

Abstract
Mitochondrial bioenergetics are critical for cellular homeostasis and stress responses. The reactive oxygen species-generating enzyme, NADPH oxidase 4 (Nox4), regulates a number of physiological and pathological processes, including cellular differentiation, host defense, and tissue fibrosis. In this study we explored the role of constitutive Nox4 activity in regulating mitochondrial function. An increase in mitochondrial oxygen consumption and reserve capacity was observed in murine and human lung fibroblasts with genetic deficiency (or silencing) of Nox4. Inhibition of Nox4 expression/activity by genetic or pharmacological approaches resulted in stimulation of mitochondrial biogenesis, as evidenced by elevated mitochondrial-to-nuclear DNA ratio and increased expression of the mitochondrial markers transcription factor A (TFAM), citrate synthase, voltage-dependent anion channel (VDAC), and cytochrome c oxidase subunit 4 (COX IV). Induction of mitochondrial biogenesis was dependent on TFAM up-regulation but was independent of the activation of the peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α). The enhancement of mitochondrial bioenergetics as well as the increase in mitochondrial proteins in Nox4-deficient lung fibroblasts is inhibited by silencing of nuclear factor erythroid-derived 2-like 2 (Nrf2), supporting a key role for Nrf2 in control of mitochondrial biogenesis. Together, these results indicate a critical role for both Nox4 and Nrf2 in counter-regulation of mitochondrial biogenesis and metabolism.
Keywords: bioenergetics, fibroblast, mitochondria, NADPH oxidase, Nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), NADPH oxidase-4, mitochondrial biogenesis, mitochondrial transcription factor A
Introduction
Regulation of mitochondrial mass is a tightly controlled process. It is key to pressure-overload adaptation in the heart (1) and to endurance training in the skeletal muscle (2–4). Stress-induced increase in mitochondrial mass results from the stimulation of mitochondrial biogenesis. The mitochondrial transcription factor A (TFAM)2 (5) ensures mitochondrial DNA replication during mitochondrial biogenesis (6–8). The TFAM promoter contains binding sites for transcription factors that are responsive to cellular energy (9), redox status (10), and hormonal status (11). Stress-induced mitochondrial biogenesis is mostly driven by the peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) (12–16), which is upstream of TFAM (5). PGC-1α is a central integrator of multiple signaling pathways that regulates transcriptional cascades involved in mitochondrial biogenesis (13, 17–19). In particular, PGC-1α co-activates nuclear respiratory factor-1 (NRF-1) activity at the TFAM promoter. Nuclear factor erythroid-derived 2-like 2 (Nrf2) has also been reported to control oxidative stress or exercise-induced mitochondrial biogenesis via promoting NRF-1 transcription after its binding to the antioxidant response element (ARE) of the NRF-1 promoter (20–22).
NADPH-oxidase 4 (Nox4) is a constitutively active enzyme that predominantly produces H2O2 (23). Nox4-mediated oxidative stress has been involved in different cellular processes such as TGF-β1-induced differentiation (24), cytoskeletal dynamics (25, 26), and transcriptional regulation (27, 28). Nox4 has been localized to the mitochondria (29) where its interaction with complex I has been reported to inhibit the activity of this complex (30). Additionally, Nox4 has been suggested to regulate urine levels of the metabolite fumarate by controlling the activity of fumarate hydratase (31); however, to date a knowledge gap remains regarding the role of Nox4 in controlling mitochondrial bioenergetics and metabolism.
Recent studies from our laboratory demonstrated that a redox imbalance underlies the persistence of lung fibrosis in response to airway injury in aged mice (32). An increase in Nox4 activity associated with deficient Nrf2-driven cytoprotective responses characterizes this redox imbalance. Nrf2 is an oxidative stress-sensitive transcription factor that controls the expression of genes involved in antioxidant defense and detoxification (33). Recent studies have suggested a fundamental role of Nrf2 in regulating mitochondrial function (34).
In this study we investigated the role of Nox4 in mitochondrial biogenesis and bioenergetics. We found that endogenously expressed Nox4 represses mitochondrial biogenesis. This effect of Nox4 is mediated downstream of PGC-1α via a mechanism that involves Nrf2. This study supports the essential and opposing roles of Nox4 and Nrf2 signaling in the control of mitochondrial biogenesis and bioenergetics.
Results
Constitutive Nox4 Expression/Activity Regulates Mitochondrial Bioenergetics
To determine whether Nox4 modulates mitochondrial bioenergetics, we analyzed oxygen consumption rate (OCR) of lung fibroblasts isolated from Nox4+/+ or Nox4−/− mice. Nox4−/− lung fibroblasts demonstrated a marked increase in maximal respiration in response to carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) compared with controls (Nox4+/+ fibroblasts). The reserve capacity, which represents the bioenergetic capacity of cells in response to increased energy demands or oxidative stress, was also significantly increased in Nox4−/− lung fibroblasts compared with controls (Fig. 1, A and B). To investigate whether Nox4 deficiency mediates its effect on OCR by up-regulating the electron transport chain (ETC) activity, we measured the oxidation of complex I and II linked substrates. In this assay Nox4+/+ or Nox4−/− lung fibroblasts were permeabilized with plasma membrane permeabilizer (PMP, a mutant recombinant perfringolysin O) (35), and OCR was measured in response to exogenous addition of complex I or II substrates (pyruvate/malate or succinate) and inhibitors of the ETC (rotenone or antimycin A) in the presence of ADP (36). Complex I and II activity was significantly increased in Nox4−/− lung fibroblasts compared with controls (Fig. 1C). Together, these data suggest that Nox4 negatively regulates mitochondrial bioenergetics, including the capacity of mitochondria to respond to increased energetic demand.
FIGURE 1.
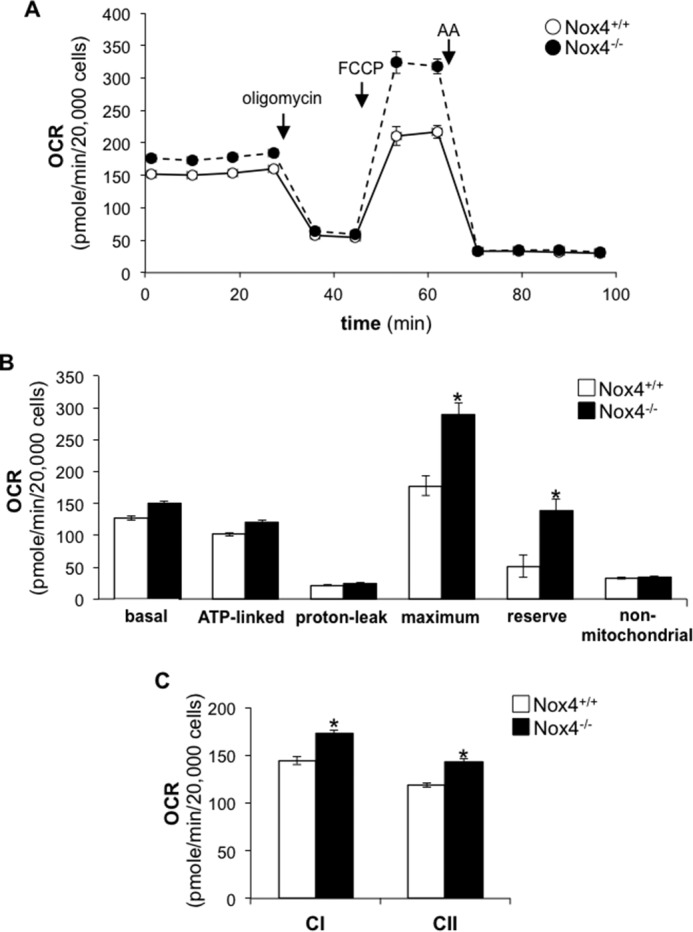
Nox4 knock-out enhances mitochondrial respiration. A, the OCR of Nox4+/+ or Nox4−/− lung fibroblasts was measured as a function of time. Oligomycin (1.0 μg/ml), FCCP (0.6 μm), or antimycin A (AA) (10 μm) was added at the indicated time point. B, bar graph corresponding to the calculation of basal, ATP-linked, proton-leak, maximum, reserve, and non-mitochondrial OCR in Nox4+/+ or Nox4−/− lung fibroblasts. C, bar graph representing the effect of Nox4 knock-out on complex I- or complex II-linked OCR in PMP-permeabilized cells in the presence of ADP (State 3). Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.005 compared with control; n = 15–18 (3 biological replicates total; 5–6 technical replicates per group per experiment).
To confirm these observations, we next investigated whether the increase in OCR observed in Nox4-deficient cells results from the stimulation of mitochondrial biogenesis. Normal human lung diploid fibroblasts (IMR-90) were transfected with non-targeting (NT) or Nox4-specific siRNA. Nox4 silencing was confirmed at the mRNA level by real-time PCR (Fig. 2A). The mitochondrial-to-nuclear DNA ratio was quantified by real-time PCR. Nox4-silenced fibroblasts demonstrated a 3-fold increase (n = 18, p < 0.005) in mitochondrial DNA copy number compared with controls (Fig. 2B). We observed similar effects on the mitochondrial-to-nuclear DNA ratio when Nox4 activity was blocked using the Nox1/Nox4 inhibitor GKT137831 (37), albeit to a lesser extent; a 1.8-fold increase compared with controls, n = 12, p < 0.005 (Fig. 2C).
FIGURE 2.
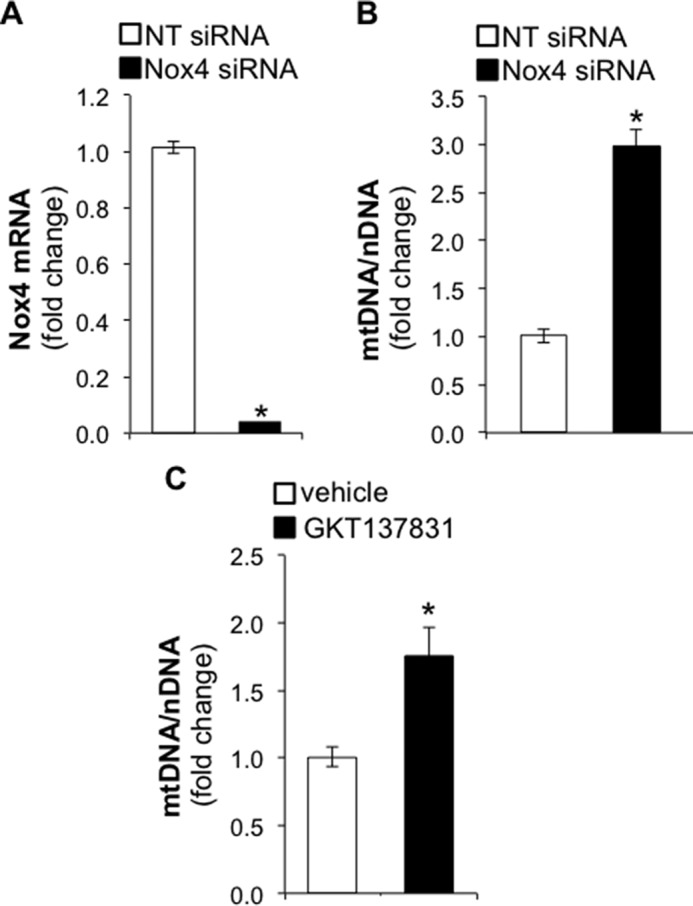
Nox4 silencing or inhibition increases the mitochondrial-to-nuclear DNA ratio. A, bar graph representing the silencing efficiency of NT or Nox4 siRNA in IMR-90 cells. Total RNA was isolated for real-time PCR analysis 3 days post siRNA transfection. B, bar graph representing the effect of Nox4 silencing on mitochondrial (mt)-to-nuclear DNA ratio in IMR-90 cells. Total DNA was isolated for real-time PCR analysis 5 days post siRNA transfection. C, bar graph representing the effect of Nox4 inhibition following GKT137831 treatment (10 μm) on the mt-to-nuclear DNA ratio. IMR-90 cells were treated with GKT137831 for 72 h before DNA isolation. Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.005 compared with control; A and B, n = 18 (6 biological replicates total, 3 technical replicates per group per experiment); C, n = 12 (4 biological replicates total, 3 technical replicates per group per experiment).
Mitochondrial biogenesis requires the up-regulation of TFAM, a transcription factor that ensures mitochondrial DNA replication during biogenesis. To determine whether Nox4 silencing mediates its effects on mitochondrial biogenesis via TFAM, we studied the effects of dual silencing of both Nox4 and TFAM on the mitochondrial-to-nuclear DNA ratio. Human lung fibroblasts were transfected with NT, Nox4, TFAM, or Nox4 plus TFAM siRNA (equal amounts of siRNA were transfected in each experimental group). Mitochondrial-to-nuclear DNA ratios were assessed for each experimental group by real-time PCR. As previously observed, Nox4 silencing led to an increase in mitochondrial-to-nuclear DNA ratios; a 2-fold increase compared with controls, n = 9, p < 0.005 (Fig. 3A). As expected, TFAM silencing decreased the mitochondrial-to-nuclear DNA ratio; additionally, dual silencing of Nox4 and TFAM failed to rescue the increase in the mitochondrial-to-nuclear DNA ratio observed in Nox4-silenced cells. The silencing efficiency of Nox4 and/or TFAM was confirmed at the mRNA level by real time-PCR for each experimental group (Fig. 3, B, C, and D). These data suggest that Nox4 silencing stimulates mitochondrial biogenesis via TFAM.
FIGURE 3.

Nox4 silencing increases mitochondrial mass via TFAM. A, bar graph representing the effect of silencing Nox4, TFAM, or Nox4 plus TFAM on the mitochondrial-to-nuclear DNA ratio in IMR-90 cells. NT siRNA was transfected in with Nox4 siRNA alone or TFAM siRNA alone to keep equal the amount of siRNA between each experimental group (final concentration of siRNA 100 nm). B–D, bar graphs representing the silencing efficiency of Nox4 or TFAM siRNA in IMR-90 cells. Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.005 compared with control; A–D, n = 9 (3 biological replicates total, 3 technical replicates per group per experiment). E, total cell lysates were isolated from IMR-90 cells transfected with NT, Nox4, TFAM, or Nox4 plus TFAM siRNA (cells were harvested 5 days post transfection). NT siRNA was transfected in with Nox4 siRNA alone or TFAM siRNA alone to keep the amount of siRNA between each experimental group equal. Then the levels of the mitochondrial proteins TFAM, citrate synthase, VDAC, COX IV, and α-tubulin were determined by Western blotting; molecular weight markers are indicated on the left side of the corresponding panel. F, densitometry analysis of the signal associated with the detection of TFAM, citrate synthase, VDAC, and COX IV in total cell lysates of IMR-90 cells deficient in Nox4, TFAM, or Nox4 and TFAM. Values represent the mean ± S.E.; error bars represent S.E.; * p < 0.05 compared with control (NT siRNA transfected cells); #, p < 0.05 compares Nox4 siRNA to Nox4 + TFAM siRNA; n = 4 (4 biological replicates total, 1 technical replicate per group per experiment).
Next, we analyzed the protein expression of mitochondrial markers, TFAM, citrate synthase (38), the voltage-dependent anion channel (VDAC) (39), and the cytochrome c oxidase subunit IV (COX IV) (40) in whole cell lysates of lung fibroblasts treated with NT, Nox4, TFAM, and Nox4+TFAM siRNA. The protein levels of these mitochondrial markers were increased in Nox4 siRNA-treated cells compared with controls, an effect that was partially reversed by silencing of TFAM in Nox4-silenced cells (Fig. 3, E and F). Together, these data indicate that Nox4 silencing increases levels of mitochondrial proteins in human lung fibroblasts via TFAM up-regulation.
TFAM Induction in Nox4-deficient Cells Is Independent of PGC-1α Activation
Mitochondrial biogenesis has been reported to be induced by the phosphorylation of PGC-1α, a co-activator of transcription that binds to NRF-1 to activate the expression of genes involved in mitochondrial biogenesis (12, 14). TFAM is a downstream target of PGC-1α (5). To determine whether Nox4 silencing mediates its effect on mitochondrial biogenesis by activating the PGC-1α/NRF-1 axis for mitochondrial biogenesis signaling, we analyzed the expression of phosphorylated PGC-1α and TFAM in total cell lysates isolated from control or Nox4-silenced human lung fibroblasts. We observed that Nox4 silencing did not induce PGC-1α phosphorylation (Fig. 4, A and B), whereas it led to a 3-fold increase in TFAM protein levels compared with controls (n = 3, p < 0.005) (Fig. 4, A and C). These data suggest that Nox4 silencing stimulates mitochondrial biogenesis by increasing TFAM protein levels independently of PGC-1α activation.
FIGURE 4.
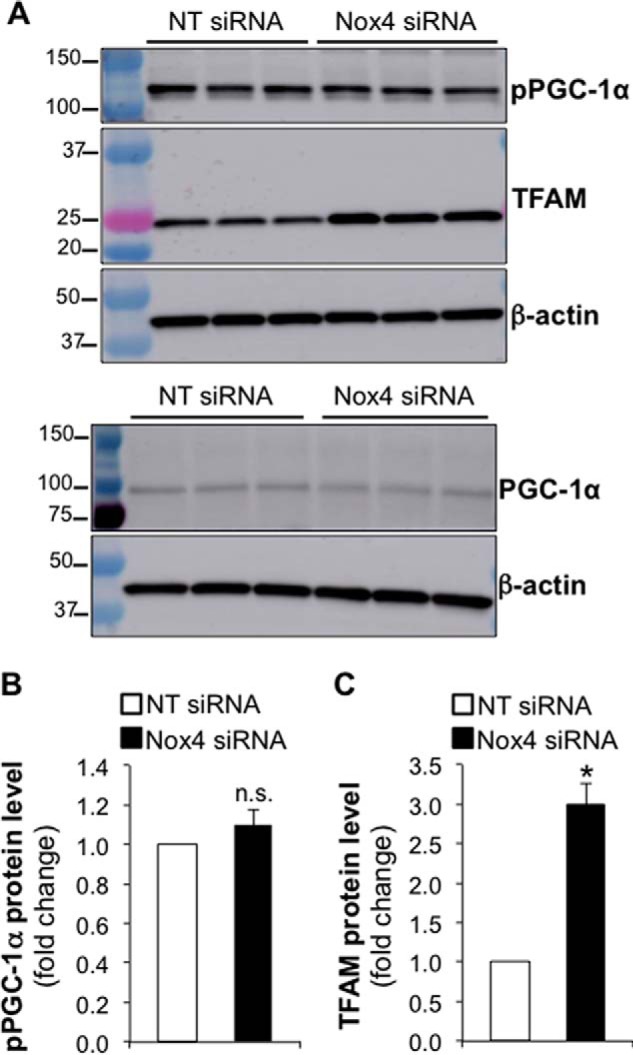
Nox4 silencing increases mitochondrial mass independently of PGC-1α activation. A, total cell lysates were isolated from IMR-90 cells transfected with NT or Nox4 siRNA. Then protein levels of phosphorylated PGC-1α (pPGC-1α), TFAM, total PGC-1α, and β-actin were determined by Western blot; molecular weight markers are indicated on the left side of the corresponding panel. Phosphorylated and total PGC-1α were analyzed in the same samples; however, two separate Western blots were run to avoid antibody-associated signal overlap. B and C, densitometry analysis of pPGC-1α or TFAM in total cell lysates from control or Nox4 silenced IMR-90 cells. Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.005 compared with control; n.s., non-significant difference between means; n = 3 (3 biological replicates total, 1 technical replicate per group per experiment).
Up-regulation of TFAM in Nox4-silenced Cells Is Mediated by Nrf2 but Is Independent of NRF1 and c-Myc
To determine whether Nox4 silencing increases TFAM protein levels through promoting TFAM transcription, we analyzed TFAM transcript levels by real-time PCR in control or Nox4-silenced cells. Nox4 silencing efficiency was assessed by real-time PCR. Nox4 silencing resulted in a 96% reduction of Nox4 mRNA level (data not shown). We observed a significant increase in TFAM transcript levels in Nox4-silenced compared with control cells (Fig. 5A). NRF-1, Nrf2, and c-Myc have been shown to activate TFAM transcription (20, 41–44). We analyzed the protein levels of these transcription factors in nuclear and cytosolic fractions isolated from control or Nox4-silenced cells. Although we did not observe any significant effect of Nox4 silencing on the levels of nuclear NRF-1 or c-Myc (n = 4), we observed a 2-fold increase (n = 4, p < 0.005) in nuclear Nrf2 levels in Nox4-deficient lung fibroblasts compared with controls (Fig. 5, B–E). These data suggest that Nox4 silencing might stimulate mitochondrial biogenesis by promoting TFAM transcription through an Nrf2-dependent mechanism in human lung fibroblasts.
FIGURE 5.

Nox4 silencing increases TFAM transcript level and nuclear Nrf2 expression. A, bar graph representing the effect of silencing Nox4 on TFAM mRNA levels in IMR-90 cells. Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.01 compared with control, n = 15 (5 biological replicates total, 3 technical replicates per group per experiment). B, nuclear or cytosolic fractions were isolated from IMR-90 cells transfected with NT or Nox4 siRNA (cells were harvested for experimentation 3 days post transfection). Then the protein levels of NRF-1, Nrf2, c-Myc, lamin A/C, and α-tubulin were determined by Western blot; molecular weight markers are indicated on the left side of the corresponding panel. C–E, densitometry analysis of NRF-1, Nrf2, and c-Myc expression in the nuclear fractions of control or Nox4-silenced lung fibroblasts. Values represent the mean ± S.E.; error bars represent S.E.; * p < 0.005 compared with control; n.s., non-significant difference between means. B–E, n = 4 (4 biological replicates total, 1 technical replicate per group per experiment).
To determine whether Nox4 silencing induces mitochondrial biogenesis via Nrf2 activation, we analyzed the effect of silencing Nox4, Nrf2, or both Nox4 and Nrf2 on the levels of mitochondrial proteins in human lung fibroblasts. We analyzed the protein levels of TFAM, citrate synthase, and COX IV in total cell lysates and mitochondrial fractions isolated from NT, Nox4, Nrf2, or Nox4+Nrf2 siRNA-transfected lung fibroblasts (equal amounts of siRNA were transfected in each experimental group). Nox4 silencing increased the levels of TFAM, citrate synthase, and COX IV in both total cell lysates and mitochondrial fractions (Fig. 6, A–C). The silencing of Nrf2 in Nox4-deficient lung fibroblasts attenuated, in part, the increase in mitochondrial markers (Fig. 6, A–C). These data suggest that Nrf2 contributes to regulation of mitochondrial biogenesis induced by Nox4 deficiency.
FIGURE 6.

Nrf2 silencing reduces mitochondrial TFAM expression. A, total cell lysates or mitochondrial fractions were isolated from NT, Nox4, Nrf2, or Nox4 plus Nrf2 siRNA-transfected IMR-90 cells (cells were harvested for experimentation 5 days post transfection). NT siRNA was transfected in with Nox4 siRNA alone or Nrf2 siRNA alone to keep the amount of siRNA between each experimental group equal (final concentration of siRNA, 100 nm). Mitochondria were isolated from the same number of cells (2.4 × 106) for each experimental group. Then the entire mitochondrial fraction was submitted to SDS-PAGE analysis for each experimental group, and the expression levels of the proteins TFAM, citrate synthase, CoxIV, and α-tubulin were analyzed by Western blot; for each signal detected, molecular weight markers are indicated on the left side of the corresponding panel. B, densitometry analysis of TFAM, citrate synthase, and CoxIV expression in total cell lysates isolated from lung fibroblasts transfected with NT, Nox4, Nrf2, or Nox4 plus Nrf2 siRNA. C, densitometry analysis of TFAM, citrate synthase, and COX IV expression in mitochondrial fractions isolated from lung fibroblasts transfected with NT, Nox4, Nrf2, or Nox4 plus Nrf2 siRNA. B and C, values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.05 compared with control (NT siRNA-transfected cells). #, p < 0.05 compares Nox4 siRNA to Nox4 + Nrf2 siRNA; n = 4 (4 biological replicates total, 1 technical replicate per group per experiment). D, bar graph representing the effect of silencing Nox4, Nrf2, or Nox4 plus Nrf2 on Nox4 mRNA expression in IMR-90 cells. E, bar graph representing the effect of silencing Nox4, Nrf2, or Nox4 plus Nrf2 on Nrf2 mRNA expression in IMR-90 cells. Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.005 compared with control, n = 9 (3 biological replicates total, 3 technical replicates per group per experiment). F, nuclear and cytosolic fractions were isolated from NT, Nox4, Nrf2, or Nox4 plus Nrf2 siRNA-transfected IMR-90 cells (cells were harvested for experimentation 3 days post transfection). Then the protein levels of NRF-1, Nrf2, c-Myc, lamin A/C, and α-tubulin were determined by Western blot.
In addition, similar to a previous study (45), we found that Nrf2 silencing led to a decrease in Nox4 mRNA expression (Fig. 6D). A redox-dependent co-regulation of these proteins was further supported by the finding that Nox4 silencing increased the levels of Nrf2 mRNA (Fig. 6E), which is consistent with the effect of Nox4 silencing in increasing nuclear Nrf2 protein (Fig. 5, B and D). The analysis of Nrf2 protein levels in nuclear fractions isolated from NT, Nox4, Nrf2, or Nox4+Nrf2 siRNA-transfected fibroblasts confirmed its efficient silencing (Fig. 6F). NRF-1 and c-Myc protein levels remained similar between controls and Nrf2-deficient cells (Fig. 6F). Together, these data support the concept that de-repression of Nrf2 by Nox4 silencing contributes to TFAM expression and mitochondrial biogenesis in human lung fibroblasts.
Constitutive Nox4 Expression Regulates Mitochondrial Bioenergetics via Nrf2
To confirm whether Nox4 modulates mitochondrial bioenergetics via a Nrf2-dependent mechanism, we investigated the effect of silencing Nox4 or Nox4+Nrf2 on OCR in human lung fibroblasts. Nox4-silenced cells exhibited a significant increase in basal, ATP-linked, maximum, and reserve OCR compared with controls (NT siRNA-transfected cells) (Fig. 7, A and B). This up-regulation of mitochondrial bioenergetics in Nox4-deficient cells was abrogated in Nox4 and Nrf2 double-deficient cells. Additionally, CI and CII activity was significantly enhanced in Nox4-deficient lung fibroblasts compared with controls (Fig. 7C). This effect of Nox4 silencing on CI and CII activity was also abrogated when Nrf2 was co-silenced.
FIGURE 7.
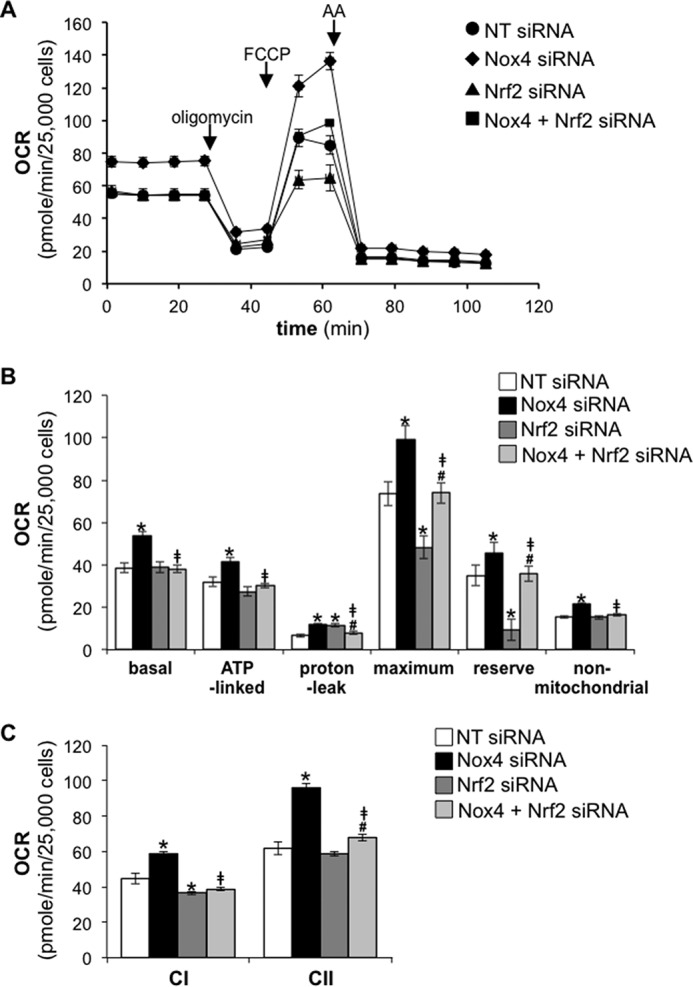
Nrf2 silencing abrogates enhanced bioenergetics in Nox4-deficient human lung fibroblasts. A, the OCR of IMR-90 cells transfected with NT, Nox4, Nrf2, or Nox4 plus Nrf2 siRNA was measured as a function of time. Oligomycin (1.0 μg/ml), FCCP (0.6 μm), or antimycin A (AA; 10 μm) was added at the indicated time point. B, bar graph corresponding to the calculation of basal, ATP-linked, proton-leak, maximum, reserve, and non-mitochondrial OCR in control, Nox4, Nrf2, or Nox4 plus Nrf2-deficient human lung fibroblasts. C, bar graph representing the effect of Nox4, Nrf2, or Nox4 plus Nrf2 silencing on complex I- or complex II-linked OCR in PMP-permeabilized cells in the presence of ADP (State 3). Values represent the mean ± S.E.; error bars represent S.E.; *, p < 0.05 compared with control; #, p < 0.05 compares Nrf2 siRNA to Nrf2 + Nox4 siRNA; ‡, p < 0.01 compares Nox4 siRNA to Nox4 + Nrf2 siRNA; n = 15–18 (3 biological replicates total; 5–6 technical replicates per group per experiment).
Of note, the maximum mitochondrial respiration and reserve capacity were significantly reduced in Nrf2-deficient cells compared with controls (Fig. 7, A and B). This effect of Nrf2 silencing on maximum mitochondrial respiration and reserve capacity was abrogated in Nox4 and Nrf2 dually silenced cells. These data indicate that repression of mitochondrial bioenergetics by endogenous Nox4 in lung fibroblast is mediated via inhibition of an Nrf2-dependent mechanism.
Discussion
The regulation of mitochondrial biogenesis and bioenergetics is a crucial adaptive response that allows organisms to survive stress or exercise (46). Although a number of studies report changes in mitochondrial bioenergetics and/or biogenesis to specific stimuli, mechanisms of the control of constitutive mitochondrial bioenergetics are unknown.
Our study is the first to demonstrate that Nox4 regulates mitochondrial biogenesis and bioenergetics. Our study also shows that this effect of Nox4 on mitochondrial bioenergetics and biogenesis is partly mediated by the control of endogenous Nrf2 and TFAM expression (Fig. 8).
FIGURE 8.
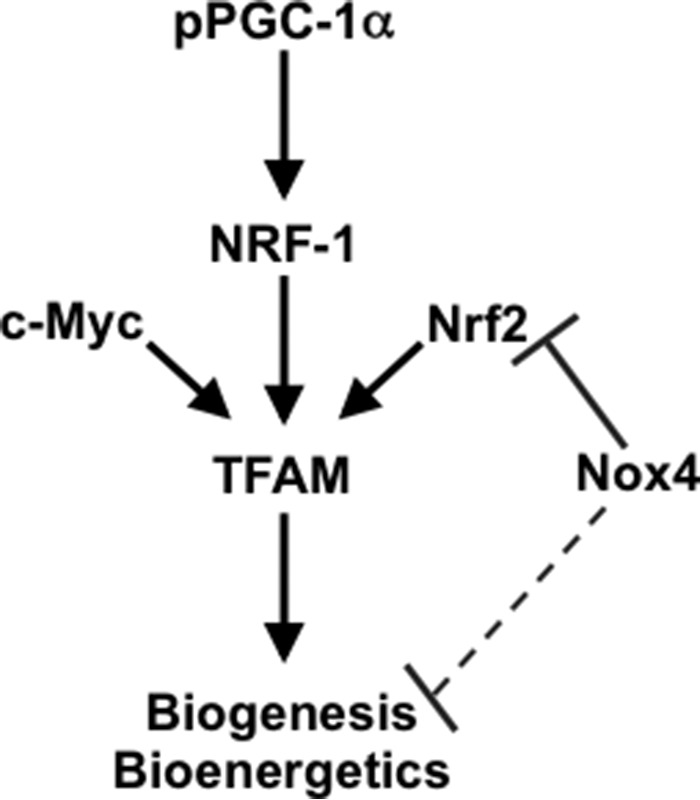
Schematic representing the co-regulation of mitochondrial biogenesis by Nox4 and Nrf2 in human lung fibroblasts.
Our data suggest that Nox4 modulates the activity of enzymatic complexes within the ETC. Work of others has shown that Nox4 is localized to the mitochondria (29, 47) where its interaction with complex I inhibits the activity of this complex (30, 48). Our data imply that, in addition to direct effects, Nox4 represses mitochondrial biogenesis, which indirectly alters bioenergetics and reserve capacity; this regulation of mitochondrial bioenergetics by Nox4 may be part of a quality-control mechanism by which lung fibroblasts preserve mitochondrial integrity at baseline by preventing function-associated damages (49–51).
Our data indicate that the regulation of mitochondrial biogenesis in human lung fibroblasts by Nox4, at baseline, is mediated via a mechanism independent of PGC-1α. Mitochondrial biogenesis in response to specific stimuli is a PGC-1α-driven process that predominantly induces TFAM expression through NRF-1 co-activation at the TFAM promoter site (13, 52). Because PGC-1α is an integrator of multiple extracellular stimuli-associated signaling pathways (nutrient status, temperature, growth hormones) (53), it is possible that PGC-1α does not participate in the regulation of mitochondrial homeostasis at baseline and may predominantly drive mitochondrial adaption in response to specific stimuli.
Our data show that Nox4 silencing increases Nrf2 levels in the nucleus of human lung fibroblasts. Because oxidative stress induces Nrf2 translocation to the nucleus (54–57), where it activates the transcription of genes involved in cytoprotective responses (58), this unexpected finding suggests oxidant stress responses independent of Nox4 or that Nrf2 can be activated by a non-redox mechanism. In support of a Nox4-independent oxidative stress pathway, it has been shown that Nox4 down-regulates the activity of complex I in the mitochondria (30). Complex I of the ETC is one of the major sites of mitochondrial ROS production (50). Therefore, the silencing of Nox4 may affect its interaction with complex I in the mitochondria and promote ROS formation at this complex. The precise mechanism(s) of how endogenous Nox4 represses Nrf2 require further study.
Our data suggest that endogenous Nox4 represses mitochondrial bioenergetics and biogenesis via decreasing Nrf2 induction and activation. This observation is consistent with reports that link Nrf2 activation to mitochondrial function (34). Nrf2 has also been reported to regulate mitochondrial membrane potential, mitochondrial respiration, and ATP production (59).
Fibroblasts are key in wound-healing processes (60). During wound repair, fibroblasts reprogram their metabolism to meet the accrued energetic needs associated with their function. The knowledge of mechanisms governing mitochondrial functions in resting conditions may help our understanding of how fibroblasts are primed to respond to lung injury.
Experimental Procedures
Animals
Nox4 knock-out mice were a gift from Karl-Heinz Krause, University of Geneva. Mice were sacrificed by CO2 inhalation. All procedures involving animals were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Alabama at Birmingham.
Cell Culture
Lung fibroblasts were isolated from Nox4+/+ or Nox4−/− mice. In brief, mouse lung tissue was collected and minced in ice-cold PBS. The minced tissue was then digested with collagenase (1 mg/ml, Worthington (Lakewood, NJ)) for 30 min at 37 °C and filtered through a 100-μm cell strainer. Collagenase was neutralized by adding 10% fetal calf serum-containing DMEM. After centrifugation (1000 rpm, 5 min), cells were plated in 100-mm dishes for culture. Primary mouse lung fibroblasts were passaged one time before experimentation.
Human fetal lung fibroblasts (IMR-90 cells) at low population doubling (PDL 7) were purchased from Coriell Cell Repositories (Camden, NJ). All cells (primary cultures and IMR-90) were cultured in DMEM (Life Technologies) supplemented with 10% fetal calf serum (HyClone Laboratories, Logan, UT), 100 units/ml penicillin, 100 μg/ml streptomycin, 1.25 μg/ml amphotericin B, and 2 mm l-glutamine at 37 °C in 5% CO2, 95% air.
Reagents
GKT137831 was a generous gift from Genkyotex (Geneva, Switzerland). Plasma membrane permeabilizer (PMP, a mutant recombinant perfringolysin O) was purchased from Seahorse Bioscience. Protease and phosphatase inhibitor mixtures (complete, Mini, EDTA-free, and PhosSTOP) were purchased from Roche Applied Science. We purchased antibodies (Ab) to β-actin (clone AC-15, mouse monoclonal Ab against N-terminal peptide, catalogue #A1978, lot #043M4840V) and α-tubulin (clone B-5–1-2, mouse mAb against C-terminal peptide, catalogue #T5168, lot #103M4773V) from Sigma; TFAM (clone D5C8 rabbit mAb against peptide containing Val-225, catalogue #8076S, lot #1), total PGC-1α (rabbit mAb against PGC-1α fusion protein, catalogue #2178S, lot #2), c-Myc (clone D3N8F, rabbit mAb against central region of human c-Myc, catalogue #12987S, lot #1), NRF-1 (rabbit Ab against peptide near Leu167, catalogue #12381, lot #1), VDAC (clone D73D12, rabbit mAb against N-terminal peptide, catalogue #4661S, lot #5), citrate synthase (clone D7V8B, rabbit mAb against residues near the C terminus of the protein, catalogue #14309S, lot #1) and COX IV (clone 3E11, rabbit mAb against synthetic peptide corresponding to residues surrounding Lys29 of human COX IV, catalogue #4850S, lot #6) from Cell Signaling Technology (Boston, MA); lamin A/C (clone 636, mouse mAb against lamin preparation of porcine origin, catalogue #sc-7292, lot #F3011) was purchased from Santa Cruz Biotechnology (Dallas, TX); Nrf2 (rabbit Ab against fusion protein from amino acids 256–605, catalogue #16396-1-AP, lot #00043173) was from Proteintech Group Inc. (Rosemont, IL); phospho-PGC-1α rabbit Ab against Ser-571, catalogue #AF6650, lot #CEBA0114011) was from R&D Systems (Minneapolis, MN).
Assessment of Bioenergetics and Mitochondrial Assay
Primary mouse lung fibroblasts were plated on Seahorse Extracellular Analyzer XF96 plates in serum-free culture media. Measurements of the OCR and the activity of complex I and II of the ETC were measured as described in Refs. 36, 61, and 62).
RNA Interference
siRNA targeting Nox4, TFAM, and Nrf2 as well as non-targeting controls were purchased from InvitrogenTM (Stealth siRNA technology; for siRNA sequences, see Table 1). siRNA were transfected into lung fibroblasts using Lipofectamine RNAiMAX (InvitrogenTM) at a final concentration of 100 nm. 48 h post transfection, lung fibroblasts were growth-arrested by replacing the transfection media with serum-free DMEM supplemented with 2 mm l-glutamine.
TABLE 1.
siRNA sequences
| Gene accession number | siRNA sequence (5′-3′) |
|---|---|
| Nox4, NM_016931 | Sense, ACAGUGAAGACUUUGUUGAACUGAA |
| Anti-sense, UUCAGUUCAACAAAGUCUUCACUGU | |
| TFAM, NM_003201.2 | Sense, GCGUUGGAGGGAACUUCCUGAUUCA |
| Anti-sense, UGAAUCAGGAAGUUCCCUCCAACGC | |
| Nrf2, NM_006164.4 | Sense, CAAACUGACAGAAGUUGACAAUUAU |
| Anti-sense, AUAAUUGUCAACUUCUGUCAGUUUG |
Western Immunoblotting
Total cell lysates were prepared using radioimmune precipitation assay buffer (150 mm NaCl, 1.0% IGEPAL® CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0; Sigma) supplemented with protease and phosphatase inhibitors. Cytosolic and nuclear fractions were isolated using the NE-PER Nuclear and Cytosolic Extraction kit (Thermo Scientific). Mitochondrial fractions were isolated using the Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific). The total protein concentration of lysates or cell fractions was quantitated using a Micro BCA Protein Assay kit (Thermo Scientific) or the DC Protein Assay kit (Bio-Rad). Lysates were then subjected to SDS-PAGE under reducing conditions, and Western immunoblotting was performed as described previously (3). Immunoblots were imaged using an Amersham Biosciences 600 Imager (GE Healthcare). Signals were quantitated using ImageQuant TL software.
Real-time PCR
Real-time PCR was performed as described in Ref. 62. Primer sequences for Nox4, TFAM, and Nrf2 are given in Table 2.
TABLE 2.
Primer sequences
| Gene accession number | Primer sequence (5′-3′) | Product size |
|---|---|---|
| bp | ||
| Nox4, NM_016931 | Forward, AGATGTTGGGGCTAGGATTG | 137 |
| Reverse, TCTCCTGCTTGGAACCTTCT | ||
| TFAM, NM_003201.2 | Forward, CATCTGTCTTGGCAAGTTGTCC | 194 |
| Reverse, CCACTCCGCCCTATAAGCATC | ||
| Nrf2, NM_006164.4 | Forward, AGTGGATCTGCCAACTACTC | 106 |
| Reverse, CATCTACAAACGGGAATGTCTG | ||
| 18S rRNA, NR_003286.2 | Forward, GTCTGCCCTATCAACTTTCG | 111 |
| Reverse, ATGTGGTAGCCGTTTCTCAG |
Quantification of the Mitochondrial-to-nuclear DNA Ratio
The ratio of mitochondrial to nuclear DNA was assessed by real-time PCR using the Human Mitochondrial DNA Monitoring Primer Set Ratio kit (Takara Bio, Mountain View, CA).
Statistical Analysis
Statistical analysis was performed using GraphPad software. The data are presented as the mean ± S.E. Statistical comparisons were made by performing unpaired Student's t tests unless otherwise indicated.
Author Contributions
K. B. and V. J. T. conceived and coordinated the study, designed the experiments, analyzed and interpreted the data, and wrote the manuscript. N. J. L., V. M., and G. A. B. performed the experiments and analyzed the data. J. Z., A. B. C., and V. M. D.-U. assisted with data analysis, data interpretation, and manuscript preparation.
Acknowledgments
We are grateful to UAB Nathan Shock Center (funded by National Institutes of Health Grant P30AG050886) for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants P01 HL114470 and R01 AG046210. The authors declare that they have no conflicts of interest with the content of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TFAM
- mitochondrial transcription factor A
- PGC-1α
- peroxisome proliferator-activated receptor-γ coactivator 1-α
- NRF-1
- nuclear respiratory factor-1
- Nrf2
- nuclear factor erythroid-derived 2-like 2
- Nox4
- NADPH-oxidase 4
- OCR
- oxygen consumption rate
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- ETC
- electron transport chain
- PMP
- plasma membrane permeabilizer
- NT
- non-targeting
- VDAC
- voltage-dependent anion channel
- COX IV
- cytochrome c oxidase subunit IV
- Ab
- antibody.
References
- 1. Tornatore T. F., Dalla Costa A. P., Clemente C. F., Judice C., Rocco S. A., Calegari V. C., Cardoso L., Cardoso A. C., Gonçalves A. Jr., and Franchini K. G. (2011) A role for focal adhesion kinase in cardiac mitochondrial biogenesis induced by mechanical stress. Am. J. Physiol. Heart Circ. Physiol. 300, H902–H912 [DOI] [PubMed] [Google Scholar]
- 2. Fitts R. H., Booth F. W., Winder W. W., and Holloszy J. O. (1975) Skeletal muscle respiratory capacity, endurance, and glycogen utilization. Am. J. Physiol. 228, 1029–1033 [DOI] [PubMed] [Google Scholar]
- 3. Davies K. J., Packer L., and Brooks G. A. (1981) Biochemical adaptation of mitochondria, muscle, and whole-animal respiration to endurance training. Arch. Biochem. Biophys. 209, 539–554 [DOI] [PubMed] [Google Scholar]
- 4. Gordon J. W., Rungi A. A., Inagaki H., and Hood D. A. (2001) Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. J. Appl. Physiol. 90, 389–396 [DOI] [PubMed] [Google Scholar]
- 5. Hood D. A. (2001) Invited review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J. Appl. Physiol. 90, 1137–1157 [DOI] [PubMed] [Google Scholar]
- 6. Larsson N. G., Oldfors A., Holme E., and Clayton D. A. (1994) Low levels of mitochondrial transcription factor A in mitochondrial DNA depletion. Biochem. Biophys. Res. Commun. 200, 1374–1381 [DOI] [PubMed] [Google Scholar]
- 7. Dong X., Ghoshal K., Majumder S., Yadav S. P., and Jacob S. T. (2002) Mitochondrial transcription factor A and its downstream targets are up-regulated in a rat hepatoma. J. Biol. Chem. 277, 43309–43318 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Kang D., Kim S. H., and Hamasaki N. (2007) Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 7, 39–44 [DOI] [PubMed] [Google Scholar]
- 9. Collu-Marchese M., Shuen M., Pauly M., Saleem A., and Hood D. A. (2015) The regulation of mitochondrial transcription factor A (Tfam) expression during skeletal muscle cell differentiation. Biosci. Rep. 35, e00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piantadosi C. A., and Suliman H. B. (2006) Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J. Biol. Chem. 281, 324–333 [DOI] [PubMed] [Google Scholar]
- 11. Zambrano A., García-Carpizo V., Gallardo M. E., Villamuera R., Gómez-Ferrería M. A., Pascual A., Buisine N., Sachs L. M., Garesse R., and Aranda A. (2014) The thyroid hormone receptor β induces DNA damage and premature senescence. J. Cell. Biol. 204, 129–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., and Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 13. Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., and Spiegelman B. M. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 [DOI] [PubMed] [Google Scholar]
- 14. Lehman J. J., Barger P. M., Kovacs A., Saffitz J. E., Medeiros D. M., and Kelly D. P. (2000) Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Terada S., Goto M., Kato M., Kawanaka K., Shimokawa T., and Tabata I. (2002) Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem. Biophys. Res. Commun. 296, 350–354 [DOI] [PubMed] [Google Scholar]
- 16. Irrcher I., Adhihetty P. J., Sheehan T., Joseph A. M., and Hood D. A. (2003) PPARγ coactivator-1α expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 284, C1669–C1677 [DOI] [PubMed] [Google Scholar]
- 17. Li X., Monks B., Ge Q., and Birnbaum M. J. (2007) Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature 447, 1012–1016 [DOI] [PubMed] [Google Scholar]
- 18. Jäger S., Handschin C., St.-Pierre J., and Spiegelman B. M. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kelly D. P., and Scarpulla R. C. (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 18, 357–368 [DOI] [PubMed] [Google Scholar]
- 20. Suliman H. B., Carraway M. S., Tatro L. G., and Piantadosi C. A. (2007) A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 120, 299–308 [DOI] [PubMed] [Google Scholar]
- 21. Piantadosi C. A., Carraway M. S., Babiker A., and Suliman H. B. (2008) Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merry T. L., and Ristow M. (2016) Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and antioxidant response in mice. J. Physiol. 594, 5195–5207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nisimoto Y., Diebold B. A., Cosentino-Gomes D., Constentino-Gomes D., and Lambeth J. D. (2014) Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53, 5111–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., and Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu T., Ramachandrarao S. P., Siva S., Valancius C., Zhu Y., Mahadev K., Toh I., Goldstein B. J., Woolkalis M., and Sharma K. (2005) Reactive oxygen species production via NADPH oxidase mediates TGF-β-induced cytoskeletal alterations in endothelial cells. Am. J. Physiol. Renal Physiol. 289, F816–F825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lyle A. N., Deshpande N. N., Taniyama Y., Seidel-Rogol B., Pounkova L., Du P., Papaharalambus C., Lassègue B., and Griendling K. K. (2009) Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 105, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gregg J. L., Turner R. M. 2nd, Chang G., Joshi D., Zhan Y., Chen L., and Maranchie J. K. (2014) NADPH oxidase NOX4 supports renal tumorigenesis by promoting the expression and nuclear accumulation of HIF2α. Cancer Res. 74, 3501–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mistry R. K., Murray T. V., Prysyazhna O., Martin D., Burgoyne J. R., Santos C., Eaton P., Shah A. M., and Brewer A. C. (2016) Transcriptional regulation of cystathionine-γ-lyase in endothelial cells by NADPH oxidase 4-dependent signaling. J. Biol. Chem. 291, 1774–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graham K. A., Kulawiec M., Owens K. M., Li X., Desouki M. M., Chandra D., and Singh K. K. (2010) NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 10, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kozieł R., Pircher H., Kratochwil M., Lener B., Hermann M., Dencher N. A., and Jansen-Dürr P. (2013) Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 452, 231–239 [DOI] [PubMed] [Google Scholar]
- 31. You Y. H., Quach T., Saito R., Pham J., and Sharma K. (2016) Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J. Am. Soc. Nephrol. 27, 466–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hecker L., Logsdon N. J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y. Y., and Thannickal V. J. (2014) Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 6, 231ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kansanen E., Kuosmanen S. M., Leinonen H., and Levonen A. L. (2013) The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 1, 45–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dinkova-Kostova A. T., and Abramov A. Y. (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 88, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Divakaruni A. S., Rogers G. W., and Murphy A. N. (2014) Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a clark-type oxygen electrode. Curr. Protoc. Toxicol. 60:25.2.1–16 [DOI] [PubMed] [Google Scholar]
- 36. Salabei J. K., Gibb A. A., and Hill B. G. (2014) Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 9, 421–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang J. X., Chen X., Serizawa N., Szyndralewiez C., Page P., Schröder K., Brandes R. P., Devaraj S., and Török N. J. (2012) Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 53, 289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Larsen S., Nielsen J., Hansen C. N., Nielsen L. B., Wibrand F., Stride N., Schroder H. D., Boushel R., Helge J. W., Dela F., and Hey-Mogensen M. (2012) Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 590, 3349–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Colombini M. (2004) VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 256, 107–115 [DOI] [PubMed] [Google Scholar]
- 40. Li Y., Park J. S., Deng J. H., and Bai Y. (2006) Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 38, 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Virbasius J. V., and Scarpulla R. C. (1994) Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. U.S.A. 91, 1309–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li F., Wang Y., Zeller K. I., Potter J. J., Wonsey D. R., O'Donnell K. A., Kim J. W., Yustein J. T., Lee L. A., and Dang C. V. (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 25, 6225–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu K. L., Wu C. W., Chao Y. M., Hung C. Y., and Chan J. Y. (2016) Impaired Nrf2 regulation of mitochondrial biogenesis in rostral ventrolateral medulla on hypertension induced by systemic inflammation. Free Radic. Biol. Med. 97, 58–74 [DOI] [PubMed] [Google Scholar]
- 44. Morrish F., and Hockenbery D. (2014) MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 4, a014225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pendyala S., Moitra J., Kalari S., Kleeberger S. R., Zhao Y., Reddy S. P., Garcia J. G., and Natarajan V. (2011) Nrf2 regulates hyperoxia-induced Nox4 expression in human lung endothelium: identification of functional antioxidant response elements on the Nox4 promoter. Free Radic. Biol. Med. 50, 1749–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barbour J. A., and Turner N. (2014) Mitochondrial stress signaling promotes cellular adaptations. Int. J. Cell Biol. 2014, 156020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Block K., Gorin Y., and Abboud H. E. (2009) Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. U.S.A. 106, 14385–14390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirschhäuser C., Bornbaum J., Reis A., Böhme S., Kaludercic N., Menabò R., Di Lisa F., Boengler K., Shah A. M., Schulz R., and Schmidt H. H. (2015) NOX4 in mitochondria: yeast two-hybrid-based interaction with Complex I without relevance for basal reactive oxygen species? Antioxid. Redox Signal. 23, 1106–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cassina A., and Radi R. (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch. Biochem. Biophys. 328, 309–316 [DOI] [PubMed] [Google Scholar]
- 50. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y., Nartiss Y., Steipe B., McQuibban G. A., and Kim P. K. (2012) ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 8, 1462–1476 [DOI] [PubMed] [Google Scholar]
- 52. Bartz R. R., Suliman H. B., and Piantadosi C. A. (2015) Redox mechanisms of cardiomyocyte mitochondrial protection. Front. Physiol. 6, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fernandez-Marcos P. J., and Auwerx J. (2011) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S–890S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang H. C., Nguyen T., and Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774 [DOI] [PubMed] [Google Scholar]
- 55. Itoh K., Wakabayashi N., Katoh Y., Ishii T., O'Connor T., and Yamamoto M. (2003) Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391 [DOI] [PubMed] [Google Scholar]
- 56. Chandel N. S., McClintock D. S., Feliciano C. E., Wood T. M., Melendez J. A., Rodriguez A. M., and Schumacker P. T. (2000) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138 [DOI] [PubMed] [Google Scholar]
- 57. Qutub A. A., and Popel A. S. (2008) Reactive oxygen species regulate hypoxia-inducible factor 1α differentially in cancer and ischemia. Mol. Cell. Biol. 28, 5106–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nguyen T., Nioi P., and Pickett C. B. (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291–13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Holmström K. M., Baird L., Zhang Y., Hargreaves I., Chalasani A., Land J. M., Stanyer L., Yamamoto M., Dinkova-Kostova A. T., and Abramov A. Y. (2013) Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2, 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., and Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bernard K., Logsdon N. J., Ravi S., Xie N., Persons B. P., Rangarajan S., Zmijewski J. W., Mitra K., Liu G., Darley-Usmar V. M., and Thannickal V. J. (2015) Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem. 290, 25427–25438 [DOI] [PMC free article] [PubMed] [Google Scholar]