hypersensitivity precluded a functional testing of our findings in existing mouse models. Future humanized mouse models should address this important issue.
Of note, ABC stimulation failed to induce a direct innate immune signal in THP1 cells and was unable to prime inflammasome activation on its own. This raises the important question about the factor(s) that may provide an adequate priming signal for ABC-induced inflammasome activation in vivo. Considering that ABC is a drug used to control HIV-1 virus infection, it is possible that the virus itself may deliver the necessary priming signal. Intriguingly, HIV-1 infection has recently been shown to potently prime for inflammasome activation by different stimuli (40) and was able to induce pro-IL-1β in a TLR8-dependent fashion in macrophages and monocytes (30, 31). Consistently, our results demonstrate that TLR8 stimulation is sufficient to prime for ABC-induced inflammasome activation. It is therefore tempting to speculate that, depending on the viral load, ABC exposition might be able to trigger sufficient innate immune activation to allow for de novo production of ABC-reactive T cells. This challenges the currently prevailing notion that ABC allergy develops by reactivation of allo-specific memory T cells cross-reacting with the drug, as concluded from the successful isolation of ABC-responsive memory T cells from drug-naive patients (9). However, even in such a scenario, inflammasome activation may still improve the success rate of memory T cell activation, which could account for the relatively high incidence of 55% ABC drug hypersensitivity (9) among HLA-B*57:01-positive individuals. It was beyond the scope of our study to clarify the issue of to which extent de novo generation of T cells or reactivation of cross-reactive allo-specific T cells contributes to ABC-induced drug hypersensitivity. However, in light of our data, we would argue that de novo production of ABC-specific T cells at least cannot categorially be ruled out, even more so because this reasonably could explain the typical late onset of ABC hypersensitivity.
Mechanistically, ABC-induced inflammasome activation appears to follow that of other non-particulate NLRP3 activators, which trigger inflammasome activation via mitochondrial damage (14). Given the striking correlation of ABC-induced cytotoxicity and IL-1β cleavage and release, it is likely that ABC-induced IL-1β secretion requires cellular injury. This notion is consistent with data from Cullen et al. (22), who recently proposed a key role of necrosis in inflammasome-mediated IL-1β release by different NLRP3-activating stimuli. Similar to their study, ABC-induced NLRP3 inflammasome activation was invariably associated with co-release of unrelated cellular proteins, in our case tubulin, along with cleaved and uncleaved forms of IL-1β, suggesting that IL-1β release was not specific but rather a result of passive cell lysis (22). Using ABC as stimulus, we could further reproduce their data that elevation of KCl levels resulted in dose-dependent reduction of cytotoxicity and IL-1β release upon ABC stimulation, placing ABC-induced cytotoxicity downstream of KCl efflux from the cell. However, in one important aspect, our results contradict Cullen's study: NLRP3 inactivation clearly prevented both ABC-induced cytotoxicity and IL-1β release in our experiments, whereas Cullen et al. (22) did not observe any cytoprotective effect upon transient knockdown of NLRP3 or other inflammasome components. The reason for this discrepancy is not clear, but we exclude an off-target effect of our shRNA because treatment of wild-type THP1 with the small-compound NLRP3 inhibitor MCC950 provided identical results. In any case, our data argue against Cullen's model that necrosis occurs independently of NLRP3 activation. We would rather suggest that ABC-induced cytotoxicity is a consequence, not a cause, of NLRP3 activation. However, despite some discrepancies, our results still largely support Cullen's recent concept that the release of cleaved IL-1β products ultimately requires cellular injury (22).
Unexpectedly, NLRP3 depletion significantly reduced ABC-induced mROS production, suggesting that mROS production is an epiphenomenon rather than causal for ABC-induced inflammasome activation. Nevertheless, our data do not contradict the prevailing hypothesis proposed by Tschopp and colleagues (19) that a common mitochondria-generated signal accounts for NLRP3 activation by structurally unrelated inflammasome activators. For instance, Iyler et al. (21) recently reported that the antibiotic linezolid similarly induced a strong mROS signal but mROS production was dispensable for linezolid-induced NLRP3 inflammasome activation. Instead, the authors demonstrated that the cytoplasmic release of mitochondrial cardiolipin induced by mitochondrial damage was involved in linezolid-induced inflammasome activation. A similar mechanism may underlie ABC-induced inflammasome activation because siRNA-mediated depletion of CRLS1 strongly decreased ABC-induced IL-1β release. Of note, we failed to observe a similar reduction of ATP-induced IL-1β production, which is known to be sensitive to mROS inhibition (18), indicating that mitochondrial cardiolipin release is unlikely to be a general mechanism of NLRP3 inflammasome activation by mitochondria-damaging signal II agents but may rather be a feature of distinct classes of stimuli.
In summary, our data identify ABC as a potent inflammasome-activating drug allergen, suggesting that, similar to allergic contact dermatitis, delayed-type drug allergy may critically depend on innate immune activation. Our results thus may serve as the basis for the design of novel concepts for prevention and treatment of potentially life-threatening drug allergies.
Experimental Procedures
Cell Culture and Reagents
Primary human monocytes were isolated from whole blood of healthy donors using the human Monocyte Isolation Kit II from Miltenyi and suspended and stimulated in X-Vivo 15 medium for the experiments (Lonza). Human THP1 monocytic leukemia cells and functionally NLRP3-deficient THP1-defNLRP3 cells (34) (InvivoGen) were cultured in RPMI 1640 GLUTAMAX® (Gibco) supplemented with 10% FCS. For inflammasome experiments, THP1 were primed with 100 nm TPA for 3 h, and then 1 × 106 cells were reseeded into 6-well dishes containing 2 ml of TPA-free medium for maturation. On the following day, cells were stimulated for 16 h in 2 ml of FCS-free RPMI 1640 GLUTAMAX® containing the respective substances. Alternatively, 5 × 105 primary monocytes or 1 × 106 THP1 were primed with 100 ng/ml LPS (Escherichia coli 055:B5, Sigma-Aldrich) for 5 h (primary monocytes) or 6 h (THP1), or the TLR8 agonists CL075 (5 μg/ml, InvivoGen) and VTX-2337 (10 μm, motolimod; MedChem Express) for 16 h, and then stimulated for an additional 16 h in 1 ml (primary monocytes) or 2 ml (THP1) of the respective serum- and TLR agonist-free culture media, respectively. Medium supplemented with an equivalent amount of diluent served as stimulation control. ABC and the NRTIs (d4T and AZT), ATP, and MCC950 were obtained from Sigma-Aldrich and used at 0.5 mg/ml, 5 mm, or 5 μm, unless otherwise indicated. Z-YVAD-fmk (Bachem), cytochalasin D (Tocris Biosciences), and alum (Thermo Fisher Scientific) were used at 10 μm, 2 μm, or 120 μg/ml, respectively.
Cytokine Detection
Human IL-8 and IL-1β in culture supernatants were detected using BD OptEIATM ELISA kits from BD Biosciences (IL-8: catalog number 555244, IL-1β: catalog number 557933) following the manufacturer's instructions.
siRNA Transfection and Quantitative-Real Time (qRT) PCR
THP1 were transfected with scrambled siRNA (sense, 5′-UUCUCCGAACGUGUCACGUdTdT-3′; antisense, 5′-ACGUGACACGUUCGGAGAAdTdT-3′) or a pool of two commercial siRNAs against CRLS1 (catalog number SI04339881, sense, 5′-CCGAAUAUGUUGUCAAUGAdTdT-3′, antisense, 5′-UCAUUGACAACAUAUUCGGdGdA-3′ and catalog number SI04955664, sense, 5′-GGAUGGAUUUAUUGCUCGAdTdT-3′, antisense, 5′-UCGAGCAAUAAAUCCAUCCdAdA-3′, Qiagen) at a final concentration of 150 nm by electroporation using the 4D-Nucleofector device from Lonza following a protocol provided by the manufacturer. On the following day, electroporated cells were primed with TPA and stimulated as above but at a final density of 2 × 105 cells/ml in 12-well dishes. 60-h post-transfection culture supernatants were collected for IL-1β ELISA, and cells were processed for RNA isolation and qRT-PCR-mediated quantification of IL-1β and CRLS1 mRNA as described (41). IL-1β mRNA levels were determined using the primer probe method employing a commercial TaqMan probe for IL-1β (Hs01555410_m1, Applied Biosystems) and a specific probe for the housekeeping gene GAPDH (Hs99999905_m1, Applied Biosystems) as normalization control. CRLS1 mRNA knockdown efficiency was assessed by SYBR Green-based qRT-PCR employing specific primers for CRLS1 (forward, 5′-CCCAGTTCTGGGCTATTTG-3′ and reverse, 5′-TCAAGAGCACTTCCCAAAGC-3′) and the GAPDH normalization control (forward, 5′-CCACCCATGGCAAATTCC-3′ and reverse, 5′-GATGGGATTTCCATTGATGACA-3′).
Immunoblotting Analysis of Secreted Proteins
For immunoblotting analysis of secreted proteins, approximately one-third of the volume of the culture supernatant (650 μl) was precipitated by mixing with an equal volume of methanol and 0.25 volumes of chloroform and centrifuged for 10 min at 20,000 × g. After removal of the upper phase, 500 μl of methanol was added to the interphase, and total proteins were precipitated by centrifugation for 10 min at 20,000 × g. Pellets were dried at 55 °C, and samples were boiled at 95 °C for 5 min in Laemmli buffer prior to loading onto a 12% SDS-PAGE gel for protein separation. Proteins were blotted onto nitrocellulose membranes for analysis of released α-tubulin or IL-1β cleavage products using mouse monoclonal antibodies against human α-tubulin (Sigma, clone B-5-1-2) or the C terminus of human IL-1β (R&D Systems, clone 8516), respectively. Cleaved human Caspase-1 protein was detected on polyvinylidene difluoride membranes using an affinity-purified rabbit polyclonal for the active Caspase-1 p10 fragment (Santa Cruz Biotechnology, C-19, Sc-515). Proteins were visualized by enhanced chemiluminescence using the appropriate horseradish-peroxidase-coupled secondary antibodies.
Cytotoxicity Assays
1 × 104 TPA-primed THP1 cells were seeded onto 96-well plates, and after 24 h of maturation, they were exposed to the respective stimuli for 16 h. Relative percentile of cytotoxicity to a lysed control (set to 100%) was assessed by colorimetric quantification of the obligate intracellular enzyme lactate dehydrogenase (LDH) in the culture supernatant using a commercial LDH detection kit (Roche Applied Science, Cytotoxicity Detection Kit) according to the supplier's suggestions.
Mitochondrial ROS Detection and Microscopic Analysis of Caspase-1 Activation
For analysis of mROS production, 1 × 106 TPA-primed cells were stimulated for 16 h in 6-well plates with the respective stimuli. Cells were scraped off, and after centrifugation at 400 × g for 20 min, cells were incubated in 2.5 μm MitoSOX (Molecular Probes) in PBS at 37 °C. After two PBS washing steps, mROS-dependent conversion of MitoSOX to its red fluorescent product was assessed by flow cytometric quantification of PE-A median fluorescence intensity of the live-gated population. Alternatively, MitoSOX positivity was determined by analyzing time-dependent increase of red fluorescence in live cells upon stimulation of in situ MitoSOX-loaded cells with 0.5 mg/ml ABC by multi-position time-lapse fluorescence microscopy using a camera- and top-stage incubator-equipped Nikon Ti-E motorized inverse fluorescence microscope. For co-detection of mROS and cleaved Caspase-1, TPA-primed THP1 cells were alternatively first stimulated with ABC, and then after 4 h, in situ co-stained for 30 min with 2.5 μm MitoSOX and a 1× solution of FAM-coupled Z-YVAD-Fmk (FAM-FLICA®, Immunochemistry Technologies), a green fluorescent Caspase-1 inhibitor. Double-positive cells were then detected by combined differential interference contrast (DIC) and dual channel fluorescence microscopy as mentioned above, and image overlays were produced using Nikon NIS Elements AR 4.2 software.
Statistical Analysis
Statistical analysis was performed using the GraphPad Prism 6 biostatistical software. At least three independent experiments were averaged, and the ± S.D. was calculated. One-sample t tests were performed to assess significant differences of single test groups to a normalized control. Multiple groups were compared by one way (one factor) or two way (two factor) analysis of variance (ANOVA) followed by Sidak's or Dunnett's multiplicity correction. Adjusted p values of <0.05 were considered significant.
Author Contributions
A. Toksoy, H. S., and C. A performed the experiments with some contribution from S. H. who performed the experiments with the NRTIs d4T and AZT. A. Toksoy, C. A., and M. S. analyzed data. A. Toksoy, A. Trautmann, M. G., and M. S. designed the experiments. M. S. managed the project, wrote the manuscript, and had overall responsibility for data interpretation. All authors discussed and commented on the manuscript.
Supplementary Material
This work was supported by the Department of Dermatology, University Hospital Würzburg, Germany. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movie 1.
- ABC-HS
- abacavir hypersensitivity syndrome
- ANOVA
- analysis of variance
- AZT
- zidovudine/azidothymidine
- d4T
- stavudine
- DAMP
- damage-associated molecular pattern
- LDH
- lactate dehydrogenase
- NLR
- NOD-like receptor
- NLRP
- NOD-like receptor protein
- NOD
- nucleotide-binding oligomerization domain
- mROS
- mitochondrial reactive oxygen species
- NRTI
- nucleoside reverse transcriptase inhibitor
- qRT-PCR
- quantitative real time PCR
- ROS
- reactive oxygen species
- SN
- supernatants
- TLR
- Toll-like receptor
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- z
- benzyloxycarbonyl
- fmk
- fluoromethylketone
- FAM
- 6-carboxyfluorescein
- DIC
- differential interference contrast
- ctrl
- control.
References
- 1. Schrijvers R., Gilissen L., Chiriac A. M., and Demoly P. (2015) Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin. Transl. Allergy 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. White K. D., Chung W. H., Hung S. I., Mallal S., and Phillips E. J. (2015) Evolving models of the immunopathogenesis of T cell-mediated drug allergy: the role of host, pathogens, and drug response. J. Allergy Clin. Immunol. 136, 219–234; quiz 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pavlos R., Mallal S., Ostrov D., Buus S., Metushi I., Peters B., and Phillips E. (2015) T cell-mediated hypersensitivity reactions to drugs. Annu. Rev. Med. 66, 439–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cutrell A. G., Hernandez J. E., Fleming J. W., Edwards M. T., Moore M. A., Brothers C. H., and Scott T. R. (2004) Updated clinical risk factor analysis of suspected hypersensitivity reactions to abacavir. Ann. Pharmacother. 38, 2171–2172 [DOI] [PubMed] [Google Scholar]
- 5. Hetherington S., Hughes A. R., Mosteller M., Shortino D., Baker K. L., Spreen W., Lai E., Davies K., Handley A., Dow D. J., Fling M. E., Stocum M., Bowman C., Thurmond L. M., and Roses A. D. (2002) Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 359, 1121–1122 [DOI] [PubMed] [Google Scholar]
- 6. Mallal S., Nolan D., Witt C., Masel G., Martin A. M., Moore C., Sayer D., Castley A., Mamotte C., Maxwell D., James I., and Christiansen F. T. (2002) Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727–732 [DOI] [PubMed] [Google Scholar]
- 7. Ostrov D. A., Grant B. J., Pompeu Y. A., Sidney J., Harndahl M., Southwood S., Oseroff C., Lu S., Jakoncic J., de Oliveira C. A., Yang L., Mei H., Shi L., Shabanowitz J., English A. M., et al. (2012) Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. U.S.A. 109, 9959–9964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Illing P. T., Vivian J. P., Dudek N. L., Kostenko L., Chen Z., Bharadwaj M., Miles J. J., Kjer-Nielsen L., Gras S., Williamson N. A., Burrows S. R., Purcell A. W., Rossjohn J., and McCluskey J. (2012) Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 486, 554–558 [DOI] [PubMed] [Google Scholar]
- 9. Lucas A., Lucas M., Strhyn A., Keane N. M., McKinnon E., Pavlos R., Moran E. M., Meyer-Pannwitt V., Gaudieri S., D'Orsogna L., Kalams S., Ostrov D. A., Buus S., Peters B., Mallal S., and Phillips E. (2015) Abacavir-reactive memory T cells are present in drug naive individuals. PLoS ONE 10, e0117160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matzinger P. (2002) The danger model: a renewed sense of self. Science 296, 301–305 [DOI] [PubMed] [Google Scholar]
- 11. Martin S. F., Esser P. R., Weber F. C., Jakob T., Freudenberg M. A., Schmidt M., and Goebeler M. (2011) Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy 66, 1152–1163 [DOI] [PubMed] [Google Scholar]
- 12. Honda T., Egawa G., Grabbe S., and Kabashima K. (2013) Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J. Invest. Dermatol. 133, 303–315 [DOI] [PubMed] [Google Scholar]
- 13. Kaplan D. H., Igyártó B. Z., and Gaspari A. A. (2012) Early immune events in the induction of allergic contact dermatitis. Nat. Rev. Immunol. 12, 114–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sutterwala F. S., Haasken S., and Cassel S. L. (2014) Mechanism of NLRP3 inflammasome activation. Ann. N.Y. Acad. Sci. 1319, 82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Latz E., Xiao T. S., and Stutz A. (2013) Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawai T., and Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 17. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., and Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., and Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou R., Yazdi A. S., Menu P., and Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 20. Shimada K., Crother T. R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V. K., Wolf A. J., Vergnes L., Ojcius D. M., Rentsendorj A., Vargas M., Guerrero C., Wang Y., Fitzgerald K. A., et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iyer S. S., He Q., Janczy J. R., Elliott E. I., Zhong Z., Olivier A. K., Sadler J. J., Knepper-Adrian V., Han R., Qiao L., Eisenbarth S. C., Nauseef W. M., Cassel S. L., and Sutterwala F. S. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cullen S. P., Kearney C. J., Clancy D. M., and Martin S. J. (2015) Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep 11, 1535–1548 [DOI] [PubMed] [Google Scholar]
- 23. Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B. L., Rajendiran T. M., and Núñez G. (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pétrilli V., Papin S., Dostert C., Mayor A., Martinon F., and Tschopp J. (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14, 1583–1589 [DOI] [PubMed] [Google Scholar]
- 25. Martinon F., Burns K., and Tschopp J. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10, 417–426 [DOI] [PubMed] [Google Scholar]
- 26. Viemann D., Schmidt M., Tenbrock K., Schmid S., Müller V., Klimmek K., Ludwig S., Roth J., and Goebeler M. (2007) The contact allergen nickel triggers a unique inflammatory and proangiogenic gene expression pattern via activation of NF-κB and hypoxia-inducible factor-1α. J. Immunol. 178, 3198–3207 [DOI] [PubMed] [Google Scholar]
- 27. Fenton M. J., Vermeulen M. W., Clark B. D., Webb A. C., and Auron P. E. (1988) Human pro-IL-1β gene expression in monocytic cells is regulated by two distinct pathways. J. Immunol. 140, 2267–2273 [PubMed] [Google Scholar]
- 28. Tsuchiya S., Kobayashi Y., Goto Y., Okumura H., Nakae S., Konno T., and Tada K. (1982) Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 42, 1530–1536 [PubMed] [Google Scholar]
- 29. Sutterwala F. S., Ogura Y., Szczepanik M., Lara-Tejero M., Lichtenberger G. S., Grant E. P., Bertin J., Coyle A. J., Galán J. E., Askenase P. W., and Flavell R. A. (2006) Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 [DOI] [PubMed] [Google Scholar]
- 30. Chattergoon M. A., Latanich R., Quinn J., Winter M. E., Buckheit R. W. 3rd, Blankson J. N., Pardoll D., and Cox A. L. (2014) HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog. 10, e1004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo H., Gao J., Taxman D. J., Ting J. P., and Su L. (2014) HIV-1 infection induces interleukin-1β production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J. Biol. Chem. 289, 21716–21726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thornberry N. A., Bull H. G., Calaycay J. R., Chapman K. T., Howard A. D., Kostura M. J., Miller D. K., Molineaux S. M., Weidner J. R., Aunins J., et al. (1992) A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature 356, 768–774 [DOI] [PubMed] [Google Scholar]
- 33. Schmidt M., and Goebeler M. (2015) Immunology of metal allergies. J. Dtsch. Dermatol. Ges. 13, 653–660 [DOI] [PubMed] [Google Scholar]
- 34. Lerner A. G., Upton J. P., Praveen P. V., Ghosh R., Nakagawa Y., Igbaria A., Shen S., Nguyen V., Backes B. J., Heiman M., Heintz N., Greengard P., Hui S., Tang Q., Trusina A., et al. (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16, 250–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coll R. C., Robertson A. A., Chae J. J., Higgins S. C., Muñoz-Planillo R., Inserra M. C., Vetter I., Dungan L. S., Monks B. G., Stutz A., Croker D. E., Butler M. S., Haneklaus M., Sutton C. E., et al. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elliott E. I., and Sutterwala F. S. (2015) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 265, 35–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eisenbarth S. C., Colegio O. R., O'Connor W., Sutterwala F. S., and Flavell R. A. (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fowler B. J., Gelfand B. D., Kim Y., Kerur N., Tarallo V., Hirano Y., Amarnath S., Fowler D. H., Radwan M., Young M. T., Pittman K., Kubes P., Agarwal H. K., Parang K., Hinton D. R., et al. (2014) Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 346, 1000–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weston J. K., and Uetrecht J. (2014) Activation of inflammasomes by agents causing idiosyncratic skin reactions: a possible biomarker. Chem. Res. Toxicol 27, 949–951 [DOI] [PubMed] [Google Scholar]
- 40. Hernandez J. C., Latz E., and Urcuqui-Inchima S. (2014) HIV-1 induces the first signal to activate the NLRP3 inflammasome in monocyte-derived macrophages. Intervirology 57, 36–42 [DOI] [PubMed] [Google Scholar]
- 41. Czymai T., Viemann D., Sticht C., Molema G., Goebeler M., and Schmidt M. (2010) FOXO3 modulates endothelial gene expression and function by classical and alternative mechanisms. J. Biol. Chem. 285, 10163–10178 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.