

Abstract
Cardiolipin (CL), the signature phospholipid of mitochondrial membranes, is important for cardiovascular health, and perturbation of CL metabolism is implicated in cardiovascular disease. Although the role of CL in mitochondrial function, biogenesis, and genome stability has been studied, recent findings indicate that it is essential for functions apart from mitochondrial bioenergetics. In this study, we report that mitophagy is perturbed in CL-deficient yeast cells. Mutants of autophagy/mitophagy genes ATG8, ATG18, and ATG32 synthetically interact with CL synthase mutant crd1Δ. CL-deficient cells exhibited decreased GFP-tagged mitochondrial proteins inside the vacuole and decreased free GFP, consistent with decreased mitophagy. Both PKC and high osmolarity glycerol (HOG) MAPK pathways were shown previously to be required for mitophagy. Activation of both MAPKs was defective in CL-deficient cells. Deletion of HOG pathway genes SHO1, SSK1, STE50, and HOG1 exacerbated crd1Δ growth. 1 m sorbitol and 0.2 m NaCl, which induce the HOG pathway, rescued growth of the mutant. Activation of the MAPK Slt2p was defective in crd1Δ cells, and up-regulation of the PKC pathway by expression of the PKC1R398P gene, which encodes constitutively activated Pkc1p, rescued crd1Δ growth and mitophagy defects. These findings indicate that loss of CL impairs MAPK pathway activation, and decreased activation of the PKC pathway leads to defective mitophagy.
Keywords: cardiolipin, mitochondria, mitogen-activated protein kinase (MAPK), mitophagy, protein kinase C (PKC)
Introduction
The anionic phospholipid cardiolipin (CL)2 is synthesized and primarily localized in the mitochondrial inner membrane. The importance of CL is underscored by the association of CL abnormalities with various cardiovascular diseases, including the life-threatening genetic disorder Barth syndrome (BTHS), diabetic cardiomyopathy, atherosclerosis, myocardial ischemia-reperfusion injury, heart failure, and Tangier disease (1). Among these, BTHS is directly linked to perturbation of CL metabolism, because it results from mutations in the CL remodeling enzyme tafazzin (TAZ/G4.5) (2, 3). BTHS manifests clinically as cardiomyopathy, skeletal myopathy, neutropenia, and growth retardation. The biochemical phenotypes at the cellular level include decreased CL levels, increased monolysocardiolipin, and altered CL fatty acyl composition (4–6). Interestingly, the clinical presentation of BTHS varies from asymptomatic to neonatal death, even among patients with identical tafazzin mutations, indicating that physiological modifiers of CL deficiency affect the clinical presentation of BTHS. Studies with yeast CL mutants indicate that CL plays an essential role in numerous cellular functions in addition to mitochondrial bioenergetics, which may be modifiers of the BTHS phenotype (7–12).
We have shown previously that the CL synthase mutant crd1Δ, which lacks CL, exhibits enlarged vacuoles, decreased vacuolar ATPase activity and proton pumping, and loss of vacuole acidification (10). The mechanism underlying vacuolar perturbation in CL-deficient cells is not understood. Autophagy, which can result in an influx of membrane and organelle contents to the vacuole, is one important route to affect the vacuolar size (13–15). Mitophagy is the selective degradation of mitochondria by autophagy in response to different stresses (16–18). Because the lack of CL leads to impaired mitochondrial function, including defective respiration (19), decreased mitochondrial membrane potential (20), and impaired mitochondrial protein import (12, 20), it is reasonable to speculate that CL deficiency might lead to increased mitophagy to eliminate damaged mitochondria, possibly resulting in enlarged vacuoles. Alternatively, perturbation of vacuolar function in CL mutant cells may deleteriously affect autophagy.
Mitophagy is regulated by two MAPK pathways, the PKC and high osmolarity glycerol (HOG) pathways (21). In response to induction of mitophagy, the PKC MAPK Slt2p is activated by phosphorylation of threonine and tyrosine residues. Deletion of the PKC pathway genes BCK1, MKK1/MKK2, and SLT2 leads to defective mitophagy. Similarly, perturbation of the HOG pathway by deletion of HOG1, PBS2, and SSK1 results in defective mitophagy. Interestingly, CL and/or the CL precursor phosphatidylglycerol (PG) are required for Slt2p phosphorylation, which is decreased in the pgs1Δ mutant, which cannot synthesize PG or CL (22).
Perturbation of vacuolar function and defective activation of the PKC pathway in CL mutants suggested that CL deficiency leads to perturbation of mitophagy as a result of defective MAPK activation. In the current study, we demonstrate that CL mutants exhibit defective mitophagy, which is rescued by up-regulation of the PKC pathway.
Results
Deletion of Autophagy/Mitophagy Genes Exacerbates crd1Δ Temperature Sensitivity
We previously observed that CL mutants exhibit vacuole defects characterized by enlarged vacuoles and loss of vacuole acidification at elevated temperature (10). To test the possibility that vacuole defects in CL-deficient cells resulted in perturbation of mitophagy, we examined genetic interactions between CRD1 and autophagy/mitophagy genes. If vacuolar defects resulted from increased mitophagy, deleting mitophagy/autophagy genes in crd1Δ would be expected to rescue temperature sensitivity of the mutant. In contrast, if crd1Δ defects resulted from decreased mitophagy, they would be exacerbated by deletion of autophagy/mitophagy genes.
Genetic interactions focused on three mitophagy ATG genes, ATG8, ATG18, and ATG32. During macromitophagy, mitochondria are brought to the phagophore assembly site (PAS), where the double membrane of the phagophore expands and closes to form a mature mitophagosome that contains mitochondria. Macromitophagy adapts the general autophagy core machinery, which involves various autophagy-related (ATG) proteins (23–29). Among these, Atg8p and Atg18p are essential components of the general autophagy core machinery. Atg8p, the yeast homolog of mammalian LC3, is conjugated to phosphatidylethanolamine (PE) (30). Atg8p-PE coats the mitophagosome membrane during membrane elongation. Atg18p binds to phosphatidylinositol 3-phosphate and regulates the retrieval of Atg9p from PAS. Normal transport of Atg9p between PAS and the peripheral site is essential for phosphatidylinositol 3-phosphate synthesis on the mitophagosome membrane (23). The mitochondrial outer membrane protein Atg32p plays a key role in selecting mitochondria as the specific cargo. When mitophagy is triggered, Atg32p is phosphorylated (31) and is involved in directing mitochondria to the PAS through interaction with other ATG machinery (31, 32). Analysis of the double mutants (crd1Δ and a mutation in ATG8, ATG18, or ATG32) indicated that deletion of these autophagy/mitophagy genes exacerbated the temperature sensitivity of crd1Δ (Fig. 1) (data not shown). Deletion of general autophagy genes ATG8 and ATG18 was more deleterious than deletion of the mitophagy-specific gene ATG32. These findings suggested that mitophagy may be decreased in crd1Δ cells.
FIGURE 1.
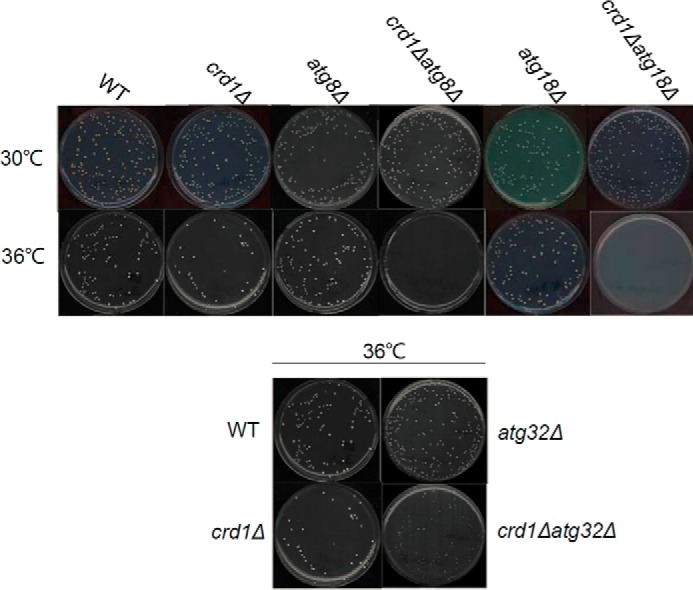
Deletion of autophagy/mitophagy genes exacerbates crd1Δ growth defect at elevated temperature. Cells in the FGY genetic background were precultured in YPD at 30 °C to the mid-log phase. About 200 cells of each strain were plated on YPD and incubated at 30 °C or 36 °C for 3 days.
Induction of Mitophagy Is Inhibited in crd1Δ Cells
We directly assayed mitophagy in WT and crd1Δ cells expressing the mitochondrial protein Idh1 tagged with GFP at the gene locus, as described previously (33). WT and crd1Δ cells expressing Idh1-GFP (WT-IDH1-GFP and crd1Δ-IDH1-GFP) were cultured in yeast extract peptone dextrose (YPD) medium to the mid-log phase at 30 °C and then switched to 39 °C for 8 h and observed under fluorescence microscopy. In WT cells, increased temperature led to vacuolar accumulation of GFP. However, translocation of Idh1-GFP was not observed in crd1Δ cells (Fig. 2).
FIGURE 2.
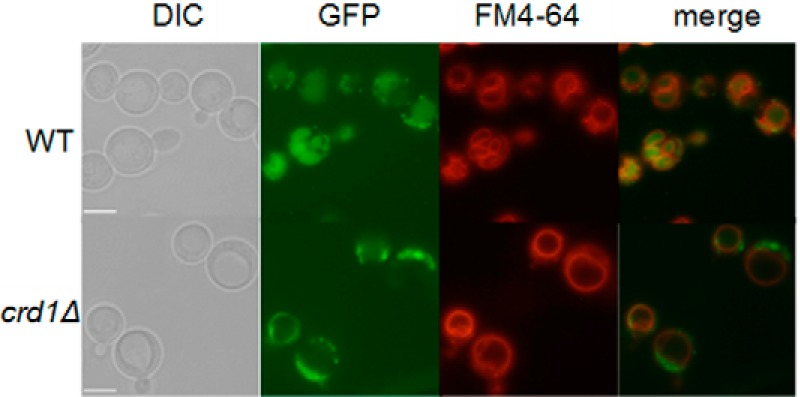
Delivery of mitochondria to the vacuole is decreased in crd1Δ cells. FGY WT and crd1Δ cells expressing a genomic copy of GFP-tagged IDH1 were cultured in YPD medium to the mid-log phase at 30 °C and then shifted to 39 °C for 8 h and observed under fluorescence microscopy. All images were taken at the same magnification (×1000). Bar, 2 μm.
Defective mitophagy was also observed in crd1Δ cells in the BY4742 genetic background, which is widely used in mitophagy studies (34). WT-IDH1-GFP and crd1Δ-IDH1-GFP cells were cultured in synthetic complete (SC) medium to the mid-log phase at 30 °C, harvested, washed twice, and resuspended in synthetic lactate (SL) medium for 18 h and observed under fluorescence microscopy. The accumulation of GFP was visualized in the vacuole of WT but not crd1Δ cells, suggesting that mitophagy was inhibited in the mutant (Fig. 3A). To exclude the possibility that the observed differences were specific to the Idh1-GFP protein, we carried out similar experiments in which the GFP-tagged mitochondrial protein Idp1p was exogenously expressed from the pCu416-IDP1-GFP plasmid. Similar to the findings with Idh1 (Fig. 3B), accumulation of Idp1-GFP was observed in the vacuole of the WT but not crd1Δ cells, consistent with decreased mitophagy in the mutant.
FIGURE 3.
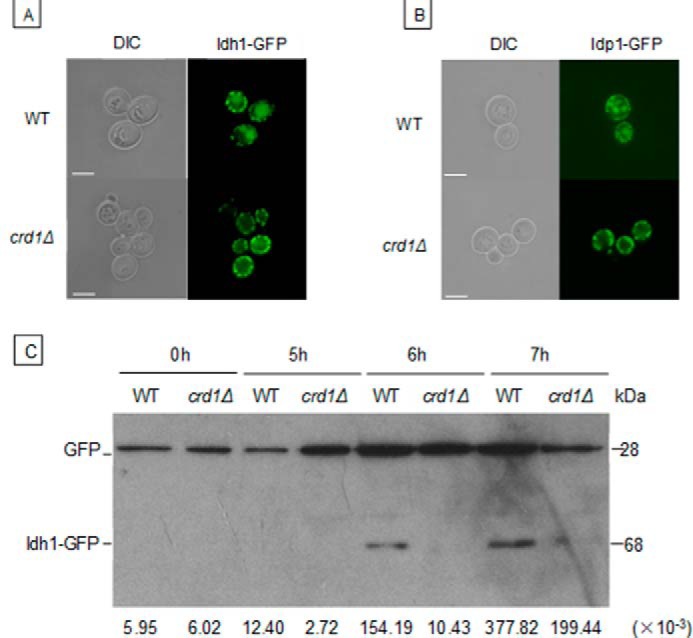
Decreased mitophagy in crd1Δ cells. WT and crd1Δ cells in the BY4742 genetic background expressing an endogenous Idh1-GFP (A) or exogenous Idp1-GFP (B) were cultured in SC medium to the mid-log phase at 30 °C and shifted to SL for 18 h. Cells were observed under fluorescence microscopy. C, WT and crd1Δ cells carrying the GFP-tagged IDH1 gene were precultured in SC medium to the mid-log phase at 30 °C and shifted to SL medium for the indicated time. Aliquots were analyzed by immunoblotting with anti-YFP antibody, and the positions of full-length Idh1-GFP and free GFP are indicated. Images in A and B were taken at the same magnification (×1000). Bar, 2 μm. Numbers at the bottom of C show the density ratios for GFP to Idh1-GFP in each lane. DIC, differential interference contrast.
Protein was extracted from WT-IDH1-GFP and crd1Δ-IDH1-GFP cells in which mitophagy was induced with SL medium, and the release of free GFP from endogenous Idh1-GFP was examined by Western blotting. A GFP band was detectable in WT cells from 6 h after transferring to SL medium but was barely detectable in crd1Δ cells (Fig. 3C). Preliminary evidence using an Idp1-GFP reporter and following a stationary phase mitophagy protocol as described previously (35) gave results consistent with a mitophagy defect in CL mutant cells (not shown). Taken together, these findings indicate that mitophagy is decreased in CL-deficient cells.
HOG Pathway Activation Is Impaired in crd1Δ Cells
The HOG pathway is an upstream regulator of mitophagy, and deletion of HOG pathway genes HOG1 and PBS2 leads to defective mitophagy (21). We investigated the possibility that the HOG pathway is perturbed in crd1Δ cells. Genetic interaction of CRD1 and HOG1 was assessed by growth of the crd1Δhog1Δ double mutant at elevated temperature. Mutants ssk1Δ, ste50Δ, sho1Δ, and hog1Δ were synthetically lethal with crd1Δ at elevated temperature (Fig. 4A). Moreover, overexpression of negative regulators of the HOG pathway, PTP2 and PTP3, were deleterious to growth of crd1Δ cells (Fig. 4B). Because constitutive activation of the HOG pathway causes lethality (36, 37), it is not feasible to determine whether up-regulating the pathway rescues the CL mutant defects. These findings indicated that down-regulation of the HOG pathway exacerbated the temperature sensitive growth defects of the CL mutant.
FIGURE 4.

Down-regulation of the HOG pathway exacerbates the crd1Δ growth defect. A, cells in the BY4742 genetic background were precultured in YPD at 30 °C to the mid-log phase. 200 cells of each strain were plated on YPD plates and incubated at the indicated temperature for 3 days. B, cells were precultured in SC leu− at 30 °C to the mid-log phase. Serial dilutions were spotted on SC leu− plates (with the most diluted spot containing 20 cells), and the plates were incubated at the indicated temperature for 2 days.
To determine whether the HOG pathway is impaired in crd1Δ cells, Hog1p activation was induced with 0.5 m NaCl for 5 min, and Hog1 phosphorylation was analyzed by Western blotting as described (8, 38). Compared with WT cells, Hog1 activation, as detected by phosphorylation, was decreased in crd1Δ in response to osmotic stress (Fig. 5A). However, translocation of phosphorylated Hog1-GFP into the nucleus was not decreased in mutant cells expressing the pPS1739 plasmid containing HOG1-GFP (Fig. 5B). We previously demonstrated that 1 m sorbitol, which induces the HOG pathway, rescued crd1Δ growth and vacuole defects (10). In agreement with this finding, 200 mm NaCl, which also activates the HOG pathway, similarly rescued crd1Δ temperature sensitivity (Fig. 5C). Taken together, these findings suggest that CL deficiency leads to decreased activation of the HOG pathway due to decreased phosphorylation of Hog1.
FIGURE 5.
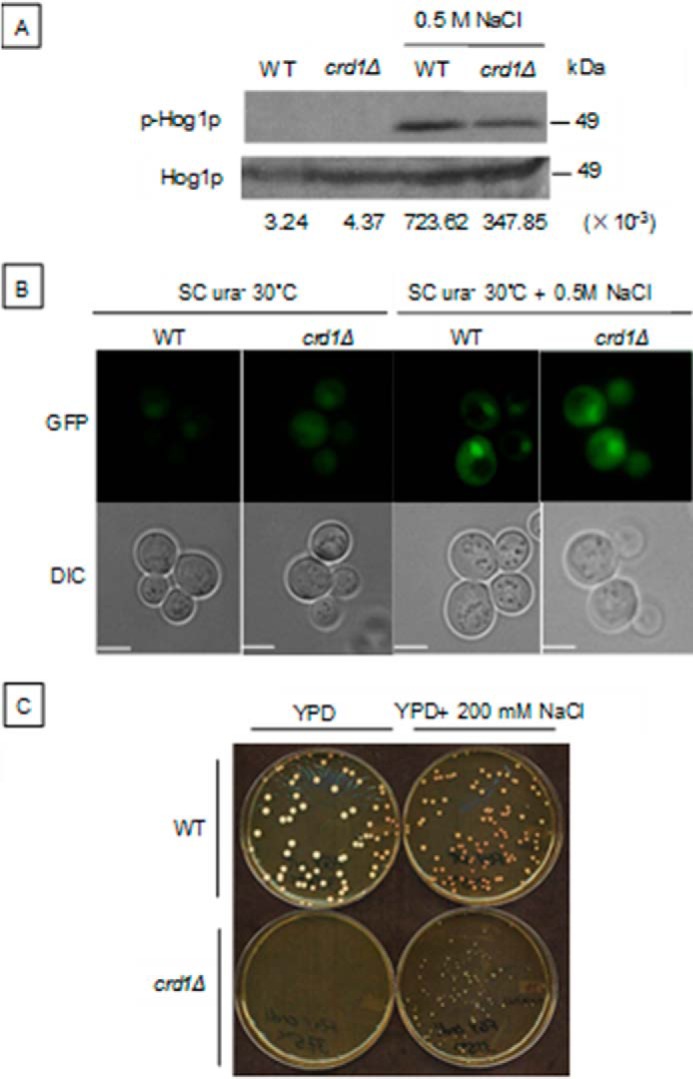
Decreased Hog1p phosphorylation in the crd1Δ mutant. A, cells in the BY4742 genetic background were precultured in YPD at 30 °C to the mid-log phase and treated with 0.5 m NaCl for 5 min. Dual phosphorylated (activated) and total Hog1 proteins were detected by Western blotting, as described previously (8). B, WT and crd1Δ cells expressing Hog1-GFP on the plasmid pPS1739 were grown in SC ura− to an A550 of 1, treated with 0.5 m NaCl for 5 min, and then observed using fluorescence microscopy. C, WT and crd1Δ cells in the FGY background were precultured in YPD liquid at 30 °C to the mid-log phase. 200 cells were plated on a YPD plate with or without 200 mm NaCl. Plates were then incubated at 37.5 °C for 3 days. Numbers at the bottom of A show the density ratios for p-Hog1p to Hog1p in each lane. Images in B were taken at the same magnification (×1000). Bar, 2 μm. DIC, differential interference contrast.
Up-regulation of the PKC Pathway Rescues Mitophagy in crd1Δ Cells
Similar to the HOG pathway, the PKC pathway is also a regulator of mitophagy (17). Deletion of PKC pathway genes BCK1, MKK1/MKK2, and SLT2 leads to defective mitophagy (21). As mentioned above, a previous study indicated that activation of Slt2p is decreased in pgs1Δ cells, which lack both PG and CL (22). We wished to determine whether PG can substitute for CL for activation (dual phosphorylation) of Slt2p. Compared with WT, dual phosphorylation of Slt2p was significantly decreased in both crd1Δ and pgs1Δ cells after 2 h at 39 °C (Fig. 6A).
FIGURE 6.
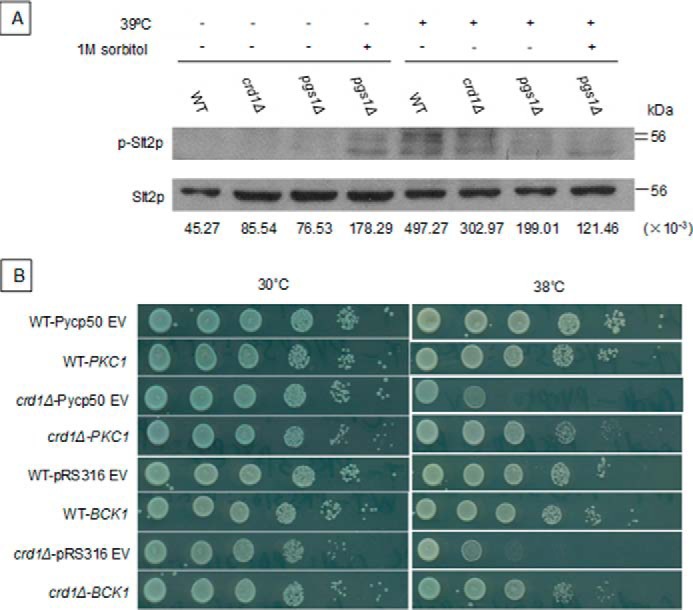
Up-regulation of the PKC pathway rescues the crd1Δ growth defect. A, cells in the BY4742 genetic background were precultured in liquid YPD at 30 °C to the mid-log phase, diluted to an A550 of 0.3, and treated with increased temperature or 1 m sorbitol for 2 h. Cell extracts were prepared and activated (dual phosphorylated), and total Slt2 protein was detected by Western blotting. B, cells expressing BCK1-20 and PKC1R398P plasmids were cultured in SC ura− medium at 30 °C to the mid-log phase, diluted, spotted on SC ura− plates, and incubated at 30 or 38 °C for 2 days. Numbers at the bottom of A show the density ratios for p-Slt2p to Slt2p in each lane.
Interestingly, up-regulation of the PKC pathway by expression of BCK1-20 and PKC1R398P genes, which encode constitutively activated proteins, rescued crd1Δ temperature sensitivity (Fig. 6B). The effect of PKC1R398P expression on delivery of mitochondria to the vacuole in response to activation of mitophagy was investigated in WT-IDH1-GFP and crd1Δ-IDH1-GFP strains. Accumulation of GFP was observed in the vacuole of WT-IDH1-GFP cells containing either the empty vector or vector expressing PKC1R398P. Consistent with the previous experiment (Fig. 3A), GFP accumulation was not observed in crd1Δ-IDH1-GFP cells containing the empty vector. However, expression of PKC1R398P led to accumulation of GFP in crd1Δ-IDH1-GFP cells, consistent with restoration of mitophagy (Fig. 7A). The release of free GFP was assessed by Western blotting. After 6 h of SL induction, free GFP was observed in crd1Δ-IDH1-GFP cells expressing PKC1R398P to a level comparable with that of WT-IDH1-GFP cells (Fig. 7B). These findings suggest that up-regulation of the PKC pathway restores mitophagy in crd1Δ cells.
FIGURE 7.
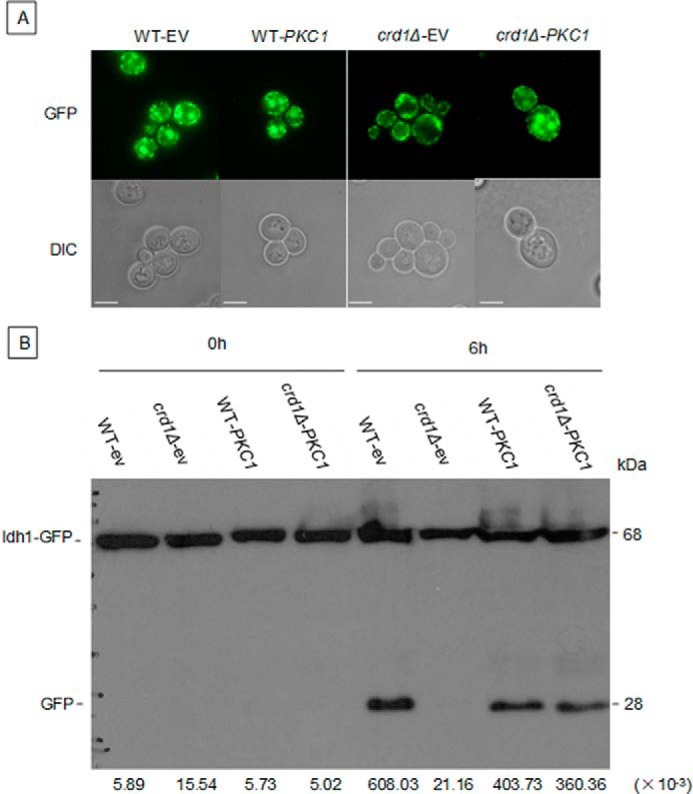
Up-regulation of the PKC pathway restores mitophagy in crd1Δ. BY WT-IDH1-GFP and crd1-IDH1-GFP cells containing either an empty vector or a vector expressing PKC1R398P were cultured in SC ura− medium to the mid-log phase at 30 °C, washed, and shifted to SL (to induce mitophagy). Aliquots were observed 18 h after shift under fluorescence microscopy (A), and full-length Idh1-GFP and free GFP were identified by Western blotting using anti-GFP antibody 6 h after shift (B). Images in A were taken at the same magnification (×1000). Bar, 2 μm. Numbers at the bottom of B show the density ratios for GFP to Idh1-GFP in each lane. DIC, differential interference contrast.
Discussion
In this study, we show that decreased activation of the MAPK Slt2 in CL-deficient cells leads to decreased mitophagy. Our specific findings indicated that 1) deletion of autophagy/mitophagy genes exacerbated crd1Δ growth defects; 2) GFP-tagged mitochondrial proteins were largely decreased in the vacuole of crd1Δ cells in conditions that induce mitophagy; 3) activation of HOG and PKC MAPK pathways required for mitophagy were defective in crd1Δ cells; and 4) increased activation of Slt2p phosphorylation rescued growth and mitophagy defects in the CL mutant.
Interestingly, deletion of general autophagy genes required for all forms of macroautophagy, including mitophagy, exacerbated crd1Δ growth defects to a greater extent than deletion of mitophagy-specific genes. As seen in Fig. 1B, crd1Δ is synthetically lethal with atg8Δ and atg18Δ but only synthetically sick with atg32Δ. This suggests that CL is required for mitophagy but not for nonselective autophagy. In this scenario, if nonselective autophagy functions normally in crd1Δ, damaged mitochondria may be delivered to the vacuole for degradation as cargo in the nonselective autophagosome. If nonselective autophagy is blocked by deletion of ATG8 or ATG18, cells will be unable to eliminate damaged mitochondria. If mitophagy is further impaired by the deletion of ATG32, a portion of damaged mitochondria may be eliminated by nonselective autophagy, resulting in a sick but not lethal phenotype. Consistent with this possibility, mitophagy is not completely absent from crd1Δ cells. A small band corresponding to free GFP was detected by Western blotting in crd1Δ-IDH1-GFP extracts after induction of mitophagy (Fig. 3C), and phosphorylation of Slt2 and Hog1 upon induction by heat or osmotic stress was decreased but not completely blocked in the mutant (Figs. 5A and 6A).
Both MAPK pathways that are required for mitophagy are perturbed in the crd1Δ mutant, which exhibited decreased phosphorylation of Hog1 (Fig. 5A) and Slt2 (Fig. 6A). Up-regulation of the PKC pathway by expressing constitutively activated PKC pathway effectors rescued the growth and mitophagy defects of crd1Δ cells (Figs. 6B and 7). Because constitutive activation of the HOG pathway causes lethality, it is not feasible to determine whether up-regulating this pathway rescues the CL mutant defects. Physiological conditions that activate the HOG pathway, including 1 m sorbitol and 0.2 m NaCl, rescue crd1Δ temperature sensitivity. However, because sorbitol and NaCl also regulate other parameters (e.g. osmotic and ion homeostasis), it cannot be concluded that perturbation of the HOG pathway causes defective mitophagy in crd1Δ cells.
Several studies suggest that tafazzin deficiency, which results in decreased CL, increased monoloyso-CL, and decreased unsaturated CL species, leads to defective mitophagy. Tafazzin-deficient yeast cells are synthetically lethal with mutants lacking the catalytic subunit of the mitochondrial inner membrane i-AAA protease (39). In tafazzin-deficient primary mouse embryonic fibroblasts, the formation of mitophagosomes is impaired (40). Kagan and co-workers (41) observed that externalization of CL to the outer mitochondrial membrane acts as an elimination signal for the mitophagic machinery in human neuronal cells. Our findings are the first to implicate CL-mediated activation of PKC in the regulation of mitophagy.
The mechanism whereby CL deficiency impairs PKC and HOG pathway activation is not known. One possibility is suggested by the finding that, in yeast mitochondria, CL and PG specifically bind to and activate inositol phosphosphingolipid phospholipase C (Isc1), which hydrolyzes complex sphingolipids to generate ceramide (39). Similarly, mitochondria-associated neutral sphingomyelinase, the mammalian orthologue of Isc1, was shown to be activated by anionic phospholipids, especially phosphatidylserine and mitochondrial CL (42). In mammals, ceramide activates PKC-ζ (43, 44) but inhibits PKC-θ and PKC-δ in mammals (45, 46). Yeast has only one PKC (Pkc1) isozyme, and its regulation by ceramide has not been studied. Thus, the role of CL in the regulation of the PKC pathway via ceramide is an exciting area for future study.
In summary, we demonstrate for the first time that loss of CL leads to decreased activation of PKC and HOG pathways, resulting in impaired mitophagy. These findings provide novel insights into the cellular functions of CL outside of mitochondrial bioenergetics.
Experimental Procedures
Yeast Strains and Growth Media
The Saccharomyces cerevisiae strains and plasmids used in this study are listed in Table 1. YPD medium contained yeast extract (1%), peptone (2%), and glucose (2%). YPDS medium is YPD medium supplemented with 1 m sorbitol. Sporulation medium contained potassium acetate (1%), glucose (0.05%), and amino acids needed for auxotrophies. SC medium contained laboratory-made vitamin-free yeast nitrogen base without amino acids (Difco protocol, 0.17%), ammonium sulfate (0.5%), glucose (2%), vitamins, and the following supplements: adenine (20.25 mg/liter), arginine (20 mg/liter), histidine (20 mg/liter), leucine (60 mg/liter), lysine (200 mg/liter), methionine (20 mg/liter), threonine (300 mg/liter), tryptophan (20 mg/liter), and uracil (20 mg/liter). Synthetic dropout medium contained all ingredients mentioned above except the amino acid identified by the selective marker. SL medium contained laboratory-made vitamin-free yeast nitrogen base without amino acids (Difco protocol, 0.17%), ammonium sulfate (0.5%), and lactate (2%). Solid medium contained agar (2%).
TABLE 1.
Yeast strains and plasmids used in this study
| Strain/plasmid | Characteristics or genotype | Source |
|---|---|---|
| FGY3 (WT) | MAT α, ura3-52, lys2-801, ade2-101, trp1Δ1, his3Δ200, leu2Δ1 | Jiang et al. (50) |
| FGY2 (crd1Δ) | MAT α, ura3-52, lys2-801, ade2-101, trp1Δ1, his3Δ200, leu2Δ1, crd1Δ::URA3 | Jiang et al. (50) |
| FGY3 atg8Δ | Derivative of FGY3, atg8Δ::KanMX4 | This study |
| FGY2 atg8Δ | Derivative of FGY2, atg8Δ::KanMX4 | This study |
| FGY3 atg18Δ | Derivative of FGY3, atg18Δ::KanMX4 | This study |
| FGY2 atg18Δ | Derivative of FGY2, atg18Δ::KanMX4 | This study |
| FGY3 atg32Δ | Derivative of FGY3, atg32Δ::KanMX4 | This study |
| FGY2 atg32Δ | Derivative of FGY2, atg32Δ::KanMX4 | This study |
| FGY3 atg21Δ | Derivative of FGY3, atg21Δ::TRP1 | This study |
| FGY3 atg18Δatg21Δ | Derivative of FGY3, atg18Δ::KanMX4, atg21Δ::TRP1 | This study |
| FGY2 atg18Δatg21Δ | Derivative of FGY2, atg18Δ::KanMX4, atg21Δ::TRP1 | This study |
| FGY3-IDH1-GFP | Derivative of FGY3, in which GFP is Inserted into the IDH1 gene locus | This study |
| FGY2-IDH1-GFP | Derivative of FGY2, in which GFP is Inserted into the IDH1 gene locus | This study |
| BY4742 (WT) | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0 | Euroscarf a |
| VGY1 (BY4742 crd1Δ) | Derivative of BY4742, crd1Δ::URA3 | Gohil et al. (51) |
| BY4742 sho1Δ | Derivative of BY4742, sho1Δ:: KanMX4 | This study |
| VGY1sho1Δ | Derivative of BY4742, crd1Δ::URA3, sho1Δ::KanMX4 | This study |
| BY4742 ssk1Δ | Derivative of BY4742, ssk1Δ:: KanMX4 | This study |
| VGY1ssk1Δ | Derivative of BY4742, crd1Δ::URA3, ssk1Δ::KanMX4 | This study |
| BY4742 ste50Δ | Derivative of BY4742, ste50Δ::KanMX4 | This study |
| VGY1ste50Δ | Derivative of BY4742, crd1Δ::URA3, ste50Δ::KanMX4 | This study |
| BY4742 hog1Δ | Derivative of BY4742, hog1Δ::KAN | This study |
| VGY1hog1Δ | Derivative of BY4742, crd1Δ::URA3, hog1Δ::KanMX4 | This study |
| BY4742 WT-IDH1-GFP | Derivative of BY4742, in which IDH1 gene is chromosomally tagged with GFP | This study |
| BY4742 crd1Δ- IDH1-GFP | Derivative of BY4742 crd1Δ, in which IDH1 gene is chromosomally tagged with GFP | This study |
| pYPGK18 | 2 μm, LEU2 | Vaz et al. (52) |
| pYPGK18-PTP2 | Derivative of pYPGK18, expresses PTP2 from PGK1 promoter | This study |
| pYPGK18-PTP3 | Derivative of pYPGK18, expresses PTP3 from PGK1 promoter | This study |
| pYCp50 | CEN, URA3 | Helliwell et al. (53) |
| pYCp50-PKC1R398P | Derivative of pYCp50, expresses PKC1R398P from PGK1 promoter | Helliwell et al. (53) |
| pRS352 | 2 μm, URA3 | Helliwell et al. (53) |
| pRS352-BCK1-20 | Derivative of pRS316, expresses BCK1-20 from PGK1 promoter | Helliwell et al. (53) |
| pPS1739 | CEN, URA3, HOG1-GFP | Ferrigno et al. (54) |
| pFA6a-GFP(S65T)-His3MX6 | Derivative of pFA6a-His3MX6 | Kanki et al. (47) |
| pCu416-IDP1-GFP | Derivative of pCu416, expresses GFP-tagged IDP1 from CUP1 promoter | Journo et al. (55) |
a Euroscarf, European Saccharomyces Cerevisiae Archive for Functional analysis.
The ATG deletion mutants were constructed as follows. The entire open reading frame of the target gene was replaced by a KanMX4 cassette via homologous recombination in WT or crd1Δ cells. The KanMX4 cassette was amplified by PCR using the pUG6 plasmid, which contains the KanMX4 cassette as the template. The amplification primers consisted of 50 nucleotides that were identical to the upstream or downstream flanking region of the target gene at the 5′ end and 21 nucleotides that annealed to the KanMX4 gene at the 3′ end. Transformants were selected on YPD plates with 200 μg/ml G418. Deletion of the target gene was confirmed by PCR using primers that amplified the original target gene.
WT and crd1Δ cells that express the mitochondrial matrix protein Idh1p tagged by GFP at the gene locus were constructed as described previously (47). The pFA6a-GFP (S65T)-HIS3MX6 plasmid that was used as the PCR template to amplify the DNA fragment encoding GFP was kindly provided by Dr. Daniel Klionsky (University of Michigan).
Plasmid Construction and Cloning
To construct the PTP2 overexpression plasmid, a sequence of 2250 base pairs that includes the entire open reading frame of PTP2 was amplified from yeast genomic DNA using KpnI-tagged forward primer PTP2fr-f (5′-GG GGTACCATTGATGGATCGCATAGCACAG-3′) and XbaI-tagged reverse primer PTP2fr-r (5′-CGCTCTAGATTAACAAGGTAACGCGTTCTTTATC-3′). Following digestion with KpnI and XbaI, the PCR product was ligated downstream of the PGK1 promoter on the pYPGK18 (2 μm, LEU2) plasmid. To construct the PTP3 overexpression plasmid, a sequence of 2751 base pairs that contains the entire open reading frame of PTP3 was amplified from yeast genomic DNA using EcoRI-tagged forward primer PTP3fr-f (5′-CCGGAATTC GAACATGAAGGACAGTGTAGACTGC-3′) and BamHI-tagged reverse primer PTP3fr-r (5′-CGCGGATCCGCCTAACTATTGTGGCAATTCTTTC-3′). Following digestion with KpnI and XbaI, the PCR product was ligated downstream of the PGK1 promoter on the pYPGK18 plasmid. The pPS1739 plasmid, which expresses HOG1-GFP, was kindly provided by Dr. Pamela Silver (Harvard Medical School). The PKC up-regulation plasmids pYcp50::PKC1R398P and pRS352::BCK1-20 and corresponding empty vectors were kindly provided by Dr. Michael Hall (Biozentrum, University of Basel). The plasmids were transformed into yeast cells using the yeast one-step transformation protocol (48).
Analysis of Synthetic Lethality
The crd1Δ mutant was crossed to sho1Δ, ssk1Δ, ste50Δ, and hog1Δ mutants obtained from the deletion collection generated in the BY4741 (MATa) background (Research Genetics). Diploids were sporulated on sporulation media, and tetrads were obtained. Haploids were then acquired via tetrad dissection. Haploids containing mutations in both CRD1 and one of the four HOG pathway genes were selected by uracil prototrophy (crd1Δ mutation) and Geneticin resistance (HOG pathway mutation). Growth of double mutants was examined at elevated temperature to identify synthetic lethality.
Fluorescence Microscopy and Analysis
All microscopic analyses were performed using an Olympus BX41 fluorescence microscope. Images were captured by an Olympus Q-Color3 digital CCD camera operated by QCapture2 software. All pictures were taken at the same magnification (×1000). To visualize vacuolar morphology in yeast, FM 4-64 staining was performed as described previously (49) with minor revisions. To monitor mitophagy, cells expressing IDH1-GFP from the gene locus or IDP1-GFP from an exogenous plasmid were treated with different mitophagy-inducing conditions, and the increase in vacuolar mito-GFP indicative of mitophagy was determined.
Western Blotting Detection of Free GFP
Whole cell extracts were prepared as described previously (47) with minor changes. Proteins from cell pellets equivalent to 1.5 A550 units were collected and loaded into a 12% SDS-polyacrylamide gel for electrophoresis. Proteins were then transferred to a PVDF membrane with 0.2-μm pores following the standard semidry Western blotting transfer procedure. Mouse anti-YFP monoclonal antibody (JL-8, Clontech) was used as primary antibody at a 1:3000 dilution, and HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, Inc.) was used as the secondary antibody at a 1:5000 dilution. After blotting, the PVDF membrane was covered with PierceTM ECL Plus Western blotting substrate reagent (ThermoFisher Scientific). The HyBlot ES® autoradiography film was exposed to the PVDF membrane and then developed. Idh1-GFP and cleaved GFP were detected as bands of 68 and 28 kDa, respectively. Phosphorylation of Hog1p was determined by Western blotting using anti-phospho-p38 rabbit IgG antibody (3D7; Cell Signaling) as described previously (8). Phosphorylation of Slt2p was determined by Western blotting using anti-phospho-p44/42 MAPK (Thr202/Tyr204) rabbit IgG antibody (catalog no. 9102, Cell Signaling) as described previously (22) with minor changes.
Author Contributions
Z. S. and M. L. G. designed the research and wrote the manuscript; Z. S., Y. L., A. N. G., and H. A. performed the experiments; Z. S., A. N. G, H. A., and M. L. G. analyzed the data. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. Michael Hall, Dr. Pamela Silver, Dr. Frederic M. Vaz, and Dr. Daniel Klionsky for kindly supplying plasmids.
This work was supported by National Institutes of Health Grant R01 HL117880. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CL
- cardiolipin
- HOG
- high osmolarity glycerol
- BTHS
- Barth syndrome
- PG
- phosphatidylglycerol
- PAS
- phagophore assembly site
- PE
- phosphatidylethanolamine
- YPD
- yeast extract peptone dextrose
- SC
- synthetic complete
- SL
- synthetic lactate.
References
- 1. Shen Z., Ye C., McCain K., and Greenberg M. L. (2015) The role of cardiolipin in cardiovascular health. Biomed Res. Int. 2015, 891707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barth P. G., Scholte H. R., Berden J. A., Van der Klei-Van Moorsel J. M., Luyt-Houwen I. E., Van 't Veer-Korthof E. T., Van der Harten J. J., and Sobotka-Plojhar M. A. (1983) An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355 [DOI] [PubMed] [Google Scholar]
- 3. Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., and Toniolo D. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 [DOI] [PubMed] [Google Scholar]
- 4. Valianpour F., Wanders R. J. A., Overmars H., Vreken P., Van Gennip A. H., Baas F., Plecko B., Santer R., Becker K., and Barth P. G. (2002) Cardiolipin deficiency in X-linked cardioskeletal myopathy and neutropenia (Barth syndrome, mim 302060): a study in cultured skin fibroblasts. J. Pediatrics 141, 729–733 [DOI] [PubMed] [Google Scholar]
- 5. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., and Blanck T. J. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 6. Schlame M., Kelley R. I., Feigenbaum A., Towbin J. A., Heerdt P. M., Schieble T., Wanders R. J. A., DiMauro S., and Blanck T. J. J. (2003) Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999 [DOI] [PubMed] [Google Scholar]
- 7. Zhong Q., Gvozdenovic-Jeremic J., Webster P., Zhou J., and Greenberg M. L. (2005) Loss of function of KRE5 suppresses temperature sensitivity of mutants lacking mitochondrial anionic lipids. Mol. Biol. Cell 16, 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou J., Zhong Q., Li G., and Greenberg M. L. (2009) Loss of cardiolipin leads to longevity defects that are alleviated by alterations in stress response signaling. J. Biol. Chem. 284, 18106–18114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S., Liu D., Finley R. L. Jr., and Greenberg M. L. (2010) Loss of mitochondrial DNA in the yeast cardiolipin synthase crd1 mutant leads to up-regulation of the protein kinase Swe1p that regulates the G2/M transition. J. Biol. Chem. 285, 10397–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen S., Tarsio M., Kane P. M., and Greenberg M. L. (2008) Cardiolipin mediates cross-talk between mitochondria and the vacuole. Mol. Biol. Cell 19, 5047–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patil V. A., Fox J. L., Gohil V. M., Winge D. R., and Greenberg M. L. (2013) Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 288, 1696–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gebert N., Joshi A. S., Kutik S., Becker T., McKenzie M., Guan X. L., Mooga V. P., Stroud D. A., Kulkarni G., Wenk M. R., Rehling P., Meisinger C., Ryan M. T., Wiedemann N., Greenberg M. L., and Pfanner N. (2009) Mitochondrial cardiolipin involved in outer membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeshige K., Baba M., Tsuboi S., Noda T., and Ohsumi Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baba M., Takeshige K., Baba N., and Ohsumi Y. (1994) Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J. Cell Biol. 124, 903–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baba M., Osumi M., and Ohsumi Y. (1995) Analysis of the membrane structures involved in autophagy in yeast by freeze-replica method. Cell Struct. Funct. 20, 465–471 [DOI] [PubMed] [Google Scholar]
- 16. Narendra D., Tanaka A., Suen D.-F., and Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frank M., Duvezin-Caubet S., Koob S., Occhipinti A., Jagasia R., Petcherski A., Ruonala M. O., Priault M., Salin B., and Reichert A. S. (2012) Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 1823, 2297–2310 [DOI] [PubMed] [Google Scholar]
- 18. Wang K., and Klionsky D. J. (2011) Mitochondria removal by autophagy. Autophagy 7, 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schlame M., and Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 [DOI] [PubMed] [Google Scholar]
- 20. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., and Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 [DOI] [PubMed] [Google Scholar]
- 21. Mao K., Wang K., Zhao M., Xu T., and Klionsky D. J. (2011) Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J. Cell Biol. 193, 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhong Q., Li G., Gvozdenovic-Jeremic J., and Greenberg M. L. (2007) Up-regulation of the cell integrity pathway in Saccharomyces cerevisiae suppresses temperature sensitivity of the pgs1Δ mutant. J. Biol. Chem. 282, 15946–15953 [DOI] [PubMed] [Google Scholar]
- 23. Reggiori F., Shintani T., Nair U., and Klionsky D. J. (2005) Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 1, 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reggiori F., Tucker K. A., Stromhaug P. E., and Klionsky D. J. (2004) The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 6, 79–90 [DOI] [PubMed] [Google Scholar]
- 25. Suzuki K., Kubota Y., Sekito T., and Ohsumi Y. (2007) Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209–218 [DOI] [PubMed] [Google Scholar]
- 26. Köfinger J., Ragusa M. J., Lee I.-H., Hummer G., and Hurley J. H. (2015) Solution structure of the Atg1 complex: implications for the architecture of the phagophore assembly site. Structure 23, 809–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakoh-Nakatogawa M., Kirisako H., Nakatogawa H., and Ohsumi Y. (2015) Localization of Atg3 to autophagy-related membranes and its enhancement by the Atg8-family interacting motif to promote expansion of the membranes. FEBS Lett. 589, 744–749 [DOI] [PubMed] [Google Scholar]
- 28. Suzuki K., Kirisako T., Kamada Y., Mizushima N., Noda T., and Ohsumi Y. (2001) The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie Z., and Klionsky D. J. (2007) Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109 [DOI] [PubMed] [Google Scholar]
- 30. Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., and Ohsumi Y. (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 147, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aoki Y., Kanki T., Hirota Y., Kurihara Y., Saigusa T., Uchiumi T., and Kang D. (2011) Phosphorylation of serine 114 on Atg32 mediates mitophagy. Mol. Biol. Cell 22, 3206–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Farré J.-C., Burkenroad A., Burnett S. F., and Subramani S. (2013) Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 14, 441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu X., and Tu B. P. (2011) Selective regulation of autophagy by the Iml1-Npr2-Npr3 complex in the absence of nitrogen starvation. Mol. Biol. Cell 22, 4124–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanki T., Wang K., Baba M., Bartholomew C. R., Lynch-Day M. A., Du Z., Geng J., Mao K., Yang Z., Yen W.-L., and Klionsky D. J. (2009) A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol. Biol. Cell 20, 4730–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tal R., Winter G., Ecker N., Klionsky D. J., and Abeliovich H. (2007) Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J. Biol. Chem. 282, 5617–5624 [DOI] [PubMed] [Google Scholar]
- 36. Maeda T., Wurgler-Murphy S. M., and Saito H. (1994) A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 369, 242–245 [DOI] [PubMed] [Google Scholar]
- 37. Wurgler-Murphy S. M., Maeda T., Witten E. A., and Saito H. (1997) Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol. Cell Biol. 17, 1289–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li S. C., Diakov T. T., Rizzo J. M., and Kane P. M. (2012) Vacuolar H+-ATPase works in parallel with the HOG pathway to adapt Saccharomyces cerevisiae cells to osmotic stress. Eukaryot. Cell 11, 282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gaspard G. J., and McMaster C. R. (2015) The mitochondrial quality control protein Yme1 is necessary to prevent defective mitophagy in a yeast model of Barth syndrome. J. Biol. Chem. 290, 9284–9298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu P., Liu X., Zhang J., Wang H.-G., Ye J.-M., and Shi Y. (2015) Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chu C. T., Ji J., Dagda R. K., Jiang J. F., Tyurina Y. Y., Kapralov A. A., Tyurin V. A., Yanamala N., Shrivastava I. H., Mohammadyani D., Qiang Wang K. Z., Zhu J., Klein-Seetharaman J., Balasubramanian K., Amoscato A. A., et al. (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu B. X., Rajagopalan V., Roddy P. L., Clarke C. J., and Hannun Y. A. (2010) Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 285, 17993–18002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fox T. E., Houck K. L., O'Neill S. M., Nagarajan M., Stover T. C., Pomianowski P. T., Unal O., Yun J. K., Naides S. J., and Kester M. (2007) Ceramide recruits and activates protein kinase C ζ (PKCζ) within structured membrane microdomains. J. Biol. Chem. 282, 12450–12457 [DOI] [PubMed] [Google Scholar]
- 44. Huwiler A., Brunner J., Hummel R., Vervoordeldonk M., Stabel S., van den Bosch H., and Pfeilschifter J. (1996) Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 93, 6959–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hage-Sleiman R., Hamze A. B., El-Hed A. F., Attieh R., Kozhaya L., Kabbani S., and Dbaibo G. (2016) Ceramide inhibits PKCθ by regulating its phosphorylation and translocation to lipid rafts in Jurkat cells. Immunol. Res. 64, 869–886 [DOI] [PubMed] [Google Scholar]
- 46. Bessa C., Pereira C., Leão M., Maciel C., Gomes S., Gonçalves J., Corte-Real M., Costa V., and Saraiva L. (2013) Using yeast to uncover the regulation of protein kinase Cδ by ceramide. FEMS Yeast Res. 13, 700–705 [DOI] [PubMed] [Google Scholar]
- 47. Kanki T., Kang D., and Klionsky D. J. (2009) Monitoring mitophagy in yeast: the Om45-GFP processing assay. Autophagy 5, 1186–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen D. C., Yang B. C., and Kuo T. T. (1992) One-step transformation of yeast in stationary phase. Curr. Genet. 21, 83–84 [DOI] [PubMed] [Google Scholar]
- 49. Conboy M. J., and Cyert M. S. (2000) Luv1p/Rki1p/Tcs3p/Vps54p, a yeast protein that localizes to the late Golgi and early endosome, is required for normal vacuolar morphology. Mol. Biol. Cell 11, 2429–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang F., Rizavi H. S., and Greenberg M. L. (1997) Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol. Microbiol. 26, 481–491 [DOI] [PubMed] [Google Scholar]
- 51. Gohil V. M., Thompson M. N., and Greenberg M. L. (2005) Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J. Biol. Chem. 280, 35410–35416 [DOI] [PubMed] [Google Scholar]
- 52. Vaz F. M., Houtkooper R. H., Valianpour F., Barth P. G., and Wanders R. J. A. (2003) Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J. Biol. Chem. 278, 43089–43094 [DOI] [PubMed] [Google Scholar]
- 53. Helliwell S. B., Schmidt A., Ohya Y., and Hall M. N. (1998) The Rho1 effector Pkc1, but not Bni1, mediates signalling from Tor2 to the actin cytoskeleton. Curr. Biol. 8, 1211–1214 [DOI] [PubMed] [Google Scholar]
- 54. Ferrigno P., Posas F., Koepp D., Saito H., and Silver P. A. (1998) Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin β homologs NMD5 and XPO1. EMBO J. 17, 5606–5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Journo D., Mor A., and Abeliovich H. (2009) Aup1-mediated regulation of Rtg3 during mitophagy. J. Biol. Chem. 284, 35885–35895 [DOI] [PMC free article] [PubMed] [Google Scholar]