

Abstract
Differentiation therapy with all-trans-retinoic acid (ATRA) improves the treatment outcome of acute promyelocytic leukemia (APL); however, the molecular mechanism by which ATRA induces granulocytic differentiation remains unclear. We previously reported that the inhibition of the NAD-dependent histone deacetylase (HDAC) SIRT2 induces granulocytic differentiation in leukemia cells, suggesting the involvement of protein acetylation in ATRA-induced leukemia cell differentiation. Herein, we show that p300/CREB-binding protein-associated factor (PCAF), a histone acetyltransferase (HAT), is a prerequisite for ATRA-induced granulocytic differentiation in leukemia cells. We found that PCAF expression was markedly increased in leukemia cell lines (NB4 and HL-60) and primary APL cells during ATRA-induced granulocytic differentiation. Consistent with these results, the expression of PCAF was markedly up-regulated in the bone marrow cells of APL patients who received ATRA-containing chemotherapy. The knockdown of PCAF inhibited ATRA-induced granulocytic differentiation in leukemia cell lines and primary APL cells. Conversely, the overexpression of PCAF induced the expression of the granulocytic differentiation marker CD11b at the mRNA level. Acetylome analysis identified the acetylated proteins after ATRA treatment, and we found that histone H3, a known PCAF acetylation substrate, was preferentially acetylated by the ATRA treatment. Furthermore, we have demonstrated that PCAF is required for the acetylation of histone H3 on the promoter of ATRA target genes, such as CCL2 and FGR, and for the expression of these genes in ATRA-treated leukemia cells. These results strongly support our hypothesis that PCAF is induced and activated by ATRA, and the subsequent acetylation of PCAF substrates promotes granulocytic differentiation in leukemia cells. Targeting PCAF and its downstream acetylation targets could serve as a novel therapeutic strategy to overcome all subtypes of AML.
Keywords: differentiation, histone acetylase, histone acetylation, leukemia, post-translational modification (PTM), ATRA, PCAF, acute promyelocytic leukemia, differentiation therapy
Introduction
Protein acetylation is a major post-translational modification involved in the regulation of gene expression (1). The acetylation of histone mediates transcription by changing the structure of chromatin, modulating the accessibility of transcription factors to their target genes, which regulates gene expression (2). The acetylation of non-histone proteins has also been shown to affect many important biological events (3). The acetylation levels of transcription factors, cellular proteins, and viral proteins are closely associated with tumorigenesis (4). The acetylation of histones and other proteins is regulated by histone acetyltransferases (HATs)2 and histone deacetylases (HDACs) (5). Because HAT and HDAC expression levels are deregulated in cancer cells, an altered protein acetylation balance has been suggested to contribute to cell transformation in cancer development (6). Furthermore, HDACs have been suggested to play an oncogenic role in cancer development (7–11), and HDAC inhibitors have been developed as anti-cancer compounds (12). In contrast, although HAT activity has previously been reported to be altered in cancer cells (13, 14), a therapeutic strategy to target HATs has not been established, perhaps due to a lack of deeper understanding of the function of each HAT in cancer.
Acute myeloid leukemia (AML) is an aggressive and fatal hematologic malignancy characterized by the accumulation of immature myeloid blasts. The treatment strategy for AML has been unchanged over the last few decades, and even today, it has yet to be based on the cytotoxic agents (15). Therefore, novel therapeutic approaches with high efficacy and less toxicity are urgently required. Previous reports have revealed that AML causes defects in granulocytic differentiation caused by mutations in cell differentiation-promoting transcription factors, such as CCAAT/enhancer-binding protein α (C/EBPα), PU.1, and globin transcription factor-1 (GATA-1) (16–18). In addition, some compounds have been proposed as differentiation-inducing drugs for AML (19–21). However, a clinically effective differentiating therapy for AML has been developed only for acute promyelocytic leukemia (APL).
In most types of APL, a disease-initiating chromosome translocation of t(15;17)(q22;q21) results in the production of the fusion gene protein promyelocytic leukemia-retinoic acid receptor α (PML-RARα), which binds to retinoic acid response elements and represses DNA transcription by recruiting HDAC co-repressors, such as nuclear receptor co-repressor (N-CoR) and silencing mediator of retinoid and thyroid hormone receptor (SMRT) complexes (22). All-trans-retinoic acid (ATRA) is clinically used to treat APL because it induces granulocytic differentiation in APL cells. Although ATRA has markedly improved the treatment outcome of APL, the precise mechanism by which it induces granulocytic differentiation in APL cells remains unclear. ATRA has been shown to separate the HDAC complex from PML-RARα; it then recruits the coactivator-HAT complex and subsequently induces granulocytic differentiation in APL cells (23). Based on this process, the blockade of HDAC activity may induce granulocytic differentiation and cell death in APL cells. Consistent with this, some classical HDAC inhibitors have been shown to induce cell death in APL and AML cells (24). In addition, valproic acid, an HDAC inhibitor, was found to induce granulocytic differentiation in an APL mouse model (25). We previously demonstrated that tenovin-6, an inhibitor of the NAD-dependent class III HDAC SIRT2, induced granulocytic differentiation in leukemia cell lines (26). Collectively, these findings indicate that protein acetylation must be involved in granulocytic differentiation in leukemia cells. However, the involvement of HATs, which promote protein acetylation, in leukemia cell differentiation remains unclear.
ATRA, at a concentration sufficient to exert anti-cancer effects, increases the expression levels of p300/CREB-binding protein-associated factor (PCAF), a HAT, in mouse kidney tissues (27). The retinoid X receptor/retinoic acid receptor (RAR) heterodimer directly recruits PCAF, and the increased expression of PCAF leads to enhanced retinoid-responsive transcription in mammalian cells (28). Additionally, RARα signaling is required for myeloid differentiation; thus, its inhibition blocks granulocytic differentiation (29). Furthermore, a previous report revealed that PCAF is a coactivator of RARα and that the PCAF-RARα complex is required for the transcriptional activation induced by a physiological concentration of retinoic acid in mammalian cells (30). However, the involvement of PCAF in ATRA-induced leukemia cell differentiation has not been elucidated. Here, we examined the involvement of PCAF and its functional role in the ATRA-induced granulocytic differentiation of leukemia cells.
Results
ATRA Induces PCAF Expression in ATRA-sensitive NB4 Cells but Not in ATRA-resistant NB4RA Cells
To investigate the role of PCAF in the ATRA-induced granulocytic differentiation of APL cells, we initially examined the effects of ATRA on the expression of PCAF. When the ATRA-sensitive APL cell line NB4 was treated with ATRA, PCAF expression levels were significantly elevated at the mRNA level (Fig. 1A) and were transiently elevated at the protein level 24 h after the ATRA treatment (Fig. 1B). In contrast with the NB4 line, in the ATRA-resistant NB4 subline NB4RA, which has a point mutation in the AF-2 domain of the PML-RARα fusion gene (31), the expression of PCAF was not induced by ATRA at either the mRNA (Fig. 1C) or the protein level (Fig. 1D). ATRA concomitantly inhibited the proliferation of NB4 cells but not NB4RA cells (Fig. 1, E and F). ATRA also enhanced nitro blue tetrazolium (NBT) reduction capacity, a hallmark of granulocyte maturation, in NB4 cells but not in NB4RA cells (Fig. 1, G and H). These results are consistent with previous findings (31, 32), showing that ATRA-treated NB4 cells differentiated into granulocytic cells, whereas ATRA-treated NB4RA cells did not. Collectively, these results indicate that ATRA-induced PCAF expression is strongly correlated with granulocytic differentiation in these cell lines.
FIGURE 1.

ATRA induces PCAF expression in ATRA-sensitive NB4 cells but not in ATRA-resistant NB4RA cells. A–D, PCAF expression in the APL cell line NB4 (A and B) and in the ATRA-resistant subline NB4RA (C and D) after the ATRA (1 μm) or ethanol (vehicle) treatment for the indicated period as analyzed by RT-qPCR (A and C) and immunoblotting (B and D). GAPDH and β-actin were used as controls for RT-qPCR and immunoblotting, respectively. The RT-qPCR results were quantified using the comparative Ct method. Experiments were performed in triplicate, and the data are shown as the mean ± S.D. (error bars). For the immunoblotting, 50-μg extracts from NB4 and NB4RA cells were loaded onto each lane. E and F, the NB4 and NB4RA cells that were viable after the ATRA treatment were counted. Cells were cultured with ATRA (1 μm) or ethanol (vehicle). The number of dead cells was determined using trypan blue dye. The viable and dead cells of three independent experiments were counted, and the data are shown as the mean ± S.D. G and H, the NBT reduction capacities of ATRA-treated NB4 and NB4RA cells were analyzed 3 and 5 days after the ATRA (1 μm) or ethanol (vehicle) treatment. The images were captured 5 days after the treatment (left). The black staining indicated that cells possessed NBT reduction capacity, a hallmark of granulocyte maturation. The graph depicts the frequency of NBT-positive cells 3 and 5 days after the treatment (right). Counting was performed in three independent microscope fields that covered at least 400 cells each. Error bars, S.E. (n = 3).
ATRA-induced PCAF Expression Is Associated with Granulocytic Differentiation
To further examine whether PCAF is associated with ATRA-induced granulocytic differentiation, we used another AML cell line, HL-60, which also has the ability to differentiate into granulocytic cells when treated with ATRA (33), and we examined the effects of ATRA on the expression of PCAF. Similar to NB4 cells, ATRA inhibited HL-60 cell proliferation, and a significant increase in NBT reduction capacity was observed in ATRA-treated HL-60 cells, indicating that ATRA induced granulocytic differentiation in these cells (Fig. 2, A and B). Consistent with the results from NB4 cells, ATRA induced the expression of PCAF in HL-60 cells at both the mRNA (Fig. 2C) and protein levels (Fig. 2D). Thus, the induction of PCAF expression after the ATRA treatment was observed not only in the APL cell line NB4 but also in the non-APL cell line HL-60. It is noteworthy that ATRA-induced PCAF expression was more evident in the non-APL cell line HL-60 than in the APL cell line NB4, and thus this implies that PML-RARα is not required for ATRA-induced PCAF expression.
FIGURE 2.

ATRA-induced PCAF expression is associated with granulocytic differentiation. A and E, live ATRA-treated AML HL-60 cells and ATRA-resistant subline HL-60-R2 cells were counted as described in Fig. 1, E and F. The data are shown as the mean ± S.E. (error bars) (n = 3). B and F, the NBT reduction capacities of ATRA-treated HL-60 and HL-60-R2 cells were also examined. The captured images represent HL-60 and HL-60-R2 cells 5 days after the ATRA (1 μm) or ethanol (vehicle) treatment (left). The graphs depict the percentage of NBT-positive cells 3 and 5 days after treatment (right). Counting was performed as described in Fig. 1, G and H. Error bars, mean ± S.E. (n = 3). PCAF expression in HL-60 (C and D) and HL-60-R2 cells (G and H) after the ATRA (1 μm) or ethanol (vehicle) treatment for the indicated period was examined by RT-qPCR (C and G) and immunoblotting (D and H). GAPDH and β-actin were used as the controls for RT-qPCR and immunoblotting, respectively. The RT-qPCR results were quantified as described in the legend to Fig. 1. For the immunoblotting, 50-μg extracts from HL-60 and HL-60-R2 cells were loaded onto each lane. I, PCAF mRNA levels in the normal granulocytes, APL primary cells, and HL-60 cells were determined using the Oncomine microarray database. The PCAF mRNA level was elevated in the normal granulocytes compared with APL primary and HL-60 cells. The data are shown as the mean ± S.E. (normal granulocytes, n = 3; APL cells, n = 2; HL-60 cells, n = 6). **, p < 0.01; ***, p < 0.001.
We then examined the correlation between ATRA-induced granulocytic differentiation and PCAF induction. We used a subline of the ATRA-resistant cell line HL-60, HL-60-R2 (34). ATRA did not induce cell growth arrest in these cells (Fig. 2E). However, NBT reduction capacity was increased by the ATRA treatment, revealing that ATRA-treated HL-60-R2 cells had differentiated into granulocytic cells (Fig. 2F). Thus, we found that ATRA has the ability to induce granulocytic differentiation in HL-60-R2 cells without inducing cell growth arrest. In accordance with the granulocytic differentiation in ATRA-treated HL-60-R2 cells, the expression of PCAF was significantly increased at the mRNA (Fig. 2G) and protein levels (Fig. 2H). Consistent with this finding, we found that the PCAF mRNA level was significantly higher in the normal granulocytes, compared with the APL and HL-60 cells (Fig. 2I) in the previous microarray data (35), available at the Oncomine database. These results strongly suggest that the ATRA-induced PCAF expression is associated with granulocytic differentiation but not with cell cycle arrest in leukemia cells.
ATRA Induces PCAF Expression in Vitro and in Vivo in Primary APL Cells
To investigate the ATRA-induced PCAF expression in primary APL cells, we isolated leukemia cells from the bone marrow of a primary APL patient (patient 1) and cultured them in vitro in the presence or absence of ATRA. Consistent with the results from the experiments using the leukemia cell lines, in primary APL cells, the ATRA-induced PCAF expression was strongly associated with granulocytic differentiation (Fig. 3A, left), judging from the increased NBT reduction capacity (Fig. 3A, middle) and the cell morphology, with segmented nuclei and lower nuclear/cytoplasmic ratios (Fig. 3A, right). These results indicated that the primary APL cells differentiated into mature granulocytic cells after the ATRA treatment, and this was accompanied by the induction of PCAF.
FIGURE 3.

ATRA induces PCAF expression in vitro and in vivo in primary APL cells. Leukemia cells were isolated from the bone marrow of APL patients (patients 1 and 2) and then cultured with ATRA (1 μm) or ethanol (vehicle). A and B (left), PCAF expression for the indicated period was examined by RT-qPCR. GAPDH was used as the control. The RT-qPCR results were quantified as described in the legend to Fig. 1. A and B (middle), granulocytic differentiation in ATRA- or ethanol-treated APL cells was evaluated by NBT reduction capacity as described in the legend to Fig. 1. The data are shown as the mean ± S.E. (error bars) (n = 3). A and B (right), morphological alterations were examined via Wright-Giemsa staining 7 days after the treatment with 1 μm ATRA or ethanol. The ATRA-treated APL cells exhibited differentiated morphologies. Scale bar, 20 μm. C, PCAF expression in leukemia cells from the additional APL patients (patients 3, 4, and 5) 5 days after the treatment with ATRA (1 μm) or ethanol (vehicle) was examined by RT-qPCR. D, the expression of PCAF was measured in the bone marrow of APL patients (patients 1 and 6) who were treated with ATRA-containing chemotherapy. Bone marrow samples were collected at the time of the diagnosis and at the end of each treatment course. In patient 1, a molecular complete response (CR) was not achieved through chemotherapy on day 30; however, molecular CR was achieved by the ATRA-containing therapy on days 58 and 105. In patient 6, morphological CR was achieved on day 41, and molecular CR was achieved on days 74 and 111. E, PCAF mRNA levels in the bone marrow of APL, AML, ALL, CML, and MDS patients and healthy donors were determined using the Oncomine microarray database. The data are shown as the mean ± S.E. (healthy donor, n = 74; APL, n = 37; AML, n = 505; ALL, n = 750; CML, n = 76; MDS, n = 206). ***, p < 0.001.
We further confirmed the ATRA-induced PCAF expression in primary APL cells from additional patients (patients 2–5) (Fig. 3B (left) for patient 2 and Fig. 3C for the remainder). The ATRA-induced granulocytic differentiation in the cells of patient 2 was confirmed by an increased NBT reduction capacity and granulocyte-like morphology (Fig. 3B, middle and right, respectively). These results indicate that ATRA induces the expression of PCAF not only in leukemia cell lines but also in primary APL cells in vitro.
Next, to prove that ATRA induces the expression of PCAF in vivo in APL patients, we measured the expression levels in the bone marrow cells of patients treated with an ATRA-containing regimen. RNA was purified from bone marrow cells taken from APL patients at the time of diagnosis (before the treatment) and at other time points, such as at the end of the treatment or during the follow-up. The quantitative RT-PCR (RT-qPCR) results in two APL patients (patients 1 and 6) showed that the expression of PCAF was markedly increased after the ATRA-containing therapy compared with before the treatment (Fig. 3D), indicating that ATRA induced the expression of PCAF in vivo. Furthermore, an analysis with the Oncomine microarray database revealed that the PCAF mRNA levels were significantly lower in bone marrow cells from APL and non-APL myeloid leukemia patients than in the cells from healthy donors (Fig. 3E) (36). Taken together, these data strongly imply that the elevated PCAF expression is closely linked to the state of normal granulocytic differentiation, which is blocked in both APL and non-APL leukemia patients.
ATRA Induces CBP/p300 but Not Tp60 Expression in NB4 and HL-60 Cells
Because we found a robust induction of PCAF expression upon ATRA treatment, we next asked whether this is unique to PCAF. To this end, we investigated the expression levels of other HATs, such as CBP, p300, and Tip60, in cells that were treated with ATRA. As the results show, the CBP and p300, but not Tip60, mRNA levels were elevated after the ATRA treatment in NB4 (Fig. 4, A–C) and HL-60 cells (Fig. 4, D-F). However, we were unable to observe an accumulation of CBP and p300 proteins in ATRA-treated NB4 cells, which may be due to a lack of a sensitive antibody or a lower accumulation of proteins (data not shown). Nevertheless, because we observed a significant accumulation of PCAF mRNA and protein in ATRA-treated cells, we decided to further investigate the involvement of PCAF in ATRA-dependent granulocytic differentiation.
FIGURE 4.
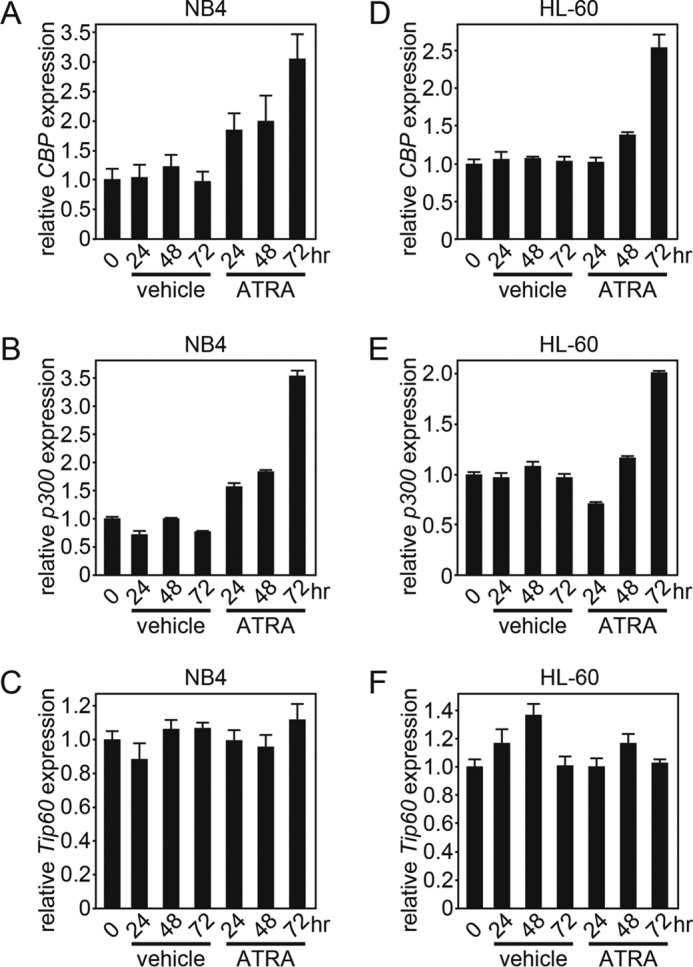
ATRA induces the PCAF coactivator CBP and p300 expression but not Tip60 expression in NB4 and HL-60 cells. CBP (A and D), p300 (B and E), and Tip60 (C and F) expression in NB4 (A–C) and in HL-60 (D–F) cells after the ATRA (1 μm) or ethanol (vehicle) treatment for the indicated period were analyzed by RT-qPCR. GAPDH was used as an internal control. The RT-qPCR results were quantified as described in the legend to Fig. 1. Error bars, S.E.
PCAF Knockdown Inhibits ATRA-induced Granulocytic Differentiation in Leukemia Cell Lines and Primary APL Cells
Based on the above mentioned results, we hypothesized that PCAF plays a critical role in ATRA-induced granulocytic differentiation in leukemia cells. To directly confirm this hypothesis, we performed a loss-of-function experiment using shRNA against PCAF. We generated the lentiviral vector pLKO2.0 with DsRed and three independent shRNA sequences against PCAF as well as non-targeting shRNA (negative control) (see “Experimental Procedures”). The expression of shRNA was induced by infecting NB4 cells with these viruses, and infected (DsRed-positive) cells were then isolated using a cell sorter. All three PCAF shRNAs knocked down PCAF at the mRNA (Fig. 5A) and the protein (Fig. 5B) levels 72 h after the infection, whereas the non-targeting shRNA did not.
FIGURE 5.

PCAF knockdown inhibits ATRA-induced granulocytic differentiation in NB4 cells and primary APL cells. A and B, cells expressing DsRed were sorted, and knockdown efficiency was then analyzed by RT-qPCR (A) and immunoblotting (B) for PCAF; GAPDH and β-actin were used as the controls for RT-qPCR and immunoblotting, respectively. Three independent sequences of shRNAs were used against PCAF. The RT-qPCR results were quantified by the comparative Ct method. Non-target control values were set as 1, and relative -fold values are depicted in the graph. Experiments were performed in triplicate, and the data are shown as the mean ± S.D. (error bars). For the immunoblotting, 10-μg extracts were loaded onto each lane. C, PCAF knockdown NB4 cells and control cells expressing non-targeting shRNA were cultured with 10 nm ATRA or ethanol (vehicle) for 72 h. DsRed and the expression of the granulocyte marker CD11b were monitored by FACS. FITC-conjugated IgG was used as the control. Infection efficiency was ∼30–60%, based on the proportion of cells expressing DsRed. D and E, the NBT reduction capacities of PCAF knockdown NB4 cells and control cells expressing non-targeting shRNA were also examined. The images were captured 72 h after treatment (D). Cells with NBT reduction capacity were counted and are depicted in the graph (E). Counting was performed as described in the legend to Fig. 1. F, a loss-of-function analysis using a lentiviral vector expressing shRNA against PCAF (gray lines) and non-targeting shRNA (black line) was performed using primary APL blast cells (patient 3). PCAF knockdown APL cells and control cells were cultured with different concentrations of ATRA (1 μm, 100 nm, and 10 nm) or ethanol (vehicle) for 96 h. DsRed and the expression of the granulocyte marker CD11b were monitored by FACS, and experiments were performed as described in the legend to Fig. 5C. Infection efficiency was ∼2–10%, based on the proportion of cells expressing DsRed.
To examine the involvement of PCAF in ATRA-induced granulocytic differentiation, PCAF or non-targeting shRNA were introduced into 30–60% of NB4 cells in the culture and treated with 10 nm ATRA or ethanol (vehicle) 48 h after the viral infection. As shown in Fig. 5C, the PCAF knockdown cells marked by Ds-Red showed no expression of CD11b, a marker for granulocytes, indicating that the knockdown of PCAF inhibited ATRA-induced granulocytic differentiation. In contrast, non-targeting shRNA-expressing cells labeled with DsRed clearly differentiated into granulocytes with a high expression level of CD11b. Furthermore, almost all cells not labeled with DsRed became CD11-positive cells (Fig. 5C). The blockade of granulocytic differentiation was also defined by the decreased levels of NBT reduction capacities, which were observed in the PCAF knockdown cells but not the control cells (Fig. 5, D and E).
Next, we assessed whether PCAF is required for ATRA-induced granulocytic differentiation in primary APL cells. PCAF or non-targeting shRNA was transduced with lentiviruses into APL primary cells freshly isolated from the bone marrow of a de novo APL patient (patient 3). The cells were then treated with ATRA and subsequently analyzed for CD11b expression levels. The PCAF knockdown APL cells and the control cells were cultured with different concentrations of ATRA (1 μm, 100 nm, and 10 nm) or ethanol (vehicle) for 96 h. FACS analysis was used to assess the expression of DsRed and CD11b. Infection efficiency ranged from ∼2 to 10% based on the proportion of cells expressing DsRed. Consistent with the results from the cell lines, the expression of CD11b was lower in the PCAF knockdown cells than in the control cells at all concentrations of ATRA (Fig. 5F). These results strongly suggest that the induction of PCAF is required for ATRA-induced granulocytic differentiation in the APL cell line NB4 and in primary APL cells. Furthermore, we found that the knockdown of PCAF blocked the ATRA-induced granulocytic differentiation in HL-60 cells (Fig. 6), implying that PCAF is required for ATRA-induced granulocytic differentiation, even in non-APL leukemia cells.
FIGURE 6.
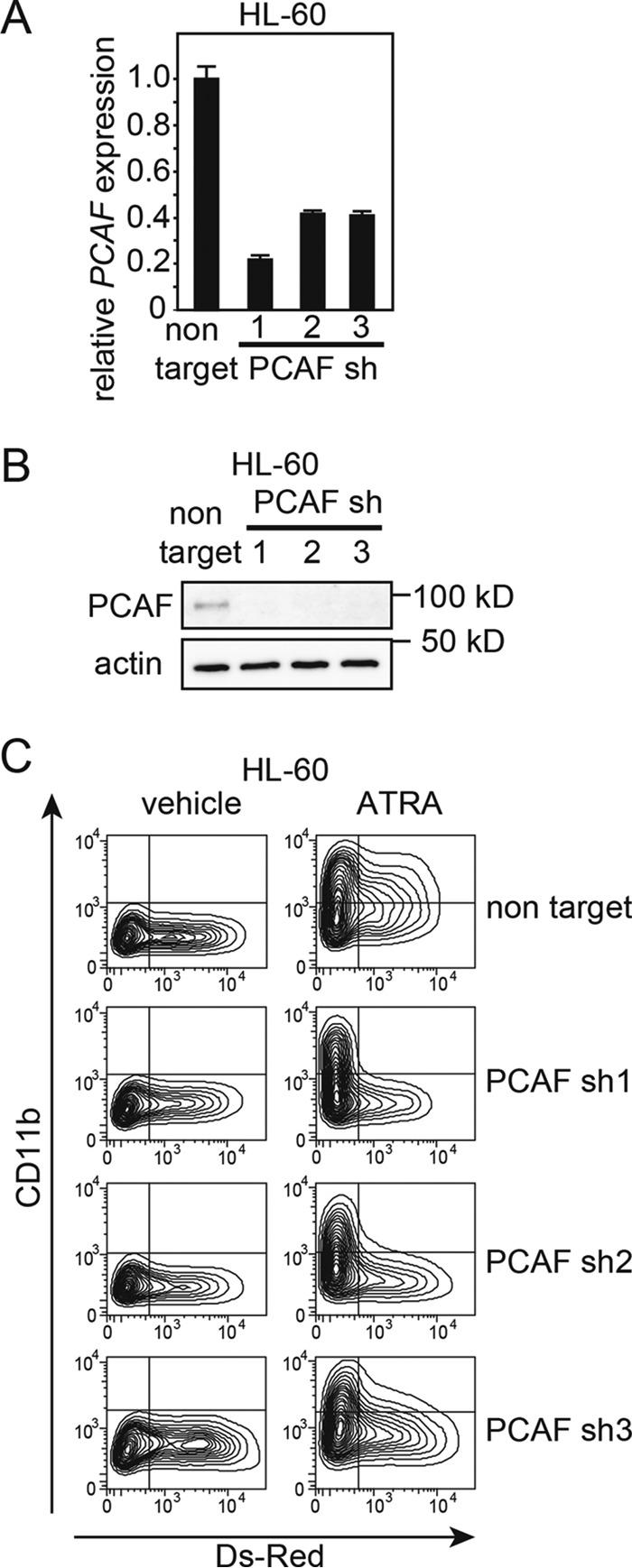
PCAF knockdown inhibits ATRA-induced granulocytic differentiation in HL-60 cells. A and B, HL-60 cells expressing DsRed were sorted, and knockdown efficiency was analyzed by RT-qPCR (A) and immunoblotting (B). Three independent sequences of shRNAs (the same sequences as in Fig. 5) were used against PCAF. The RT-qPCR results were quantified as described in the legend to Fig. 5. Experiments were performed in triplicate, and the data are shown as the mean ± S.D. (error bars). For the immunoblotting, 4.2 μg of extract was loaded into each lane. C, PCAF knockdown HL-60 cells and control cells expressing non-targeting shRNA were cultured with 10 nm ATRA or ethanol (vehicle) for 72 h. FACS analysis for monitoring DsRed and CD11b expression was then performed. Infection efficiency was ∼20–40% based on the proportion of cells expressing DsRed.
PCAF Overexpression Induces CD11b Expression in NB4 Cells
Although the loss-of-function assay using shRNA against PCAF revealed that PCAF is required for ATRA-induced granulocytic differentiation in leukemia cells, it is unclear whether the induction of PCAF expression is sufficient for granulocytic differentiation. To assess this, we examined the effects of the overexpression of PCAF in NB4 cells. NB4 cells were transiently transfected with FLAG-tagged PCAF or empty vector as a control by electroporation. We harvested cells at 8 and 24 h after transfection and subsequently extracted total RNA for RT-qPCR analysis. As shown in Fig. 7A, we confirmed the overexpression of PCAF at the mRNA level in NB4 cells after transfection. As expected, the PCAF overexpression induced CD11b at the mRNA level in NB4 cells (Fig. 7B). Contrary to our expectation, PCAF overexpression increased neither the NBT reduction capacity nor CD11b expression (data not shown), indicating that PCAF expression was not sufficient for the induction of granulocytic differentiation in NB4 cells, presumably due to a lack of an additional factor for granulocytic differentiation in NB4 cells (see “Discussion”).
FIGURE 7.
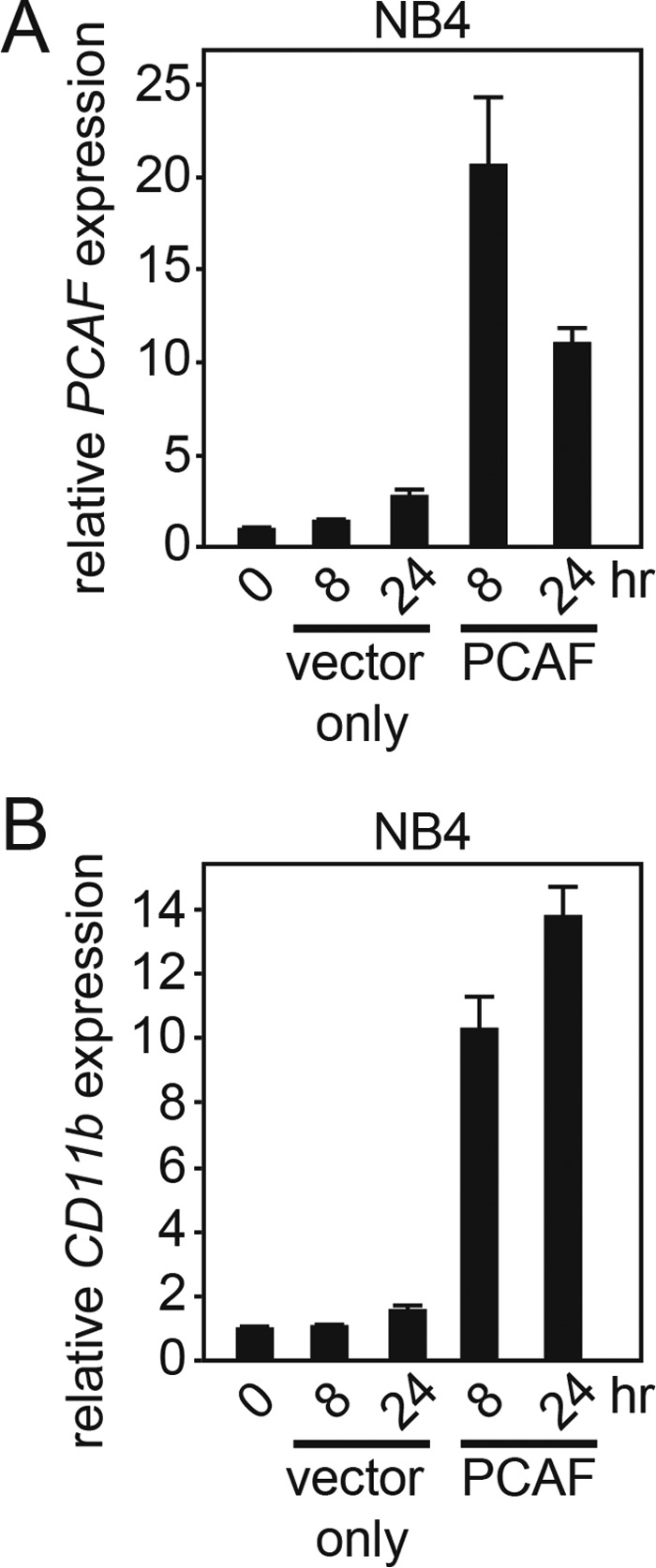
PCAF overexpression induces CD11b expression in NB4 cells. A and B, FLAG-PCAF or vector only (control) were transfected into NB4 cells by electroporation. PCAF and CD11b expression levels in these cells were analyzed by RT-qPCR at the indicated time points. The control values obtained from the NB4 cells before the transfection were set to 1, and relative -fold values are shown in the graph. Compared with vector-only-expressing NB4 cells, PCAF-overexpressing NB4 cells showed elevated CD11b expression at the mRNA level. Experiments were performed in triplicate, and the data are shown as the mean ± S.D. (error bars).
Identification of the Downstream Targets of PCAF in Response to ATRA Treatment
We attempted to identify the downstream targets of PCAF during ATRA-induced granulocytic differentiation in leukemia cells. Because ATRA-induced PCAF expression was more evident in HL-60 cells, we used HL-60 cells for the identification of PCAF acetylation targets. We initially investigated the subcellular localization of PCAF after the ATRA treatment. We separated the nuclear and cytoplasmic fractions of the cells and found that PCAF exclusively accumulated in the nuclear fraction after the ATRA treatment (Fig. 8A). To further confirm this, we performed an immunofluorescence staining analysis for ATRA-treated cells. However, none of the antibodies that we tried gave results consistent with the biochemical fraction (data not shown). The specificity of the antibody used for the immunoblot was verified by the shRNA experiments, and thus, we concluded that PCAF accumulated in the nucleus when the cells were treated with ATRA. Nevertheless, these data suggested that PCAF acetylates nuclear proteins in the course of granulocytic differentiation.
FIGURE 8.
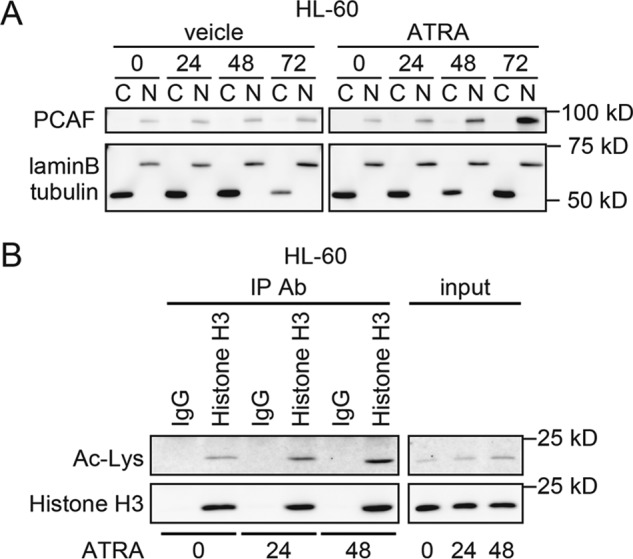
ATRA promotes the nuclear accumulation of PCAF and the acetylation of histone H3, a PCAF substrate in nuclear fractions. A, HL-60 cells were separated into nuclear and cytoplasmic fractions. PCAF expression after the ATRA (1 μm) or ethanol (vehicle) treatment for the indicated period was analyzed by immunoblotting. Lamin-B and α-tubulin were used to confirm cell fractionation. For the immunoblotting, 1.5-μg extracts were loaded onto each lane. B, the acetylation of histone H3 in HL-60 cells was analyzed after the ATRA (1 μm) treatment for the indicated period. 500 μg of whole lysates from mock- or ATRA-treated HL-60 cells were immunoprecipitated (IP) with an anti-histone H3 antibody (Ab) (1-μg input in each lane) and immunoblotted with an anti-acetyl-lysine antibody. IgG was used as the control. The total histone H3 levels were unchanged.
To identify the PCAF target in response to the ATRA treatment, an acetylome analysis was performed using mass spectrometry. Cell extracts were prepared from mock- or ATRA-treated HL-60 cells. Acetylated proteins were immunoprecipitated with an anti-acetyl lysine antibody and then analyzed using mass spectrometry (supplemental Table 1). We compared a list of acetylated proteins at 0, 3, and 24 h after the ATRA treatment. Of those compared, the following five proteins were significantly acetylated by the ATRA treatment: thymosin β-4 (TMSB4X), putative heat-shock protein HSP90-β2 (H90B2), asparagine-tRNA ligase, cytoplasmic (NARS), histone H3 (HIST1H3A), and probable ATP-dependent RNA helicase DDX17 (DDX17). Histone H3 and probable ATP-dependent RNA helicase DDX17 are nuclear proteins, and histone H3 is known to be an acetylation substrate of PCAF; thus, we focused on histone H3 and investigated whether ATRA acetylated histone H3 in HL-60 cells. Consistent with the mass spectrometry results, the ATRA treatment led to an accumulation of acetylated histone H3 (Fig. 8B), suggesting that ATRA directly acetylates the PCAF acetylation substrate histone H3, whereas ATRA induces granulocytic differentiation in leukemia cells.
PCAF Regulates the Expression of ATRA Target Genes
Because we have shown that PCAF is required for ATRA-induced granulocytic differentiation in leukemia cells, we investigated whether PCAF is directly involved in the regulation of expression of ATRA downstream target genes. To assess this, we initially confirmed the induction of chemokine (C-C motif) ligand 2 (CCL2) and Gardner-Rasheed feline sarcoma viral (v-fgr) oncogene homolog (FGR) by ATRA in NB4 and HL-60 cells (Fig. 9, A and B) (37–40). Then we investigated the expression levels of CCL2 and FGR mRNA in PCAF knockdown NB4 cells treated with or without ATRA. PCAF knockdown or control NB4 cells were first sorted with DsRed, and the PCAF knockdown efficiency at the mRNA level was subsequently confirmed (Fig. 9C). Then the cells were cultured in the presence of 10 nm ATRA or ethanol (vehicle) for 72 h. As shown previously in Fig. 5, the expression of CD11b was significantly lower in the PCAF knockdown cells than in the control cells, thus confirming that PCAF knockdown inhibited ATRA-induced granulocytic differentiation in NB4 cells (Fig. 9D). As shown in Fig. 9, E and F, in the PCAF knockdown cells but not the control cells, the induction of CCL2 and FGR expression by ATRA was greatly suppressed. This demonstrated that PCAF is involved in the expression of ATRA target genes.
FIGURE 9.

PCAF is required for the expression of ATRA target genes. A and B, the expression of ATRA downstream target genes CCL2 and FGR were analyzed by RT-qPCR in NB4 (A) and HL-60 (B) cells 48 h after ATRA (1 μm) or ethanol (vehicle) treatment. The RT-qPCR results were quantified as described in the legend to Fig. 1. Experiments were performed in triplicate, and the data are shown as the mean ± S.D. (error bars). C–F, PCAF knockdown NB4 cells expressing PCAF shRNA and control cells expressing non-targeting shRNA were sorted with DsRed (the same PCAF shRNA1 and non-targeting shRNA sequences as those used in Figs. 5 and 6). These cells were cultured with 10 nm ATRA or ethanol (vehicle) for 72 h. PCAF knockdown efficiency (C) and the expression of the granulocyte marker CD11b (D) and ATRA target genes CCL2 (E) and FGR (F) were then analyzed by RT-qPCR. Experiments were performed in triplicate, and the data are shown as the mean ± S.D. G and H, the ChIP assay was conducted using an anti-acetyl histone H3 antibody or IgG antibody as a negative control and examined via qPCR. By this assay, the acetylation status of histone H3 on the CCL2 (G) and the FGR (H) promoters in NB4 cells after the ATRA (1 μm) or ethanol (vehicle) treatment for 48 h was determined. The distal regions of CCL2 and FGR were used as negative controls. The data are shown as the mean ± S.D.
To further investigate the more direct involvement of PCAF in the regulation of ATRA-downstream target gene expression, the level of acetylation of PCAF substrate histone H3 on the promoter region of ATRA target genes was examined in ATRA-treated cells. By ChIP-quantitative PCR (qPCR) with anti-acetyl histone H3 antibody, the levels of acetylated histone H3 on the CCL2 and the FGR promoters were significantly elevated by the ATRA treatment (Fig. 9, G and H). These results suggest that, upon ATRA treatment, PCAF acetylates histone H3 on the promoters of its downstream target genes and subsequently promotes the expression of these genes for the induction of granulocytic differentiation.
Discussion
We showed that the induction of PCAF correlated with ATRA-induced granulocytic differentiation in the leukemia cell lines NB4, HL-60, and HL-60-R2 as well as in primary APL cells (Figs. 1–3). In contrast, ATRA did not induce the expression of PCAF in ATRA-resistant NB4RA cells (Fig. 1). PCAF knockdown inhibited ATRA-induced granulocytic differentiation in NB4, HL-60, and primary APL cells (Figs. 5 and 6). Furthermore, the overexpression of PCAF induced the expression of the granulocytic differentiation marker CD11b at the mRNA level in NB4 cells (Fig. 7). In agreement with this, we showed the elevation of acetylated histone H3, a substrate of PCAF, in the ATRA-treated cells (Fig. 8) and showed that acetylated histone H3 had accumulated on the promoters of genes that were induced by a PCAF-dependent manner upon ATRA treatment (Fig. 9). Based on these results, we propose a model in which ATRA promotes granulocytic differentiation in leukemia cells through the induction of PCAF expression, the subsequent acetylation of histone H3 or other PCAF targets, and the activation of downstream transcription (Fig. 10). Collectively, the enhanced expression of PCAF and the acetylation of its downstream substrates are promising strategies to induce granulocytic differentiation in leukemia cells for therapy.
FIGURE 10.
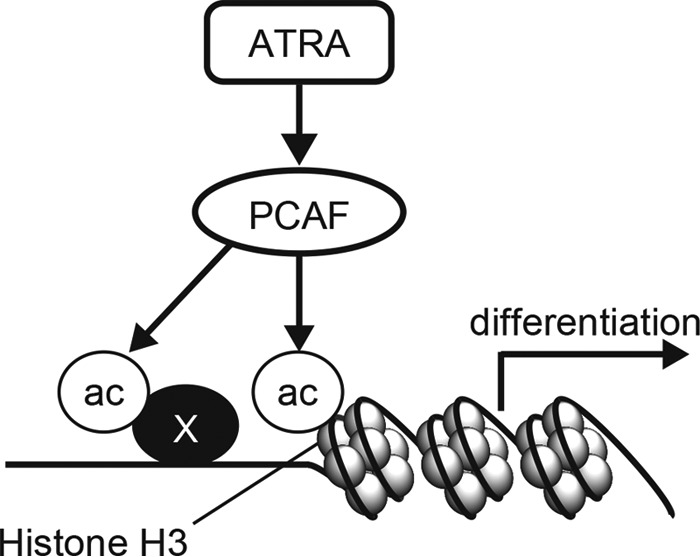
A hypothetical model in which ATRA induces granulocyte differentiation by promoting PCAF expression in APL cells. Based on our findings, we propose a model in which PCAF promotes the acetylation of histone H3 on the promoter of ATRA target genes and subsequently mediates the induction of the expression of these genes required for granulocytic differentiation in ATRA-treated leukemia cells. Although PCAF expression itself induced expression of some of the ATRA target genes, the cells did not differentiate into granulocytes (see “Results”; PCAF Overexpression Induces CD11b Expression in NB4 Cells). Thus, other mechanisms are involved in the completion of granulocytic differentiation in leukemia cells.
PCAF is a transcriptional coactivator that exhibits intrinsic histone acetylase activity, which contributes to transcriptional activation through the modification of histones and other proteins. Schiltz et al. (41) previously reported that PCAF predominantly acetylates histone H3 and, to a lesser extent, histone H4. Down-regulated PCAF has been reported in some types of cancer. The acetylation of histones H3 and H4 is virtually undetectable or very weak in acute leukemia cells (42, 43). Because PCAF is the HAT for histones H3 and H4, PCAF was predicted to be down-regulated in acute leukemia.
In agreement with this hypothesis, the expression of PCAF mRNA in the bone marrow cells in APL patients was low, but it was then elevated severalfold over the course of their treatment with ATRA-containing chemotherapy (Fig. 3D). Due to a limitation of clinical samples, we could not confirm the accumulation of PCAF protein in ATRA-treated APL primary cells. In addition, an analysis with the Oncomine microarray database revealed that the PCAF mRNA levels were significantly lower in bone marrow cells from APL and non-APL myeloid leukemia patients than in those from healthy donors (Fig. 3E). However, the down-regulated expression of PCAF was not observed in chronic myeloid leukemia (CML) or myelodysplastic syndrome (MDS), in which cancerous cells do not exhibit a strong defect in differentiation. Conversely, the down-regulation of PCAF expression may be important for maintaining the undifferentiated phenotype in acute leukemia.
In this study, we used two pairs of ATRA-sensitive and ATRA-resistant cell lines, NB4 and NB4RA, and HL-60 and HL-60R2 (Figs. 1 and 2). When comparing the cell proliferation rates between ATRA-sensitive and ATRA-resistant cells, we found that they were diverse (compare vehicle in Figs. 1 (E and F) and 2 (A and E)). Because these resistant cell lines were originally established from sensitive cell lines, the genetic background of the cell lines in each pair should be the same, except the part related to the ATRA-resistance, as is the case for the cellular property. Therefore, differential proliferation rates among isogenic cell lines were presumably established by the long duration culture in a different environment since establishment.
In addition to the loss-of-function assay (Figs. 5 and 6), we attempted to clarify the roles of PCAF HAT activity in ATRA-induced leukemia cell differentiation by using the PCAF inhibitor embelin. Contrary to our expectation, 30 μm embelin neither blocked the granulocytic differentiation nor inhibited the CCL2 and FGR expression by ATRA in NB4 cells (data not shown). Because embelin was highly toxic to the NB4 and HL-60 cells, it induced cell death, presumably inhibiting other molecules (44–47) before reaching an optimal concentration for the blockade of PCAF. The development of more specific compounds for PCAF is required to prove that the PCAF enzymatic activity is involved in the ATRA-induced granulocytic cell differentiation.
Although we found that the overexpression of PCAF induced the CD11b expression at the mRNA level in NB4 cells (Fig. 7), we failed to detect the elevation of CD11b by flow cytometry (data not shown). In addition, PCAF overexpression did not result in the increase of NBT reduction capacity in NB4 cells (data not shown). These data suggest that the induction of the granulocytic differentiation of leukemia cells requires additional components, despite the fact that PCAF is required for ATRA-induced leukemia cell differentiation (Figs. 5 and 6).
We demonstrated that the PCAF knockdown lowered the expression of the ATRA target genes CCL2 and FGR (Fig. 9). CCL2 is a member of the CC chemokine family, of which the secretion is increased upon the differentiation of monocytes into macrophages (48). FGR is a member of the Src family of non-receptor tyrosine kinases (49), of which the expression is restricted to myeloid cells and elevated in AML leukemic blasts that differentiate along the monocytic or granulocytic lineages (50). Although ATRA-induced PCAF promoted the expression of CCL2 and FGR, the knockdown of these genes did not inhibit ATRA-induced granulocytic differentiation (data not shown). Recently, the prolyl isomerase Pin1 has been identified as a key target of ATRA in APL cells. Pin1 plays a critical role in the eradication of APL-initiating cells but not in the granulocytic differentiation of APL cells (51). Therefore, although it is speculative at the present time, Pin1 may not be a downstream target molecule of PCAF. A more comprehensive genome-wide analysis is required to identify the downstream target genes of ATRA-induced PCAF, which play critical roles in granulocytic differentiation in leukemia cells.
For the induction of granulocytic differentiation in APL cells, ATRA binds to PML-RARα and wild type RARα, promoting the degradation of PML-RARα occupying RARα-binding sites of target genes (52) and modulating the wild type RARα for transcriptional activation (23), respectively. The regulated interaction of RARα and PCAF by a physiological concentration of retinoic acid in non-cancer cells was previously reported (30). Therefore, although acetylome analysis did not identify RARα as an acetylated protein after the ATRA treatment, it is conceivable that ATRA induces the formation of the PCAF-RARα complex and subsequently promotes the granulocytic differentiation in leukemia cells.
Other than RARα, PCAF may acetylate other transcription factors for the induction of granulocytic differentiation. These transcription factors include p53, which was previously reported to act with PCAF in the activation of downstream gene expression (53). However, it is unlikely that p53 is a substrate for PCAF in ATRA-treated cells because ATRA does not alter the acetylation level of p53 in NB4 cells (data not shown) and APL primary cells (54). More detailed analysis of PCAF substrates may lead to an identification of proteins where acetylation by PCAF plays crucial roles in ATRA-induced granulocytic differentiation in leukemia cells.
To understand the molecular mechanism underlying how PCAF is induced by ATRA, we analyzed the presumptive binding sites of transcription factors on the PCAF promoter by MATCH (55). We first hypothesized that RARα directly promoted PCAF expression upon ATRA treatment; however, we failed to find the RARα-binding motif on the PCAF promoter. Instead, we found sequences to which signal transducer and activator of transcription 3 (STAT3), activator protein 1 (AP-1), myogenic differentiation 1 (MyoD), and C/EBP may bind. Among these candidates, C/EBP is associated with normal myeloid development (56). Moreover, ATRA restored the activity of C/EBPα and induced the expression level of C/EBPβ and C/EBPϵ in APL cells, whereas ATRA induced granulocytic differentiation (57). Therefore, C/EBP is one of the potent transcription factors that may induce PCAF expression. Furthermore, the aberrant overexpression of microRNA (miR)-181a/b was detected in APL cells, and ATRA suppressed the miR-181a/b levels during ATRA-induced leukemia cell differentiation (58). Because PCAF expression is negatively regulated by miR-181a/b (59), the induction of PCAF by ATRA may be due to the reduction of miR-181a/b in APL cells.
In summary, we showed that ATRA induced the expression and accumulation of PCAF and the acetylation of histone H3, one of the PCAF substrates, on the promoters of ATRA target genes during granulocytic differentiation in leukemia cells. Despite the fact that PCAF expression is not sufficient for the granulocytic differentiation in leukemia cells, we demonstrated that PCAF is required for the ATRA-induced granulocytic differentiation. Because we cannot exclude the possibility that the acetylation of an as yet unidentified PCAF target other than histone H3 may play a crucial role in ATRA-induced granulocytic differentiation in leukemia cells, further study is required for a more comprehensive understanding of ATRA-induced differentiation in leukemia cells. A clearer understanding of the mechanism responsible for the actions of ATRA will lead to the development of a new therapeutic strategy to overcome all subtypes of AML.
Experimental Procedures
Cell Culture and Reagents
NB4 (32), NB4RA (a generous gift from Dr. Tomoki Naoe, Nagoya University) (31), HL-60 (obtained from the American Type Culture Collection), HL-60-R2 (obtained from the RIKEN BRC Cell Bank through the National Bio-Resource Project of MEXT, Japan) (34), and APL primary cells were cultured in Iscove's modified Dulbecco's medium (Invitrogen) with 10% (cell line) or 30% (primary cells) heat-inactivated fetal calf serum (HyClone), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified, 5% CO2 atmosphere. In accordance with the Declaration of Helsinki, informed consent was obtained from all of APL patients, and approval was obtained from the ethics committee of the Juntendo University School of Medicine (institutional review board approval 2012195). 293T cells were cultured in DMEM (Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich) at 37 °C in a humidified, 5% CO2 atmosphere. ATRA (Sigma-Aldrich) was dissolved in absolute ethanol at 1 mm. Dissolved compounds were stored at −20 °C until they were used.
Immunoblotting
Cell extraction and immunoblotting were performed as described previously (26). The chemiluminescence reaction was performed with Pierce ECL West Femto substrate (Thermo Fisher Scientific), and images were captured using an LAS-4000 system (Fuji Film, Tokyo, Japan). The following primary antibodies were used for immunoblotting: anti-PCAF (Cell Signaling, catalog no. 3378), anti-β-actin (Cell Signaling, catalog no. 4967), anti-acetyl-lysine (Cell Signaling, catalog no. 9441), anti-histone H3 (Cell Signaling, catalog no. 3638), anti-lamin-B (Santa Cruz Biotechnology, Inc., catalog no. sc-6216), and anti-α-tubulin (Sigma-Aldrich, catalog no. T5168). The following horseradish peroxidase-conjugated secondary antibodies were used: polyclonal goat anti-mouse IgG (Santa Cruz Biotechnology, catalog no. sc-2005), for anti-α-tubulin and anti-histone H3, and polyclonal goat anti-rabbit IgG (Santa Cruz Biotechnology, catalog no. sc-2004), for the other primary antibodies.
RT-qPCR
Total RNA was isolated from cells using a PureLink RNA minikit (Life Technologies, Inc.) and an RNeasy minikit (Qiagen, Hilden, Germany). cDNA was synthesized from 200–300 ng of RNA using a ReverTra Ace RT-qPCR kit (Toyobo, Osaka, Japan) according to the manufacturer's protocol. The synthesized cDNA (2 μl) was analyzed by RT-qPCR performed with THUNDERBIRD SYBR qPCR mix (Toyobo) in a CFX96 real-time system (Bio-Rad). In the experiments represented in Fig. 9, total RNA was extracted from the cells using TRIzol reagent (Life Technologies) followed by phenol-chloroform extraction and isopropyl alcohol precipitation. cDNA was synthesized from 400 ng of total RNA according to the protocol above. RT-qPCR was performed in a 7500 Fast real-time PCR system (Life Technologies). The following primers were used for RT-qPCR: PCAF (forward, AGGAAAACCTGTGGTTGAAGG; reverse, CAGTCTTCGTTGAGATGGTGC), CBP (forward, CAACCCCAAAAGAGCCAAACT; reverse, CCTCGTAGAAGCTCCGACAGT); p300 (forward, AGCCAAGCGGCCTAAACTC; reverse, TCACCACCATTGGTTAGTCCC), Tip60 (forward, GGGGAGATAATCGAGGGCTG; reverse, TCCAGACGTTTGTTGAAGTCAAT); CD11b (forward, CCCTGGTTCACCTCCTTC; reverse, CATGACATAAGGTCAAGGCTG); CCL2 (forward, CAGCCAGATGCAATCAATGCC; reverse, TGGAATCCTGAACCCACTTCT); FGR (forward: ACTATGAGGCTCGAACTGAGG, reverse: TCAGCTTGGATTGAGTCAACAG); and GAPDH (forward, AGCCACATCGCTCAGACAC; reverse, GCCCAATACGACCAAATCC). Quantification was performed using the relative standard curve method against the control, GAPDH.
Cell Proliferation and Morphological Analysis
The cell proliferation rate and viability were determined by a trypan blue dye exclusion assay using a TC10 automated cell counter (Bio-Rad). To examine cellular morphology, cells were cytospun, stained with Wright-Giemsa, and observed using a BX51 microscope (Olympus, Tokyo, Japan).
Granulocytic Differentiation
The NBT reduction capacity assay was performed to examine granulocytic differentiation as described previously (26). To investigate the expression of CD11b, cells were stained with a fluorescein isothiocyanate (FITC)-conjugated anti-CD11b antibody (Beckman-Coulter) and analyzed using a Navios Flow Cytometer (Beckman-Coulter). Dead cells were excluded with forward scatter/side scatter. The data obtained were further analyzed using FlowJo software (Tree Star).
Lentiviral shRNA Expression
To perform the knockdown experiments, the following shRNA sequences obtained from the RNAi consortium database (Broad Institute) and siDirect version 2.0 database were subcloned into the AgeI and EcoRI sites of the lentiviral vector pLKO2.0.DsRed, bearing DsRed in place of puromycin-resistance sequences: non-targeting sh, CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTG; PCAFsh1, CCGGGCAGATACCAAACAAGTTTATCTCGAGATAAACTTGTTTGGTATCTGCTTTTT; PCAFsh2, CCGGTGGCATGTCCATTAGCTATTTCTCGAGAAATAGCTAATGGACATGCCATTTTTG; and PCAFsh3, CCGGGGTACTACGTGTCTAAGAAATCTCGAGATTTCTTAGACACGTAGTACCTTTTT. Lentivirus production and concentration were performed as described previously (60). Approximately 100,000 cells were infected with the concentrated virus and then cultured for 48 h. Cells expressing DsRed were then sorted using MoFlo XDP flow cytometry (Beckman-Coulter). Hoechst dye (Life Technologies, catalog no. 33258) was used to stain and exclude dead cells. The sorted cells were used for the immunoblotting assay and real-time quantitative PCR following the procedures above, and the PCAF knockdown level was confirmed.
Acetylome Analysis
Acetylome analysis was performed as described previously (61) with minor modifications. In brief, cell lysate was precipitated with acetone, and the pellet was then dissolved in 50 mm NH4HCO3. Proteins were reduced by incubation with 20 mm DTT at 56 °C for 30 min and subsequently alkylated by being treated with 2-iodoacetamide at 37 °C for 30 min. Proteins were then digested by being incubated with 0.25 μg/ml trypsin (Promega) at 37 °C overnight. After dialysis with NETN buffer (50 mm Tris-HCl (pH 7.6), 150 mm NaCl, 1 mm EDTA, and 0.1% Nonidet P-40), acetylated peptides were collected by immunoprecipitation with a mixture of three different anti-acetyl-lysine polyclonal antibodies (Rockland and Cell Signaling Technology) using protein A beads. The beads were washed with NETN buffer and subsequently with NETN buffer without Nonidet P-40. Then the acetylated peptides were eluted with 0.1% trifluoroacetic acid and subsequently with 0.1% trifluoroacetic acid, 75% acetonitrile. The eluted solutions were combined, cleaned with ZipTip SCX (Millipore) according to the manufacturer's instructions, and then processed by a liquid chromatograph (EASY-nLC 1000) (Thermo Fisher Scientific) coupled to a Q Exactive hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific) with a nanospray ion source in positive mode. The acetylated peptides were separated with a nano-HPLC C18 capillary column (0.075 × 150 mm, 3 mm) (Nikkyo Technos, Tokyo, Japan). A 60-min gradient was used at a flow rate of 300 nl/min, including 5–35% B in 48 min and then 35–65% B in 12 min (solvent A, 0.1% formic acid; solvent B, 100% CH3CN, 0.1% formic acid). The resulting MS and MS/MS data were used to search the Swiss-Prot database using Proteome Discoverer (Thermo Fisher Scientific) with MASCOT search engine software (Matrix Science, London, UK).
Subcellular Fractionation
To separate cells into nuclear and cytoplasmic fractions, cells were washed with PBS containing 2 mm orthovanadate and 1 μm TSA and then solubilized in 10 mm HEPES, 3 mm MgCl2, 10 mm KCl, 5% glycerol, and 0.5% Nonidet P-40. After the samples were centrifuged, the supernatants containing cytosolic proteins were collected. The pellets, which contained the nuclear proteins, were then sonicated in radioimmune precipitation assay buffer (Cell Signaling) supplemented with 10 mm nicotinamide, 1 μm TSA, 1 mm orthovanadate, 20 mm sodium fluoride, 1 μg/ml aprotinin (Sigma-Aldrich), 2 μg/ml E-64 (Roche Applied Science), 1 μg/ml leupeptin (Roche Applied Science), 0.67 μg/ml bestatin (Calbiochem), 0.67 μg/ml pepstatin (Roche Applied Science), and 43.5 μg/ml PMSF (Sigma-Aldrich).
Immunoprecipitation
To obtain cell extracts for immunoprecipitation, cells were washed with PBS containing 0.1% Triton X-100, 5 mm EDTA, 43.5 μg/ml PMSF, 2 mm sodium vanadate, 10 mm β-glycerophosphate, 10 mm nicotinamide, and 1 mm sodium butyrate and were then solubilized in PBS containing 0.1% Triton X-100, 5 mm EDTA, 1 μg/ml aprotinin, 2 μg/ml E-64, 1 μg/ml leupeptin, 0.67 μg/ml bestatin, 0.67 μg/ml pepstatin, 2 mm sodium vanadate, 10 mm β-glycerophosphate, 10 mm nicotinamide, and 1 μm TSA. An equal amount of protein lysate was incubated with either normal rabbit IgG (Santa Cruz Biotechnology, catalog no. sc-2027) or anti-histone H3 antibody (Abcam (Cambridge, UK), catalog no. ab-1791) on ice, followed by an incubation with Dynabeads Protein G (Life Technologies). Immune complexes were analyzed by an immunoblotting analysis with anti-acetyl-lysine antibody (Cell Signaling, catalog no. 9441) and anti-histone H3 antibody (Cell Signaling, catalog no. 3638).
ChIP-qPCR
ChIP was performed as described previously (62) with minor modifications. In brief, cells were cross-linked with 1% formaldehyde (Thermo Fisher Scientific) in PBS at 25 °C for 10 min, and 2.5 m glycine at room temperature was added to stop the reaction. The fixed cells were then sequentially treated with three kinds of lysis buffer (buffer 1: 50 mm HEPES-KOH, 140 mm NaCl, 1 mm EDTA, 10% glycerol, 0.5% Nonidet P-40, and 0.25% Triton X-100; buffer 2: 10 mm Tris-HCl, 200 mm NaCl, 1 mm EDTA, and 0.5 mm EGTA; buffer 3: 50 mm Tris-HCl, 1% SDS, and 10 mm EDTA) with a proteinase inhibitor mixture (1 μg/ml aprotinin, 2 μg/ml E-64, 1 μg/ml leupeptin, 0.67 μg/ml bestatin, 0.67 μg/ml pepstatin, and 43.5 μg/ml PMSF) to obtain nuclear extracts. Chromatin samples were sheared by treatment with Covaris M220 (Covaris). Then 10 μg of sonicated samples was immunoprecipitated with 5 μg of antibodies against anti-acetyl-histone H3 (Millipore, catalog no. 06–599) or normal rabbit IgG (Santa Cruz Biotechnology, catalog no. sc-2027). The immunoprecipitates were eluted, and the cross-link was reversed. DNA fragments were purified using a PCR purification kit (Qiagen). The DNA samples were analyzed by qPCR using the ABI 7500 Fast system. The following primer sets were used for qPCR: CCL2 promoter region (+105 to +293 bp; forward, GCTCATAGCAGCCACCTTCA; reverse, GGCTCACCCCTTGTCCTTTT), CCL2 distal region (−11258 to −11123 bp; forward, GCAGATCAGACGGGTGGTTT; reverse, CCCCAAAGACTCTGCCTCAC), FGR promoter region (−251 to −135 bp; forward, GCAGAATTGGGCTCCAGGTA; reverse, TCAGGCTTCTCTTCCCGTGT-3′), and FGR distal region (+11538 to +11655 bp; forward, CCAGCGGAACTATGCCAACT; reverse, GGAGCGTGTCAGGGGTTCTA).
PCAF Overexpression
FLAG-tagged PCAF expression plasmid has been described previously (63, 64). A pcDNA3.1 vector was used as a control. Transient NB4 cell transfection with FLAG-PCAF or empty vectors was accomplished by using the Amaxa Nucleofector system and the Cell Line Nucleofector kit T (Lonza, Basel, Switzerland) with program T-001 for high viability, according to the manufacturer's recommendations; 5 μg of plasmid was used for each transfection. The transfection efficiency was typically 70–80%, as determined by the transfection of cells with a green fluorescent protein plasmid vector. Successful transfection was confirmed with RT-qPCR.
Statistical Analysis
Significance levels for comparisons between samples were determined by Student's t test. p < 0.05 was considered significant. All experiments were performed at least three times.
Author Contributions
Y. S., M. A., A. I., M. I., S. M., and N. K. conceived of and designed the projects. Y. S., M. A., Y. H., and S. K. performed the experiments. A. I. performed the acetylome analysis. Y. S., M. A., A. I., M. I., S. M., A. O., and N. K. analyzed the data. Y. S., M. A., A. I., and N. K. wrote the manuscript. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
We thank Mitsue Saito (Department of Breast and Endocrine Surgery, Juntendo University School of Medicine) for support of the Oncomine microarray database; Michiaki Koike (Department of Hematology, Juntendo University Shizuoka Hospital) for providing patient samples; Erina Hayashi for technical assistance; and Kaori Otsuki, Masaya Usui, and Aya Abe (Support Unit for Bio-Material Analysis, RIKEN BSI Research Resources Center) for technical help with the MS analysis. We also acknowledge the Division of Molecular and Biochemical Research, Proteomics and BioMolecular Science and Cell Biology, Juntendo University School of Medicine.
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grants 26893264 and 25118725; the Juntendo University Young investigator joint project award 2014 (Grant 2608); and the Ministry of Education, Culture, Sports, Science, and Technology (MEXT)-supported Program for the Strategic Research Foundation at Private Universities, and the MEXT Promotion Plan for the Platform of Human Resource Development for Cancer project. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1.
- HAT
- histone acetyltransferase
- ATRA
- all-trans-retinoic acid
- APL
- acute promyelocytic leukemia
- AML
- acute myeloid leukemia
- HDAC
- histone deacetylase
- PCAF
- p300/CREB-binding protein-associated factor
- PML
- promyelocytic leukemia
- RARα
- retinoic acid receptor α
- C/EBP
- CCAAT/enhancer-binding protein
- RT-qPCR
- quantitative RT-PCR
- qPCR
- quantitative PCR
- NBT
- nitro blue tetrazolium
- MDS
- myelodysplastic syndrome
- CML
- chronic myeloid leukemia
- miR
- microRNA
- CR
- complete response.
References
- 1. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 2. Roth S. Y., and Allis C. D. (1996) Histone acetylation and chromatin assembly: a single escort, multiple dances? Cell 87, 5–8 [DOI] [PubMed] [Google Scholar]
- 3. Spange S., Wagner T., Heinzel T., and Krämer O. H. (2009) Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 41, 185–198 [DOI] [PubMed] [Google Scholar]
- 4. Glozak M. A., Sengupta N., Zhang X., and Seto E. (2005) Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23 [DOI] [PubMed] [Google Scholar]
- 5. Ropero S., and Esteller M. (2007) The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh B. N., Zhang G., Hwa Y. L., Li J., Dowdy S. C., and Jiang S. W. (2010) Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 10, 935–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sakuma T., Uzawa K., Onda T., Shiiba M., Yokoe H., Shibahara T., and Tanzawa H. (2006) Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 29, 117–124 [PubMed] [Google Scholar]
- 8. Cang S., Feng J., Konno S., Han L., Liu K., Sharma S. C., Choudhury M., and Chiao J. W. (2009) Deficient histone acetylation and excessive deacetylase activity as epigenomic marks of prostate cancer cells. Int. J. Oncol. 35, 1417–1422 [DOI] [PubMed] [Google Scholar]
- 9. Halkidou K., Gaughan L., Cook S., Leung H. Y., Neal D. E., and Robson C. N. (2004) Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59, 177–189 [DOI] [PubMed] [Google Scholar]
- 10. Song J., Noh J. H., Lee J. H., Eun J. W., Ahn Y. M., Kim S. Y., Lee S. H., Park W. S., Yoo N. J., Lee J. Y., and Nam S. W. (2005) Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 113, 264–268 [DOI] [PubMed] [Google Scholar]
- 11. Sharma S., Kelly T. K., and Jones P. A. (2010) Epigenetics in cancer. Carcinogenesis 31, 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan O., and La Thangue N. B. (2012) HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 90, 85–94 [DOI] [PubMed] [Google Scholar]
- 13. Sun W. J., Zhou X., Zheng J. H., Lu M. D., Nie J. Y., Yang X. J., and Zheng Z. Q. (2012) Histone acetyltransferases and deacetylases: molecular and clinical implications to gastrointestinal carcinogenesis. Acta Biochim. Biophys. Sin. 44, 80–91 [DOI] [PubMed] [Google Scholar]
- 14. Muraoka M., Konishi M., Kikuchi-Yanoshita R., Tanaka K., Shitara N., Chong J. M., Iwama T., and Miyaki M. (1996) p300 gene alterations in colorectal and gastric carcinomas. Oncogene 12, 1565–1569 [PubMed] [Google Scholar]
- 15. Grimwade D., Ivey A., and Huntly B. J. (2016) Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 127, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith M. L., Cavenagh J. D., Lister T. A., and Fitzgibbon J. (2004) Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 351, 2403–2407 [DOI] [PubMed] [Google Scholar]
- 17. Mueller B. U., Pabst T., Osato M., Asou N., Johansen L. M., Minden M. D., Behre G., Hiddemann W., Ito Y., and Tenen D. G. (2002) Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood 100, 998–1007 [DOI] [PubMed] [Google Scholar]
- 18. Xu G., Nagano M., Kanezaki R., Toki T., Hayashi Y., Taketani T., Taki T., Mitui T., Koike K., Kato K., Imaizumi M., Sekine I., Ikeda Y., Hanada R., Sako M., et al. (2003) Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood 102, 2960–2968 [DOI] [PubMed] [Google Scholar]
- 19. Waxman S. (2000) Differentiation therapy in acute myelogenous leukemia (non-APL). Leukemia 14, 491–496 [DOI] [PubMed] [Google Scholar]
- 20. Miller W. H. Jr., and Waxman S. (2002) Differentiation induction as a treatment for hematologic malignancies. Oncogene 21, 3496–3506 [DOI] [PubMed] [Google Scholar]
- 21. Gocek E., and Marcinkowska E. (2011) Differentiation therapy of acute myeloid leukemia. Cancers 3, 2402–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeisig B. B., Kwok C., Zelent A., Shankaranarayanan P., Gronemeyer H., Dong S., and So C. W. (2007) Recruitment of RXR by homotetrameric RARα fusion proteins is essential for transformation. Cancer Cell 12, 36–51 [DOI] [PubMed] [Google Scholar]
- 23. de Thé H., and Chen Z. (2010) Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783 [DOI] [PubMed] [Google Scholar]
- 24. De Bellis F., Carafa V., Conte M., Rotili D., Petraglia F., Matarese F., Françoijs K. J., Ablain J., Valente S., Castellano R., Goubard A., Collette Y., Mandoli A., Martens J. H., de Thé H., et al. (2014) Context-selective death of acute myeloid leukemia cells triggered by the novel hybrid retinoid-HDAC inhibitor MC2392. Cancer Res. 74, 2328–2339 [DOI] [PubMed] [Google Scholar]
- 25. Leiva M., Moretti S., Soilihi H., Pallavicini I., Peres L., Mercurio C., Dal Zuffo R., Minucci S., and de Thé H. (2012) Valproic acid induces differentiation and transient tumor regression, but spares leukemia-initiating activity in mouse models of APL. Leukemia 26, 1630–1637 [DOI] [PubMed] [Google Scholar]
- 26. Sunami Y., Araki M., Hironaka Y., Morishita S., Kobayashi M., Liew E. L., Edahiro Y., Tsutsui M., Ohsaka A., and Komatsu N. (2013) Inhibition of the NAD-dependent protein deacetylase SIRT2 induces granulocytic differentiation in human leukemia cells. PLoS One 8, e57633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar P., Periyasamy R., Das S., Neerukonda S., Mani I., and Pandey K. N. (2014) All-trans-retinoic acid and sodium butyrate enhance natriuretic peptide receptor a gene transcription: role of histone modification. Mol. Pharmacol. 85, 946–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blanco J. C., Minucci S., Lu J., Yang X. J., Walker K. K., Chen H., Evans R. M., Nakatani Y., and Ozato K. (1998) The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 12, 1638–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kastner P., Lawrence H. J., Waltzinger C., Ghyselinck N. B., Chambon P., and Chan S. (2001) Positive and negative regulation of granulopoiesis by endogenous RARα. Blood 97, 1314–1320 [DOI] [PubMed] [Google Scholar]
- 30. Korzus E., Torchia J., Rose D. W., Xu L., Kurokawa R., McInerney E. M., Mullen T. M., Glass C. K., and Rosenfeld M. G. (1998) Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science 279, 703–707 [DOI] [PubMed] [Google Scholar]
- 31. Kitamura K., Kiyoi H., Yoshida H., Saito H., Ohno R., and Naoe T. (1997) Mutant AF-2 domain of PML-RARα in retinoic acid-resistant NB4 cells: differentiation induced by RA is triggered directly through PML-RARα and its down-regulation in acute promyelocytic leukemia. Leukemia 11, 1950–1956 [DOI] [PubMed] [Google Scholar]
- 32. Lanotte M., Martin-Thouvenin V., Najman S., Balerini P., Valensi F., and Berger R. (1991) NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 77, 1080–1086 [PubMed] [Google Scholar]
- 33. Breitman T. R., Selonick S. E., and Collins S. J. (1980) Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 77, 2936–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mori J., Suzuki S., Hara M., Kaneko A., Yamashita K., Kumagai M., Sakuma T., Kakizawa T., Yamazaki M., Takeda T., Miyamoto T., Ichikawa K., and Hashizume K. (1999) Characterization of two novel retinoic acid-resistant cell lines derived from HL-60 cells following long-term culture with all-trans-retinoic acid. Jpn. J. Cancer Res. 90, 660–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stegmaier K., Ross K. N., Colavito S. A., O'Malley S., Stockwell B. R., and Golub T. R. (2004) Gene expression-based high-throughput screening (GE-HTS) and application to leukemia differentiation. Nat. Genet. 36, 257–263 [DOI] [PubMed] [Google Scholar]
- 36. Haferlach T., Kohlmann A., Wieczorek L., Basso G., Kronnie G. T., Béné M. C., De Vos J., Hernández J. M., Hofmann W. K., Mills K. I., Gilkes A., Chiaretti S., Shurtleff S. A., Kipps T. J., Rassenti L. Z., et al. (2010) Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 28, 2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee K. H., Chang M. Y., Ahn J. I., Yu D. H., Jung S. S., Choi J. H., Noh Y. H., Lee Y. S., and Ahn M. J. (2002) Differential gene expression in retinoic acid-induced differentiation of acute promyelocytic leukemia cells, NB4 and HL-60 cells. Biochem. Biophys. Res. Commun. 296, 1125–1133 [DOI] [PubMed] [Google Scholar]
- 38. Burn T. C., Petrovick M. S., Hohaus S., Rollins B. J., and Tenen D. G. (1994) Monocyte chemoattractant protein-1 gene is expressed in activated neutrophils and retinoic acid-induced human myeloid cell lines. Blood 84, 2776–2783 [PubMed] [Google Scholar]
- 39. Liu T. X., Zhang J. W., Tao J., Zhang R. B., Zhang Q. H., Zhao C. J., Tong J. H., Lanotte M., Waxman S., Chen S. J., Mao M., Hu G. X., Zhu L., and Chen Z. (2000) Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood 96, 1496–1504 [PubMed] [Google Scholar]
- 40. Katagiri K., Yokoyama K. K., Yamamoto T., Omura S., Irie S., and Katagiri T. (1996) Lyn and Fgr protein-tyrosine kinases prevent apoptosis during retinoic acid-induced granulocytic differentiation of HL-60 cells. J. Biol. Chem. 271, 11557–11562 [DOI] [PubMed] [Google Scholar]
- 41. Schiltz R. L., Mizzen C. A., Vassilev A., Cook R. G., Allis C. D., and Nakatani Y. (1999) Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J. Biol. Chem. 274, 1189–1192 [DOI] [PubMed] [Google Scholar]
- 42. Xiao L., Huang Y., Zhen R., Chiao J. W., Liu D., and Ma X. (2010) Deficient histone acetylation in acute leukemia and the correction by an isothiocyanate. Acta Haematol. 123, 71–76 [DOI] [PubMed] [Google Scholar]
- 43. Lee S. M., Bae J. H., Kim M. J., Lee H. S., Lee M. K., Chung B. S., Kim D. W., Kang C. D., and Kim S. H. (2007) Bcr-Abl-independent imatinib-resistant K562 cells show aberrant protein acetylation and increased sensitivity to histone deacetylase inhibitors. J. Pharmacol. Exp. Ther. 322, 1084–1092 [DOI] [PubMed] [Google Scholar]
- 44. Modak R., Basha J., Bharathy N., Maity K., Mizar P., Bhat A. V., Vasudevan M., Rao V. K., Kok W. K., Natesh N., Taneja R., and Kundu T. K. (2013) Probing p300/CBP associated factor (PCAF)-dependent pathways with a small molecule inhibitor. ACS Chem. Biol. 8, 1311–1323 [DOI] [PubMed] [Google Scholar]
- 45. Nikolovska-Coleska Z., Xu L., Hu Z., Tomita Y., Li P., Roller P. P., Wang R., Fang X., Guo R., Zhang M., Lippman M. E., Yang D., and Wang S. (2004) Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J. Med. Chem. 47, 2430–2440 [DOI] [PubMed] [Google Scholar]
- 46. Schaible A. M., Traber H., Temml V., Noha S. M., Filosa R., Peduto A., Weinigel C., Barz D., Schuster D., and Werz O. (2013) Potent inhibition of human 5-lipoxygenase and microsomal prostaglandin E(2) synthase-1 by the anti-carcinogenic and anti-inflammatory agent embelin. Biochem. Pharmacol. 86, 476–486 [DOI] [PubMed] [Google Scholar]
- 47. Lin Z., Jensen J. K., Hong Z., Shi X., Hu L., Andreasen P. A., and Huang M. (2013) Structural insight into inactivation of plasminogen activator inhibitor-1 by a small-molecule antagonist. Chem. Biol. 20, 253–261 [DOI] [PubMed] [Google Scholar]
- 48. Fantuzzi L., Borghi P., Ciolli V., Pavlakis G., Belardelli F., and Gessani S. (1999) Loss of CCR2 expression and functional response to monocyte chemotactic protein (MCP-1) during the differentiation of human monocytes: role of secreted MCP-1 in the regulation of the chemotactic response. Blood 94, 875–883 [PubMed] [Google Scholar]
- 49. Miranda M. B., and Johnson D. E. (2007) Signal transduction pathways that contribute to myeloid differentiation. Leukemia 21, 1363–1377 [DOI] [PubMed] [Google Scholar]
- 50. Willman C. L., Stewart C. C., Longacre T. L., Head D. R., Habbersett R., Ziegler S. F., and Perlmutter R. M. (1991) Expression of the c-fgr and hck protein-tyrosine kinases in acute myeloid leukemic blasts is associated with early commitment and differentiation events in the monocytic and granulocytic lineages. Blood 77, 726–734 [PubMed] [Google Scholar]
- 51. Wei S., Kozono S., Kats L., Nechama M., Li W., Guarnerio J., Luo M., You M. H., Yao Y., Kondo A., Hu H., Bozkurt G., Moerke N. J., Cao S., Reschke M., Chen C. H., Rego E. M., Lo-Coco F., Cantley L. C., Lee T. H., Wu H., Zhang Y., Pandolfi P. P., Zhou X. Z., and Lu K. P. (2015) Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 21, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ablain J., and de The H. (2011) Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 117, 5795–5802 [DOI] [PubMed] [Google Scholar]
- 53. Liu L., Scolnick D. M., Trievel R. C., Zhang H. B., Marmorstein R., Halazonetis T. D., and Berger S. L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell Biol. 19, 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ablain J., Rice K., Soilihi H., de Reynies A., Minucci S., and de Thé H. (2014) Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat. Med. 20, 167–174 [DOI] [PubMed] [Google Scholar]
- 55. Kel A. E., Gössling E., Reuter I., Cheremushkin E., Kel-Margoulis O. V., and Wingender E. (2003) MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31, 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Friedman A. D. (2007) Transcriptional control of granulocyte and monocyte development. Oncogene 26, 6816–6828 [DOI] [PubMed] [Google Scholar]
- 57. Jing Y. (2004) The PML-RARα fusion protein and targeted therapy for acute promyelocytic leukemia. Leuk. Lymphoma 45, 639–648 [DOI] [PubMed] [Google Scholar]
- 58. Su R., Lin H. S., Zhang X. H., Yin X. L., Ning H. M., Liu B., Zhai P. F., Gong J. N., Shen C., Song L., Chen J., Wang F., Zhao H. L., Ma Y. N., Yu J., and Zhang J. W. (2015) MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene 34, 3226–3239 [DOI] [PubMed] [Google Scholar]
- 59. Zhao J., Gong A. Y., Zhou R., Liu J., Eischeid A. N., and Chen X. M. (2012) Downregulation of PCAF by miR-181a/b provides feedback regulation to TNF-α-induced transcription of proinflammatory genes in liver epithelial cells. J. Immunol. 188, 1266–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Araki M., Yang Y., Masubuchi N., Hironaka Y., Takei H., Morishita S., Mizukami Y., Kan S., Shirane S., Edahiro Y., Sunami Y., Ohsaka A., and Komatsu N. (2016) Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127, 1307–1316 [DOI] [PubMed] [Google Scholar]
- 61. Kosono S., Tamura M., Suzuki S., Kawamura Y., Yoshida A., Nishiyama M., and Yoshida M. (2015) Changes in the acetylome and succinylome of Bacillus subtilis in response to carbon source. PLoS One 10, e0131169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee T. I., Johnstone S. E., and Young R. A. (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1, 729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ito A., Lai C. H., Zhao X., Saito S., Hamilton M. H., Appella E., and Yao T. P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 20, 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ishfaq M., Maeta K., Maeda S., Natsume T., Ito A., and Yoshida M. (2012) Acetylation regulates subcellular localization of eukaryotic translation initiation factor 5A (eIF5A). FEBS Lett. 586, 3236–3241 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.