

Abstract
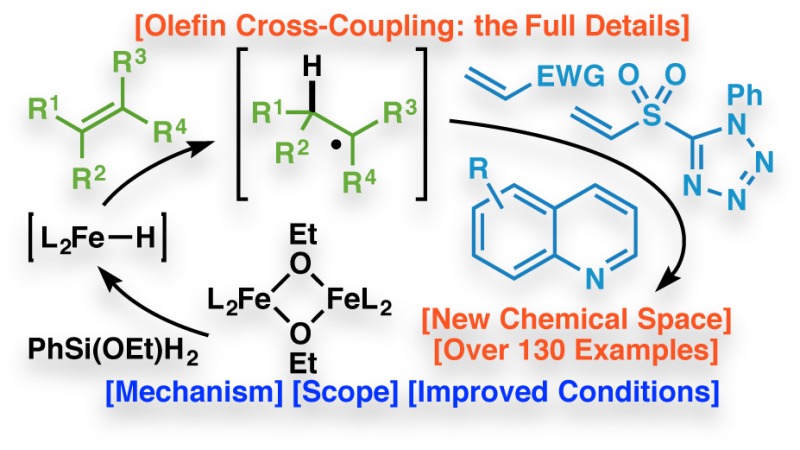
This Article details the development of the iron-catalyzed conversion of olefins to radicals and their subsequent use in the construction of C–C bonds. Optimization of a reductive diene cyclization led to the development of an intermolecular cross-coupling of electronically-differentiated donor and acceptor olefins. Although the substitution on the donor olefins was initially limited to alkyl and aryl groups, additional efforts culminated in the expansion of the scope of the substitution to various heteroatom-based functionalities, providing a unified olefin reactivity. A vinyl sulfone acceptor olefin was developed, which allowed for the efficient synthesis of sulfone adducts that could be used as branch points for further diversification. Moreover, this reactivity was extended into an olefin-based Minisci reaction to functionalize heterocyclic scaffolds. Finally, mechanistic studies resulted in a more thorough understanding of the reaction, giving rise to the development of a more efficient second-generation set of olefin cross-coupling conditions.
1. Introduction
A salient characteristic of the cyclase phase of terpene biosynthesis is the chemo-, regio-, and stereoselectivity with which olefins are cyclized, activated, and manipulated to make new C–C linkages.1 This allows Nature to avoid many of the functional group interconversions that plague chemical synthesis2,3 and is illustrated in the key steps in the cyclase phase of the eudesmanes (e.g., 1),4 the taxanes (e.g., 2),5 and the biosynthetic precursors to the ingenanes (e.g., 3)6 and tiglianes:7 the casbanes8 (Figure 1). In eudesmane biosynthesis, protonation of one of the trisubstituted olefins in germacrene A (4) triggers a cyclization to form the decalin core of the eudesmyl cation (5) and its various family members. Similarly, the taxane pathway involves an intramolecular proton transfer to a trisubstituted olefin in the verticillyl cation (7), leading to an olefin cyclization to form the taxenyl cation (8). Finally, an intramolecular cyclopropanation of the terminal olefin of geranylgeranyl pyrophosphate (10) generates casbene (12), which is converted to other casbanes, ingenanes, and tiglianes through further cyclizations and oxidations. In all three cases, our research group accomplished two-phase total syntheses of representative members of these families (e.g., 1,92,10 and 3(11)), featuring relatively mundane C–C bond-forming events tied to more daring C–H oxidations.12 Whereas this strategy can succeed in reducing step counts to complex targets,13 it largely falters in recapitulating Nature’s synthetic efficiency that arises from olefin manipulation.14 As a feedstock and ubiquitous functional group, olefins represent an ideal starting material for creating new connections.15,16
Figure 1.
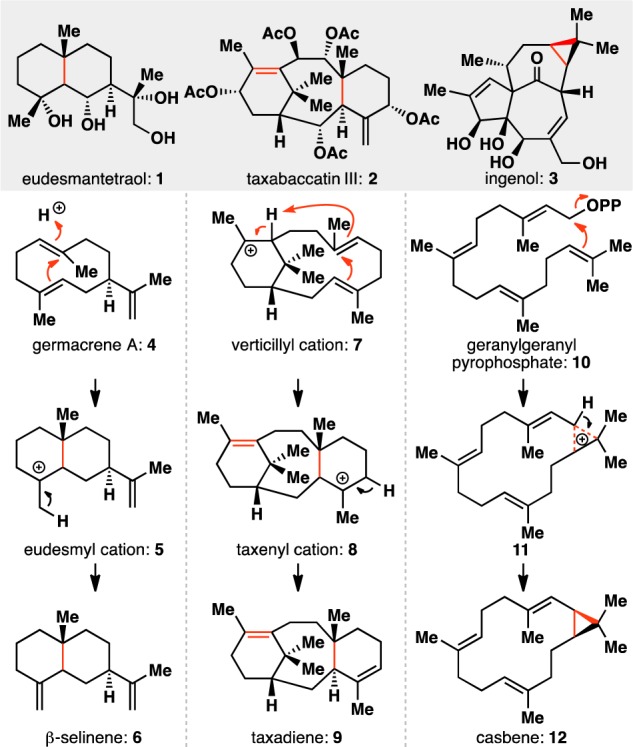
Cyclase phase of terpene biosynthesis is enabled by olefin chemistry.
Interest in replicating this aspect of terpene chemistry in the laboratory led to the discovery of a unique method for the generation of radicals from numerous classes of electron-neutral or -rich olefins and their subsequent capture with electron-deficient olefins.17,18 Building on the findings of Mukaiyama and others in this area,19 simple Fe-based catalysts and an inexpensive silane are employed. In this way, access to new chemical space is enabled in a practical fashion. This Article details the historical context, discovery, development, scope, and mechanism of this useful reaction. Several new aspects of this general transformation are reported herein for the first time, such as the development of a versatile sulfone acceptor for homologation, a Minisci-type functionalization, and a second-generation set of reaction conditions that allows for lower catalyst loadings and near equimolar quantities of both coupling partners.
2. Background
Prior work using steviol, an ent-kaurane, as a cyclase phase end point20 for an “aza-oxidase” phase of various ent-atisane and related diterpenes21 stimulated interest in pursuing the two-phase synthesis of other ent-kaurane family members.22 Plant extracts containing these natural products have been used for centuries in traditional Chinese medicine to treat inflammation, bacterial infections, malaria, and cancer.22 Over 500 ent-kauranes have been identified to date, with each family member harboring a unique oxidation barcode,23 making this class of natural products ideally suited for two-phase terpene total synthesis. Representative members belonging to the ent-kaurane family of diterpenoids (13–16) are shown in Figure 2A. By applying two-phase terpene synthesis logic12 to these natural products, a suitable cyclase phase end point was determined to be ketone 17, which contains a motif that could be disconnected through an intramolecular conjugate addition transform to give enone 18.24
Figure 2.

Impetus to develop an olefin cross-coupling.
Given the difficulty associated with using polar conjugate additions25 to generate sterically congested quaternary carbon centers,26 alternative radical processes were considered.27 Giese’s radical conjugate additions have been shown to excel in these situations,28,29 as σ bond formation occurs at relatively long lengths (calculated to be 2.55 Å for the addition of t-Bu• to methyl vinyl ketone) due to the reaction’s early transition state.30 This renders these transformations less susceptible to steric effects than the analogous polar manifolds. Using this transformation as a proposed key step in the ent-kaurane cyclase phase resulted in the identification of precursor 18, where homolysis of the C–X bond would give tertiary radical 19, which would then add into the enone to forge the desired ring system.
Examination of the literature revealed several functionalities that could be used as tertiary radical precursors (Figure 2B) to provide model systems, such as xanthate 21,31 iodide 22,32 and borane 23.33 However, such precursors are either unstable or require multiple functional group interconversions to access in certain cases.
An attractive alternative for radical generation uses an olefin such as 25 to generate the same nucleophilic radical intermediate 24 as 21–23. This obviates the need for multistep preparations of the traditional radical progenitors depicted in Figure 2B. Olefins are prevalent in feedstock chemicals, and numerous methods have been developed for their introduction into small-molecule scaffolds.15,16 Additionally, olefins are typically bench-stable, in contrast to other alternative radical precursors.
A series of mild, Markovnikov olefin hydrofunctionalizations demonstrated the feasibility of using olefins as radical precursors and were initiated by a report from Mukaiyama that detailed the use of a cobalt catalyst, O2, and Et3SiH34 or PhSiH335 to achieve hydration of an unactivated olefin, such as 26 (Figure 2C). This was later extended to an olefin hydroperoxidation36 and hydronitrosylation.37 Our repeated exposure to these powerful reactions was in the context of key steps used for the preparation of complex molecules such as (+)-cortistatin A,38,39 ouabagenin,40,41 (−)-methyl atisenoate,21 fumitremogin A,42 (+)-phorbol,43 and various polyoxypregnanes.23 Several different methods for the net hydrogenation of olefins using cobalt and manganese catalysts have been disclosed by Magnus,44 Shenvi,45 and Herzon.46,47 Furthermore, a Co(salen) complex was used by Girijavallabhan to effect a hydrothioetherification of olefins.48 Both Carreira and Boger have developed systems that are amenable to trapping with a wide variety of electrophiles, with Carreira focusing on Co and Mn catalysts49−57 and Boger utilizing Fe2(ox)3·6H2O as a stoichiometric mediator.58,59 Furthermore, Krische has discovered a related Co-based system that results in reductive aldol and Michael cycloreductions of electron-deficient olefins.60 Other groups have developed transformations involving additional redox manipulations of the intermediate radical 27, which have been reviewed elsewhere.19
Shenvi has suggested that these Mukaiyama-type transformations proceed via an initial hydrogen atom transfer (HAT) from a transition metal hydride to an olefin,19 a process that has been well-documented in the reduction of various olefins by stoichiometric transition metal hydrides (e.g., Mn(CO)5H, CpW(CO)3H, CpCr(CO)3H, and CpFe(CO)2H).61 This forms a nucleophilic radical intermediate 27, which is then trapped by a suitable electrophile. The use of a Michael acceptor as an electrophile would allow for a coupling of two electronically-differentiated olefins (e.g., 26 and 34), which forms the foundation of this report.
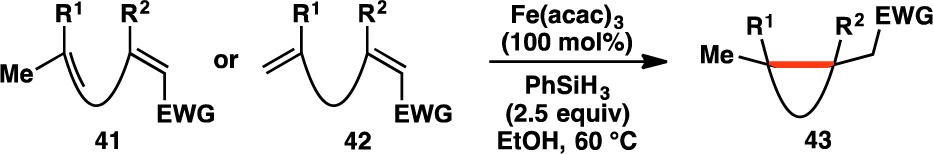
3. Olefin Cross-Coupling
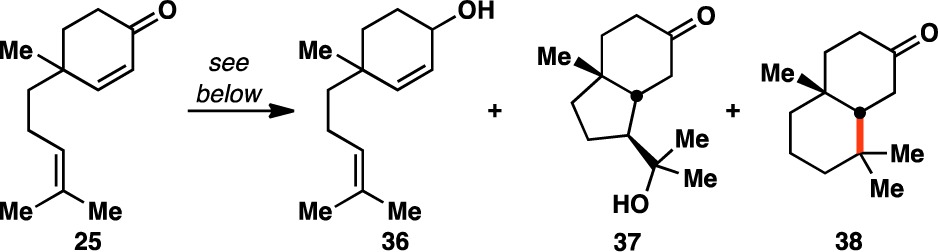
In surveying the field of radical-based Markovnikov olefin hydrofunctionalizations, we found the conditions developed by Boger to be an attractive starting point.58 As their system utilized a wide variety of electrophiles (i.e., NaN3, KSCN, O2, ArSO2Cl, KOCN, TsCN, TEMPO, NaNO2, and Selectfluor), it stood to reason that electron-deficient olefins might be employed as radical traps as well. Using diene 25 as a model system for the ent-kaurane cyclization phase end point 17, application of Boger’s conditions led to the formation of decalin 38 (Table 1, entry 1). However, this was accompanied by reduction of the enone moiety of 25 to give allylic alcohol 36.62 Given the facility with which NaBH4 reduces ketones, other milder terminal reductants were examined (entries 2–5). Although NaBH(OAc)3 and PhSiH3 provided the desired decalin 38 without forming 36, their use led to the formation of the hydroxy indanone 37. This byproduct was exclusively formed with (TMS)3SiH and Et3SiH, suggesting that these silanes are incapable of forming the requisite Fe hydride (vide infra).
Table 1. Optimization of the Reductive Diene Cyclization.
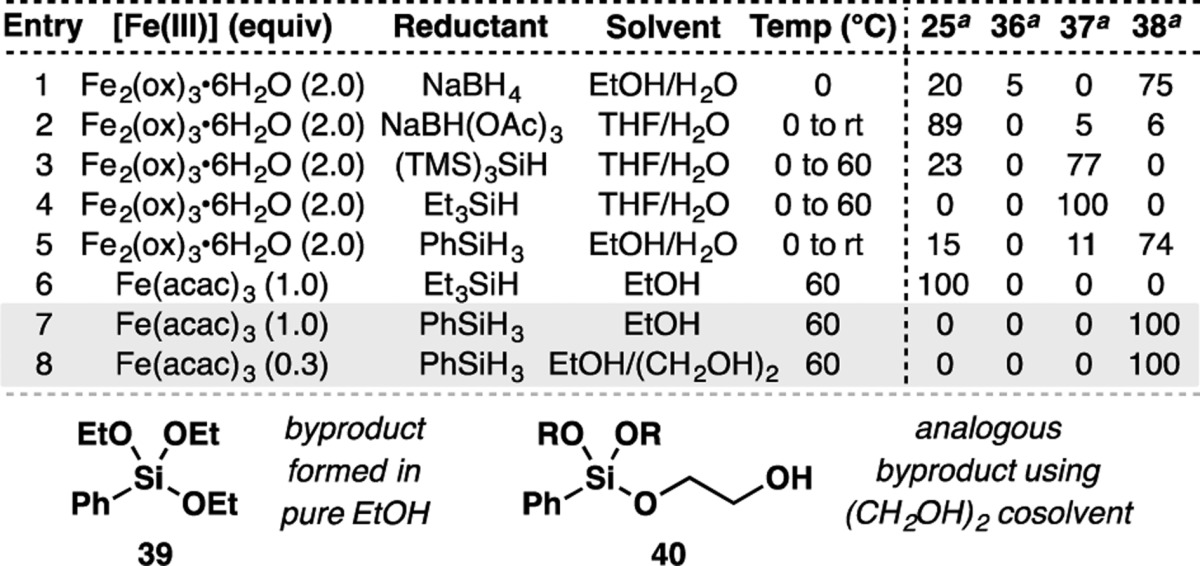
Ratios determined by GC/MS.
The generation of hydroxy indanone 37 presumably arises from a vinylogous Prins addition of the trisubstituted olefin into the enone, followed by trapping of the resultant tertiary carbocation with a molecule of water from the reaction solvent. Snider has shown that the conversion of 25 to scaffolds like 37 can be promoted by Lewis acids.63 Reasoning that competitive formation of 37 was caused by the Lewis acidity of Fe2(ox)3·6H2O, alternative Fe(III) sources were screened. Similar to the case of Fe2(ox)3·6H2O, the use of Et3SiH in concert with Fe(acac)3 gave no reactivity (entry 6), whereas PhSiH3 was found to facilitate the desired transformation without the formation of indanone 37 (entry 7).
However, even after purification by silica gel chromatography, samples of products made using the conditions shown in entry 7 were frequently contaminated with PhSi(OEt)3 (39). As this byproduct was formed from the EtOH solvent, ethylene glycol was added as a cosolvent to favor the analogous formation of the more polar and easily separable 40. It was also found that the reaction could also be run with a substoichiometric loading of Fe(acac)3 (30 mol%) without altering the reaction outcome (entry 8).
Using the reaction conditions developed in entries 7 and 8, various terpene and terpenoid scaffolds were cyclized (Table 2). Decalin 38 was isolated in 60% yield. The skipped diene moiety of α-ionone (44) could be cyclized to cyclopropane 45 nearly quantitatively on a small scale and in 81% yield on gram scale. Additionally, (+)-nootkatone (46) could be cyclized to the fused bicyclo[2.2.1]hepane 47, which bears three contiguous quaternary carbon centers. Mixtures of geranyl and neryl derivatives 48, 50, and 52 provided cyclopentanes where two vicinal quaternary carbon centers are generated (49, 51, and 53), demonstrating that esters, amides, and aldehydes could be tolerated under the reaction conditions. As previously theorized, the early transition state of the radical-based reaction allowed for the facile construction of the sterically congested environments present in 38, 45, 47, 49, 51, and 53.30
Table 2. Scope of the Intramolecular Reductive Olefin Cyclization.
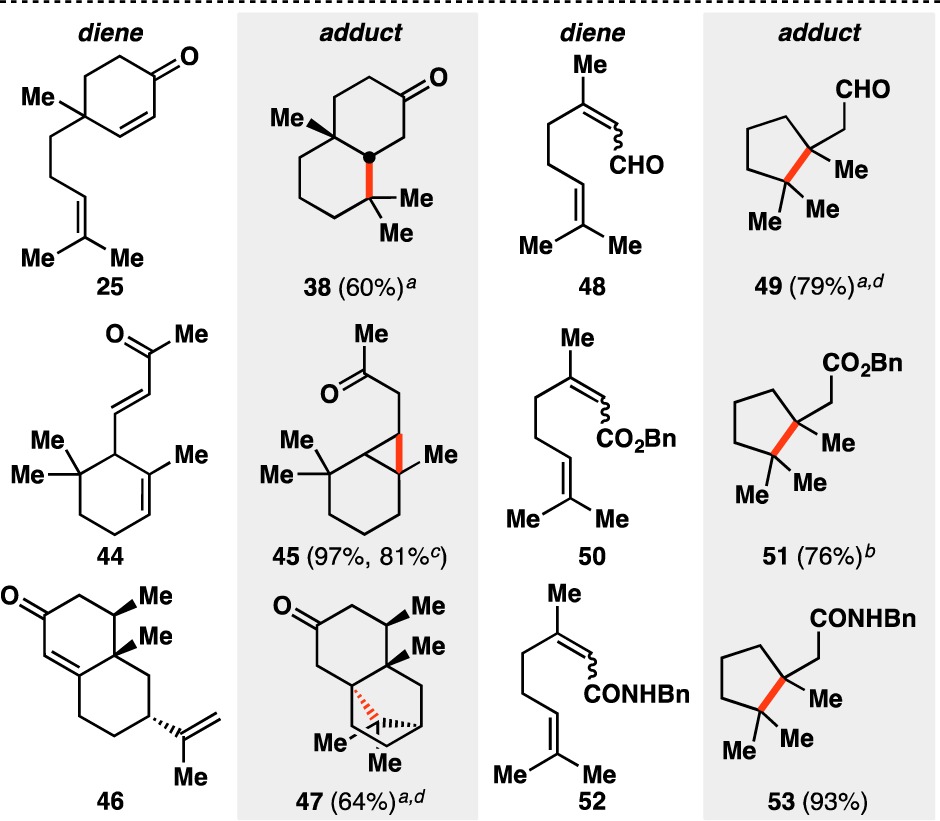
Yields in parentheses are isolated yields. Data were originally reported in ref (17).
30 mol% Fe(acac)3 in 5:1 EtOH/(CH2OH)2 used.
20 mol% Fe(acac)3 in 5:1 EtOH/(CH2OH)2 used.
Run on gram scale.
1.5 equiv PhSiH3 used.
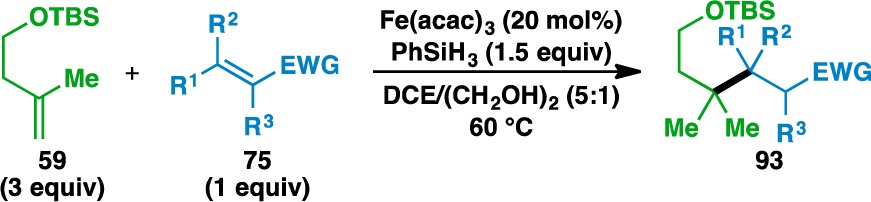
The expansion of this reaction to an intermolecular setting (Tables 3–5) was pursued next and complemented by a report from Overman and co-workers detailing the activation of tertiary alcohols as the corresponding N-phthalimiodyl oxalates for tertiary radical conjugate addition.64 This would allow the unification of two separate olefin coupling partners: an electron-rich donor olefin (shown in green) and an electron-deficient acceptor olefin (shown in blue). The scope of the donor olefin component was first examined using methyl vinyl ketone (55) as an acceptor (Table 3). As with the intramolecular cyclizations, the intermolecular coupling is relatively insensitive to sterics and can generate quaternary carbon centers from trisubstituted olefins (e.g., 57, 67, and 71) and geminally disubstituted olefins (e.g., 59, 61, and 73). In the case of 73, the corresponding adduct 74 bears two vicinal quaternary carbon centers. The reaction proceeded in the presence of silyl-protected alcohols and Boc-protected amines to generate 60 and 62, respectively. Despite its tendency to polymerize under free radical reactions,65 styrene (63) could be used in the reaction to give ketone 64.
Table 3. Donor Scope of the Olefin Cross-Coupling.
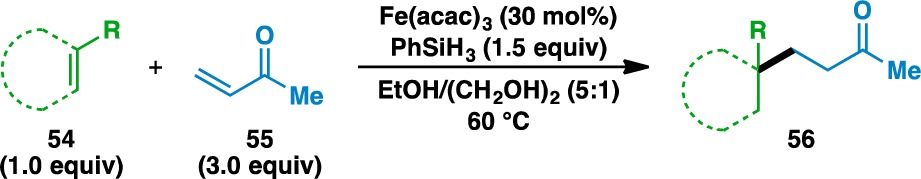
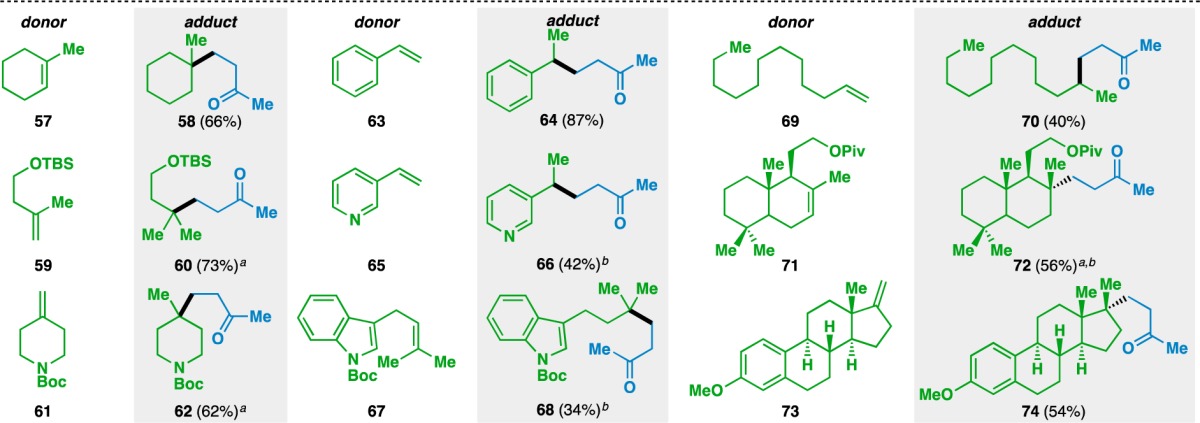
Yields in parentheses are isolated yields. Data were originally reported in ref (17).
2.5 equiv PhSiH3 used.
100 mol% Fe(acac)3 used.
Table 5. Acceptor Scope of the Olefin Cross-Coupling, Continued.
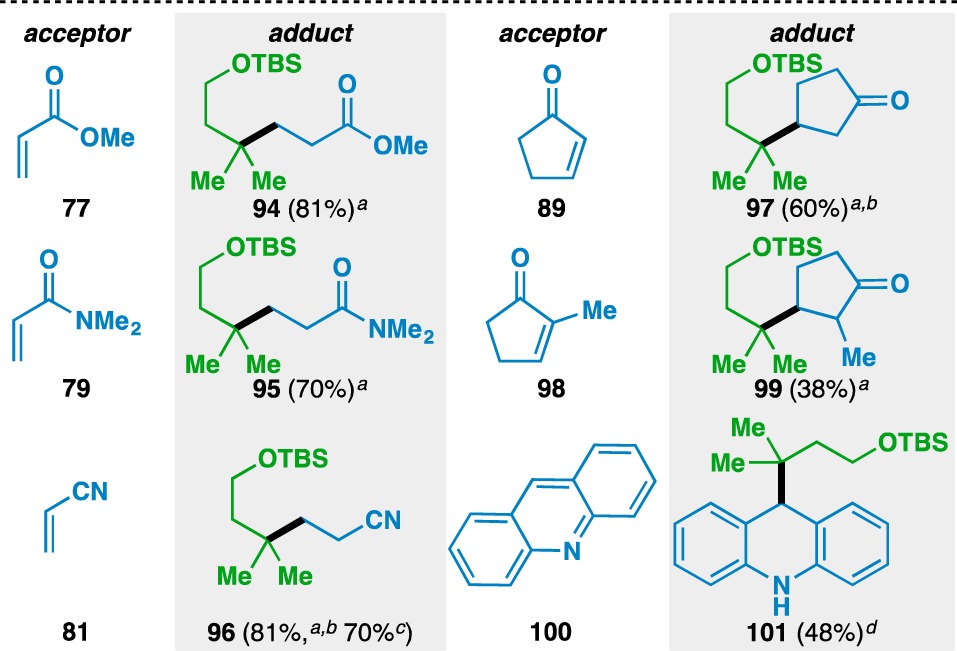
Yields in parentheses are isolated yields. Data were originally reported in ref (17).
1 equiv donor and 3 equiv acceptor used.
40 mol% Fe(acac)3 used.
Run on gram scale.
1 equiv donor and 1.1 equiv acceptor used.
Heteroaromatic motifs such as pyridines (e.g., 65) and indoles (e.g., 67) could also be present in the donor olefin, although their use required stoichiometric amounts of Fe(acac)3. Although the majority of the substrates generated either tertiary or benzylic radicals, this was not a requirement for the reaction, as monosubstituted olefins could also be used (e.g., 69). Finally, the use of natural product scaffolds in the reaction was demonstrated by the formation of 72 from scalerolide derivative 71 and that of 74 from estrone derivative 73. In each of these cases, the stereoselectivity of the reaction was controlled by the rigid, polycyclic substrate.66
The scope of the acceptor olefins was next probed using 1-methylcyclohexene (57) as the donor (Table 4). In addition to ketones, esters (e.g., 77) and amides (e.g., 79) could be used to activate the acceptor olefin. Nitriles (e.g., 81) and sulfones (e.g., 83) were also competent electron-withdrawing groups in the reaction. Acyclic (e.g., 85) and cyclic (e.g., 87, 89, and 91) disubstituted olefins could also be used, although five-membered-ring enones (e.g., 89) gave higher yields than the corresponding six-membered-ring enones (e.g., 91).
Table 4. Acceptor Scope of the Olefin Cross-Coupling.
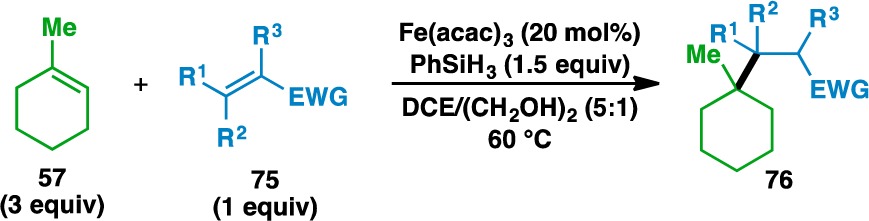
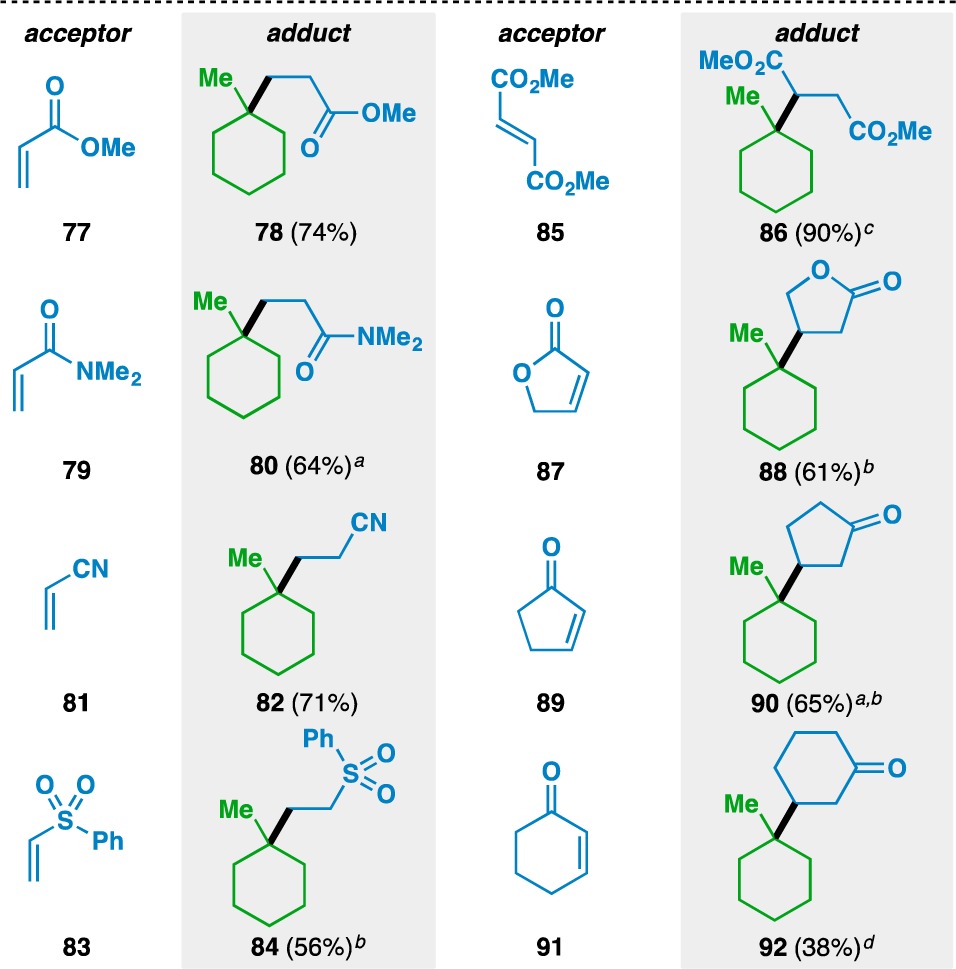
Yields in parentheses are isolated yields. Data were originally reported in ref (17).
1 equiv donor and 3 equiv acceptor used.
40 mol% Fe(acac)3 used.
DCE/(CH2OH)2 (1:1) used.
100 mol% Fe(acac)3 used.
Additional insights into the scope of the acceptor olefin were gained using geminally disubstituted 59 as the donor component (Table 5). As in the case with 1-methylcyclohexene, methyl acrylate (77), N,N-dimethylacrylamide (79), and acrylonitrile (81) could all be used as acceptor olefins. Alkyl substitution adjacent to the electron-withdrawing group in the acceptor olefin decreased the reaction yield, as evidenced by the yield using 2-cyclopentenone (89, 60%) vs 2-methyl-2-cyclopentenone (98, 38%). Finally, acridine (100) could be used as an acceptor to give the reductively functionalized 101 in 48% yield, which prompted the development of an olefin-based Minisci reaction67 (vide infra).
4. Functionalized Olefin Cross-Coupling
Although the olefin cross-coupling had demonstrated that olefins can serve as convenient radical precursors, those same intermediates could already be accessed through other means. Specifically, many of the products shown in Tables 3–5 have been prepared using chemistry recently developed by Overman.64 However, substitution of the donor olefin with functionality based on heteroatoms such as oxygen, nitrogen, sulfur, silicon, boron, and the halogens would lead to a group of functionalized donor olefins represented by 102 (Figure 3A). HAT to these donor olefins would generate the corresponding α-heteroatom-bearing radical 103, which would add into an acceptor olefin (e.g., 55) to give 104.
Figure 3.
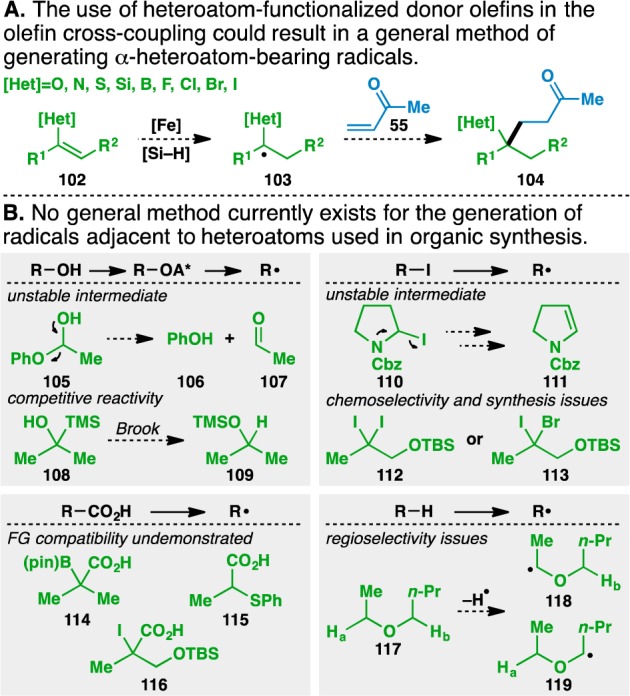
Implications of the creation of a functionalized olefin cross-coupling.
Unlike the previous donor olefins used, a general method of accessing the requisite α-heteroatom-bearing radicals is currently unavailable (Figure 3B). Existing chemistry does allow for the formation of these radicals in certain cases, but these methods are not universally applicable to all the heteroatoms shown in Figure 3A. For example, alcohol activation and homolytic fragmentation is limited by the ability to form the requisite alcohol. Application of these types of methods to form α-oxy radicals would require the intermediacy of hemiketal 105, which would likely undergo facile elimination to phenol (106) and acetaldehyde (107).68 Accessing α-silyl radicals through similar means necessitates the activation of alcohol 108, which would likely undergo a competitive Brook rearrangement69 to give 109 instead of the desired α-silyl radical precursor.
Similarly, using alkyl iodide homolysis to give α-heteroatom-bearing radicals can be complicated by the presence of neighboring oxygen, nitrogen, and sulfur atoms. Thus, the unstable iodocarbamate 110(70) can readily eliminate to give olefin 111. Additionally, using this method to generate α-iodo and α-bromo radicals can lead to chemoselectivity issues, as in the case of geminal dihalides 112 and 113. Furthermore, it is unclear how to access these dihalide motifs using existing methodology.
Although decarboxylative radical generation provides an attractive means of generating radicals adjacent to oxygen and nitrogen atoms, its compatibility with the functionalities contained in boronic ester 114, thioether 115, and iodide 116 has not been demonstrated.71 Furthermore, while this method works well to incorporate structures from commercially available carboxylic acids, the incorporation of more complex motifs is limited by the ability to synthesize the corresponding carboxylic acids.
Additionally, hydrogen atoms adjacent to oxygen and nitrogen atoms have a lower bond dissociation energy than the parent alkanes due to the radical stabilizing effect of these heteroatoms. Thus, abstraction of these hydrogen atoms provides direct access to α-oxy72 and α-amino73,74 radicals. However, in the case of unsymmetrical molecules such as butyl ethyl ether (117), direct hydrogen atom abstraction can provide regioisomeric mixtures of radicals (e.g., 118 and 119). Furthermore, the conditions used in these transformations can be harsh, and this method is generally inapplicable to form radicals adjacent to heteroatoms other than oxygen and nitrogen.
As suitably functionalized olefins could conceivably serve as universal precursors for a wide variety of α-heteroatom-bearing radicals,75 optimization of the heteroatom-functionalized olefin coupling was undertaken using either silyl enol ether 120 or 121 as the donor component (Figure 4A). Although Fe(acac)3 provided a 53% yield of 122 (Figure 4C, entry 1), it was only able to provide 123 in trace quantities. Competing side reactions in each of these cases involved reduction of the olefin starting materials to their saturated alkyl counterparts 124–126 (Figure 4B). Ketals 127 and 128 were also obtained as byproducts in the reaction, presumably arising through α-protonation of the silyl enol ether, followed by trapping of the resultant oxocarbenium ion by EtOH from the reaction solvent. Diketone 129 was also isolated from the reaction mixture, which indicated that the same oxocarbenium ion could also be trapped by one of the ligands from Fe(acac)3.76
Figure 4.
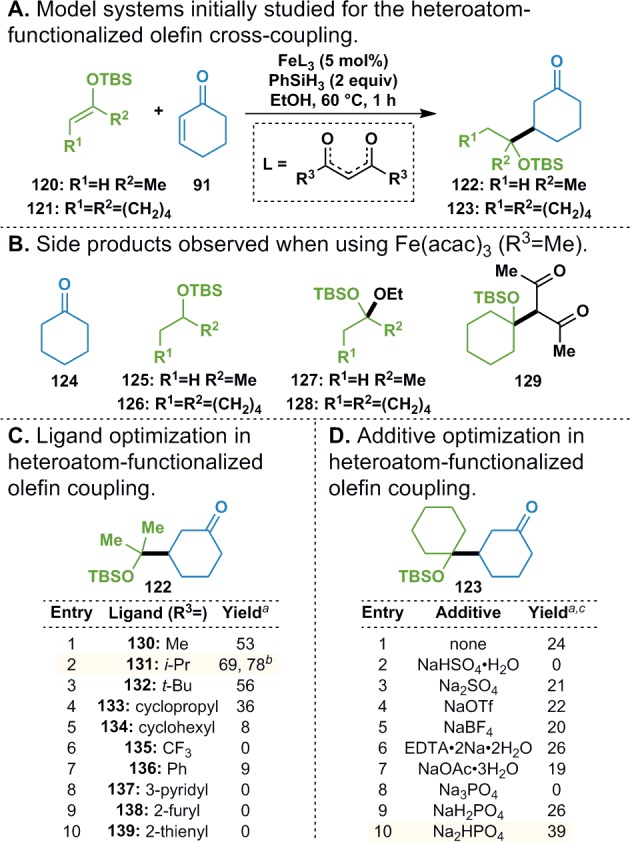
Functionalized olefin cross-coupling optimization. aYield determined by GC/MS. bWith the addition of 1 equiv Na2HPO4. cUsing 5 mol% Fe(dibm)3 (131).
To improve the efficiency of the reaction, the effects of the ligand structure on the reaction outcome were probed. It was found that changing to a slightly bulkier diisobutyromethane77 (dibm) ligand (131) improved the yield of 122 to 69%. Further increasing the size of the R3 group led to diminished yields (entries 3–5). The added steric bulk of the isopropyl group may decrease the rate of catalyst decomposition, but larger substituents slow down the desired reactivity and result in lower yields of 122.
Altering the electronics of the ligand by switching to the more electron-deficient Fe(hfac)3 (135) completely ablated the desired reactivity (entry 6), presumably due to the increased Lewis acidity of the catalyst. Aryl diketone ligands were next pursued in the hopes that their electronic properties would be more tunable; however, switching to a dibenzoylmethane ligand (136) decreased the reaction yield to 9% (entry 7). Analogous ligands bearing pyridyl (137), furyl (138), and thienyl (139) groups inhibited the formation of any desired product (entries 8–10).
Although Fe(dibm)3 catalyzed the formation of 123 in 24% yield (Figure 4D, entry 1), a significant amount of the donor olefin was converted into 128 and 129. As these byproducts presumably arose from an oxocarbenium ion, it was hypothesized that inhibiting silyl enol ether protonation would increase the yield of the desired adduct. A Brønsted acidic complex of Fe(dibm)3 and EtOH formed in situ could conceivably cause this protonation. As the addition of external Brønsted acids (e.g., NaHSO4·H2O, entry 2) provided evidence for this notion by inhibiting reactivity, efforts were taken to buffer the reaction system. Examination of a wide variety of Na salts led to negligible effects on the reaction outcome (entries 3–7 and 9), except for Na3PO4 (entry 8), which resulted in no product formation. Interestingly, the use of Na2HPO4 increased the yield of the reaction to 39% (entry 10). Although this phenomenon can be attributed to the ability of Na2HPO4 to buffer the reaction system, it is also likely due in part to the insolubility of Na2HPO4 in EtOH. If an aqueous phosphate buffer solution is used or if enough water is added to the reaction mixture to solubilize the Na2HPO4, yields of the desired product decrease.
This beneficial effect was also observed on the formation of 122, where addition of Na2HPO4 increased the yield from 69% to 78% (Figure 4C). The optimized conditions used 5 mol% Fe(dibm)3, 2 equiv PhSiH3, and 1 equiv Na2HPO4 to couple the silyl enol ether donor olefins with a threefold excess of cyclohexenone at 60 °C in EtOH.
The scope of the functionalized olefin cross-coupling was next examined using a wide range of acceptor olefins (Figure 5). Starting with (silyl) enol ether donor olefins (Table 6), it was found that acyclic (120) or cyclic (141, 143, 121, and 145) silyl enol ethers could be used. The yields of the silyl ether products tended to decrease as the size of the silyl group increased. For example, the TMS silyl enol ether 141 gave 142 in 46% while the TIPS silyl enol ether 145 gave 146 in 9% yield. Simple enol ethers, such as 2,3-dihydrofuran (147) and 3,4-dihydro-2H-pyran (149) could also be used, where the adducts 148, 150, and 151 were isolated as single regioisomers. As HAT to either carbon of the olefin would result in the formation of a 2° radical in both cases, the regioselectivity of the reaction is governed by the additional stability that the oxygen atom imparts on the adjacent radical.78 The use of alkyl (e.g., 152) and aryl (e.g., 154) vinyl ethers as donor olefins generated the branched adducts 153 and 155–157.
Figure 5.
Acceptor olefins used in the heteroatom-functionalized olefin cross-coupling.
Table 6. Scope of the (Silyl) Enol Ether Donor Olefins.
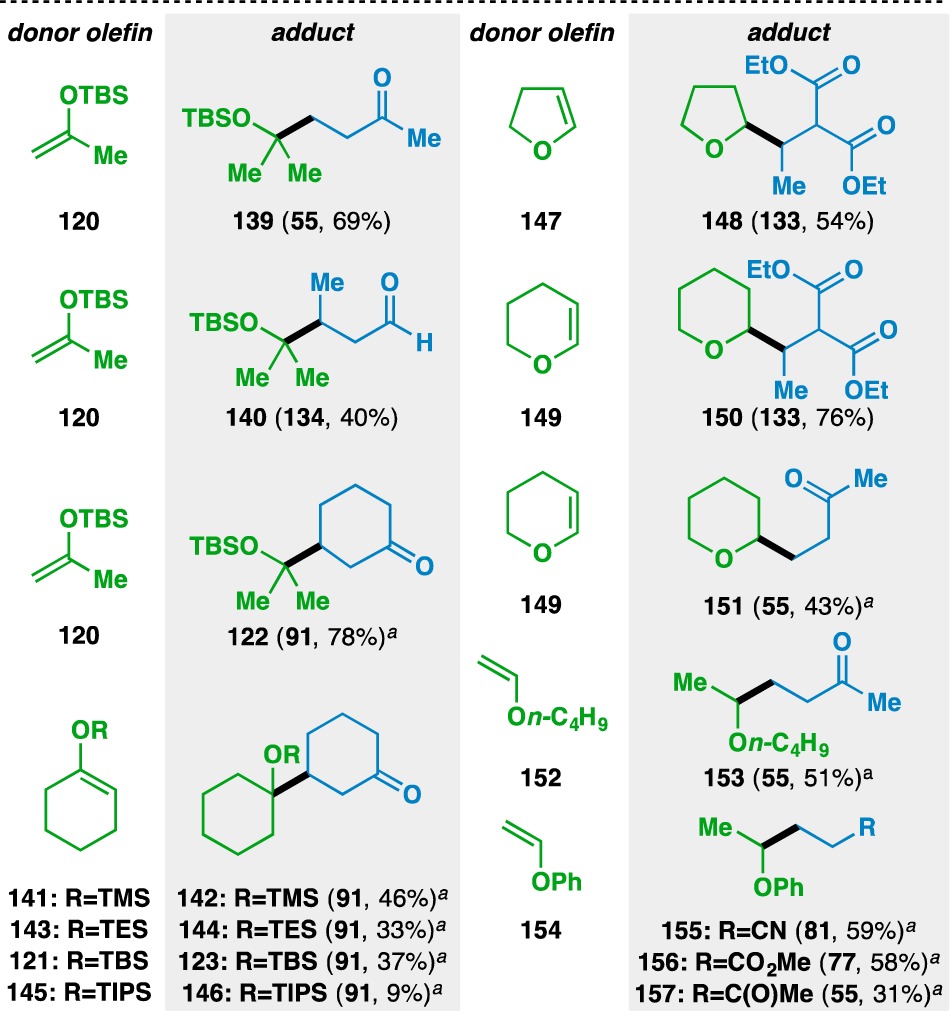
Yields in parentheses are isolated yields. Compound labels in parentheses indicate the acceptor olefin used. Data were originally reported in ref (18).
3 equiv donor and 1 equiv acceptor used.
As enamines bear electronic properties similar to those of (silyl) enol ethers, they were also examined in the functionalized olefin cross-coupling (Table 7). Early experiments revealed that the nitrogen atom of the enamines needed to be protected with an electron-withdrawing group to suppress conventional Stork enamine Michael addition.79 Similar to the case of the enol ethers, bond formation occurred adjacent to the nitrogen atom when using Cbz-protected 2,3-dihydropyrrole (111) and 3,4-dihydro-2H-pyridine (164), generating adducts 160–163 and 165–166. The nitrogen atom could also be protected as an amide (e.g., 167) and various acyclic enecarbamates could also be used in the reaction (e.g., 169, 171, 173, 176, and 178). Attempts to render the reaction stereoselective using (−)-α-phenylethylamine as a chiral auxiliary80 provided the desired 177 and 179 in only 1:1.5 and 1:1.4 dr, respectively. Recently, Fu has shown that high levels of diastereoselectivity in the olefin cross-coupling can be achieved through an acceptor-based chiral auxiliary approach.81
Table 7. Scope of the Enecarbamate and Enamide Donor Olefins.
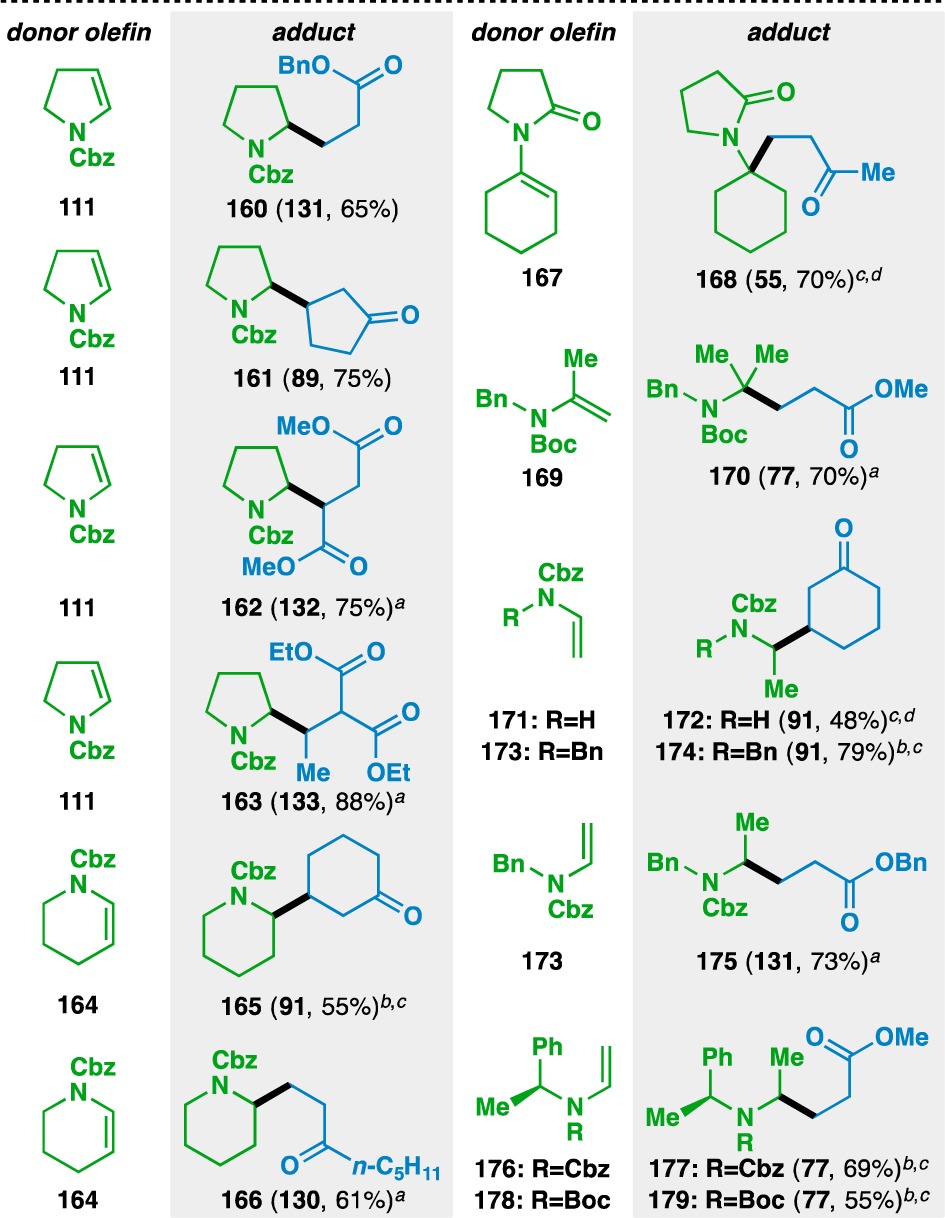
Yields in parentheses are isolated yields. Compound labels in parentheses indicate the acceptor olefin used. Data were originally reported in ref (18).
2 equiv PhSiH3 used.
6 equiv acceptor used.
15 mol% Fe(dibm)3 used.
Second portion of Fe(dibm)3, acceptor, and PhSiH3 added after 1 h.
The reaction was then extended to thioenol donor olefins (Table 8). Isopropenyl and vinyl motifs attached to the sulfur atom in 182, 185, 187, and 197 could be coupled to acrylonitrile (81) and N,N-dimethylacrylamide (79) to provide 183, 184, 186, 188, 189, and 198. The donor olefin could also be endocyclic, as evidenced by the formation of ketone 191 and nitrile 192 from 190. The bond formation in the case of 1,2-disubstituted olefins selectively occurs adjacent to the sulfur atom, as shown by the formation of 194 and 196. Heterocyclic functionalities, such as a benzothiazole, could also be incorporated in the donor olefins (e.g., 197). Notably, the vast majority of the reactions using thioenols as the donor component proceeded at room temperature.
Table 8. Scope of the Thioenol Donor Olefins.
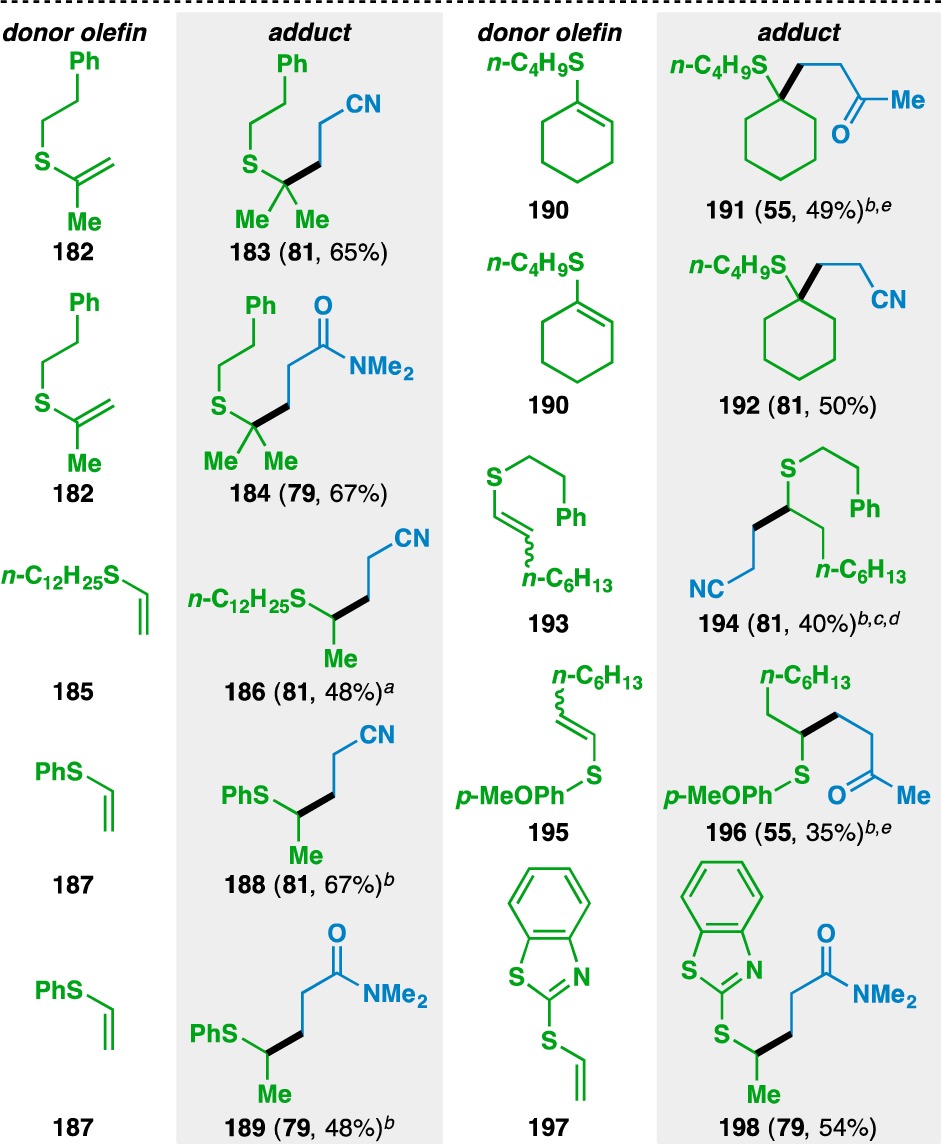
Yields in parentheses are isolated yields. Compound labels in parentheses indicate the acceptor olefin used. Data were originally reported in ref (18).
Heated at 60 °C.
6 equiv PhSiH3 used.
15 mol% Fe(dibm)3 used.
Second portion of Fe(dibm)3, acceptor, and PhSiH3 added after 1 h.
6 equiv acceptor used.
With the finding that oxygen, nitrogen, and sulfur-based functionalities could adorn the donor olefin, other heteroatom substitution about the donor olefin was pursued. Over the course of these explorations, it was determined that Fe(acac)3 provided higher yields than Fe(dibm)3 when the heteroatom did not bear Lewis basic electron pairs. Donor olefins containing boron-based functionality participated in the functionalized olefin cross-coupling (Table 9). Pinacol boronic esters (e.g., 201, 208, and 210), N-methyliminodiacetic acid esters (e.g., 203), and 1,8-diaminonaphthalene boronamides (e.g., 205) were all found to be competent coupling partners. Allylic silyl ethers and carbamates were also tolerated, as demonstrated by the formation of 209 and 211.
Table 9. Scope of the Alkenyl Boronate and Boronamide Donor Olefins.
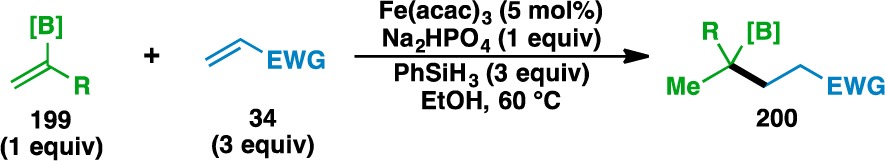
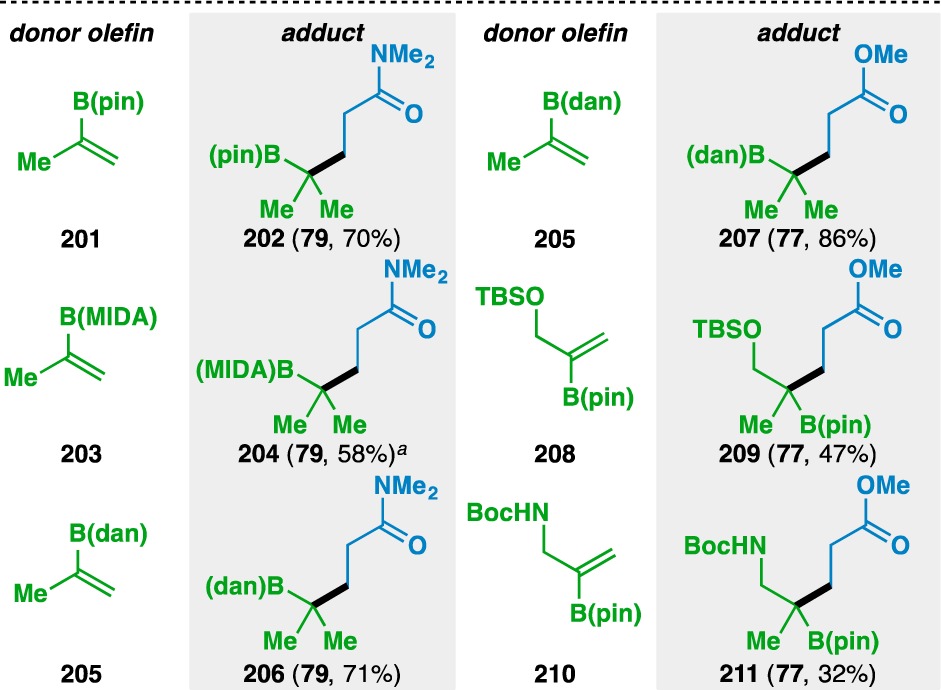
Yields in parentheses are isolated yields. Compound labels in parentheses indicate the acceptor olefin used. Data were originally reported in ref (18).
THF used as a cosolvent.
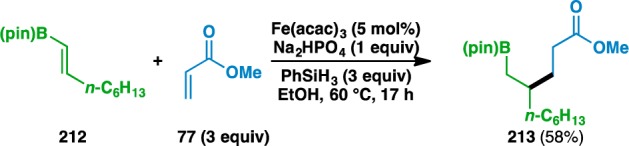
Like in the previous cases, bond formation occurred adjacent to the heteroatom, although it was found that the regioselectivity of the bond formation was controlled by the geminal disubstitution about the donor olefin, not by the location of the heteroatom. Specifically, the use of 1,2-disubstituted olefin 212 led to 213, where bond formation occurred distal to the boron atom (Scheme 1). This result was surprising, as the pinacol boronic ester was expected to stabilize an adjacent radical by delocalization into the empty p orbital.82 Although the regiochemistry of the olefin cross-couplings is typically thought to be governed by the stability of the intermediate radical, the formation of 213 suggests that in certain instances, other factors might influence the regioselectivity of the initial HAT.
Scheme 1. Unexpected Regiochemical Outcome of an Alkenyl Boronic Ester Coupling.
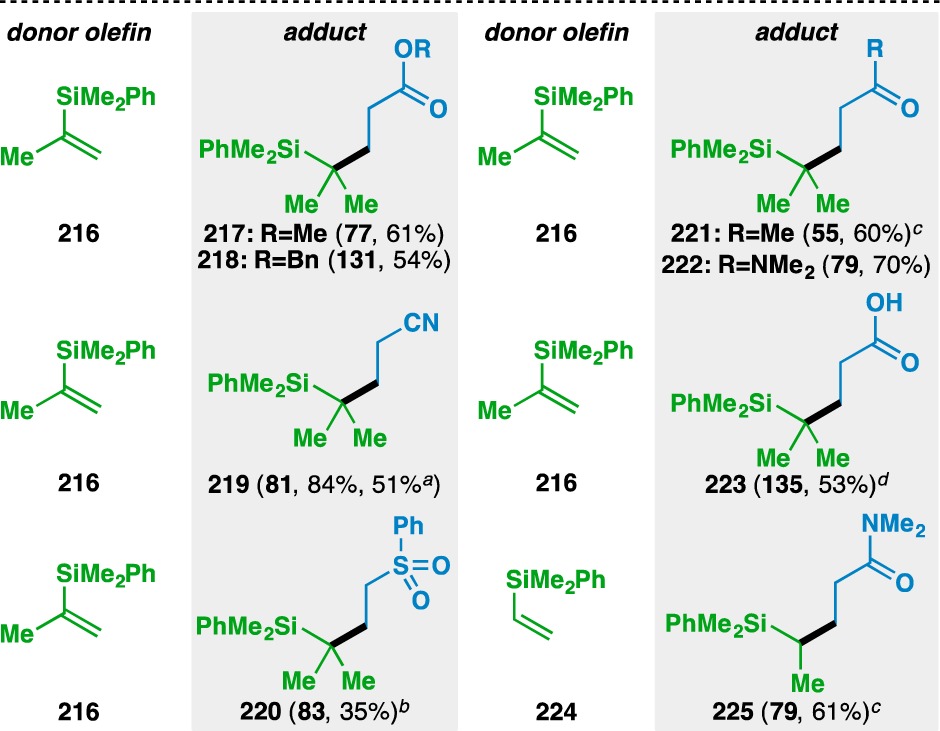
Alkenyl silanes were also found to undergo the olefin cross-coupling to deliver various alkyl silanes (Table 10). Preliminary studies revealed that switching the reaction solvent to n-PrOH aided the purification of the desired adducts by rendering the alkoxysilane byproducts less polar. Isopropenyl silane 216 was coupled with a wide variety of electron-deficient olefins to provide 217–223. In addition to the conventional carbonyl-based functionality that activates Michael acceptors, acrylic acid (135) could also be used if Na2HPO4 was omitted from the reaction mixture to access the free carboxylic acid 223. The inclusion of Na2HPO4 inhibited the desired reactivity, where the formation of the corresponding carboxylate salt presumably renders the acceptor olefin less electrophilic. Furthermore, vinyl silane 224 could be used to fashion 225 in 61% yield.
Table 10. Scope of the Alkenyl Silane Donor Olefins.
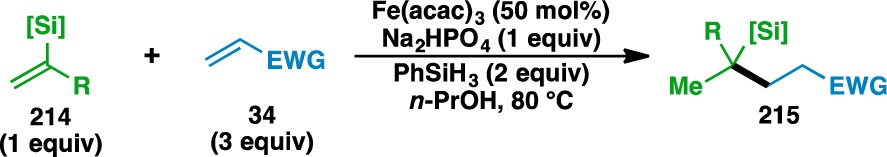
Yields in parentheses are isolated yields. Compound labels in parentheses indicate the acceptor olefin used. Data were originally reported in ref (18).
Run on gram scale.
100 mol% Fe(acac)3 used.
6 equiv acceptor used.
Na2HPO4 omitted.
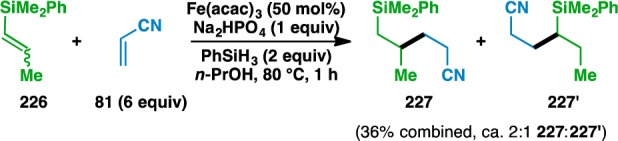
Partial β-selectivity for bond formation in the coupling of 1,2-disubstituted alkenyl silane 226 was observed to give a ca. 2:1 mixture of 227:227′ (Scheme 2). Although not completely selective, it is conceivable that this regiochemical outcome is a manifestation of the β-silicon effect83 since the corresponding intermediate radical is an electron-deficient species.
Scheme 2. Unexpected Regiochemical Outcome of an Alkenyl Silane Coupling.
As a final testament to the scope and chemoselectivity of the reaction, alkenyl halides were found to be competent donor olefins under the reaction conditions (Table 11). Functionalized olefin cross-coupling opens the possibility of incorporating alkyl fluoride motifs into molecules,84 as evidenced by the formation of 231 from alkenyl fluoride 230. 2-Chloroallyl alcohol derivatives 232 and 235 could be coupled smoothly to N,N-dimethylacrylamide (79) and to acrylic acid (135) in the absence of Na2HPO4. Hydroxyl groups did not need to be protected, as evidenced by the formation of 238. The reaction even allowed for the synthesis of tertiary alkyl bromide 240 and iodide 242. Such motifs are sensitive to various decomposition pathways, such as elimination and C–X bond homolysis,85 and their tolerance demonstrates the reaction’s mildness and chemoselectivity.
Table 11. Scope of the Alkenyl Halide Donor Olefins.
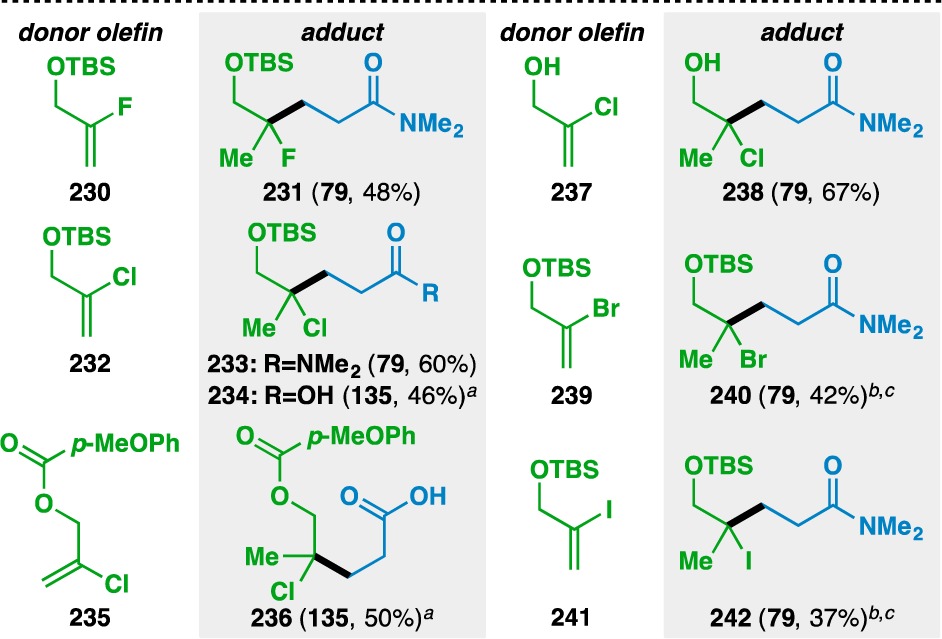
Yields in parentheses are isolated yields. Compound labels in parentheses indicate the acceptor olefin used. Data were originally reported in ref (18).
Na2HPO4 omitted.
6 equiv acceptor used.
6 equiv PhSiH3 used.
The functionalized olefin cross-coupling could also be used to modify natural products scaffolds (Scheme 3). For example, the silyl enol ether 243 derived from estrone could be used to fashion a fully substituted neopentyl carbon center. Additionally, glucal derivatives 245 and 247 could be used to access ketones 246 and 248. The diastereoselectivity of the coupling is controlled by an anomeric effect, which stabilizes the axial α-oxy radical.86 The previously disclosed synthesis of 246 entails a three-step process involving chlorination of the anomeric position of 245, followed by lithiation, two transmetalations, and finally conjugate addition into methyl vinyl ketone (55).87
Scheme 3. Functionalized Olefin Cross-Coupling of Natural Product Scaffolds.
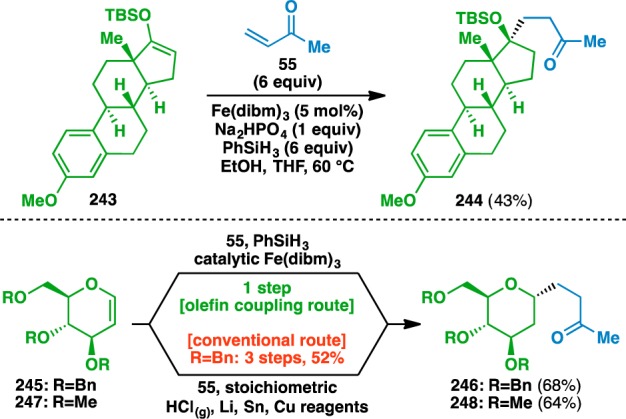
Overall, the observed regioselectivity in these reactions appears, with few exceptions, to be dictated by the initial HAT, where C–C bond formation occurs at the site of the more stable radical intermediate.88
5. Functionalized Olefin Cross-Coupling: Implications for Retrosynthetic Analysis
Many of the adducts generated using this method represent new chemical entities. Although it could be argued that umpolung strategies could be used to access motifs like those depicted in Tables 6 and 7,89 the various substitution patterns on the oxygen or nitrogen atom at the γ position would likely need to be incorporated after the C–C bond formation. Thus, olefin cross-coupling offers a direct means of accessing these exotic substitution patterns in a single step, leading to a higher degree of convergence.
Thioenols have not been as privileged as enol ethers and enamines in organic synthesis, but their use as donor olefins has led to the formation of adducts that would be difficult to construct using conventional chemistry. Like the hydrothiolation developed by Girijavallabhan,48 olefin cross-coupling gives rise to tertiary thioethers. However, conventional thiol alkylation90 to access such motifs would likely be difficult to perform, as it would require tertiary alkyl halides.91 Additionally, although addition of a thiyl radical across an olefin could conceivably be used to construct the products depicted in Table 8, this would proceed with the opposite regiochemistry to give linear products instead of the branched analogues.92
Alternative formation of the adducts generated from the boron- and silicon-substituted donor olefins would likewise be difficult. Conventional hydroboration93 or hydrosilylation94 approaches of an olefin would deliver the alternative anti-Markovnikov regioisomers, where the heteroatom would no longer be attached to the more hindered carbon atom. Borylation95 or silylation96 of an alkyl organometallic species could potentially give the same products, but this would require metal–halogen exchange of the corresponding alkyl iodide, which itself would be difficult to prepare using methods other than the developed olefin cross-coupling. Because the adducts depicted in Tables 9 and 10 are arduous to access using existing methodology, they represent “alien” entities, which can now be easily created under the manifold of olefin cross coupling.
In a similar vein to alkenyl silicon and boron species, the reactivity of alkenyl halides is typically limited to the realm of transition metal-catalyzed cross couplings, where the halogen atom is used as disposable handle for C(sp2)–C(sp2 or sp3) bond formation.97 Olefin cross-coupling instead conserves this halogen atom in the products and generates two new sp3 carbon centers. Additionally, existing methodology does not allow for a convergent approach to the types of structures depicted in Table 11.98 Similar to the case of the alkenyl boranes and silanes, the use of alkenyl halides opens an area of chemical space that has hitherto been underexplored.
The reactivity of a heteroatom-functionalized olefin is typically controlled by the identity of the attached heteroatom. This leads to structurally dissimilar products, such as 259–262 (Figure 6A). Olefin cross-coupling removes the heteroatom’s influence on the double bond and elicits the same reactivity from 249–254. In each of these cases, C–C bond formation occurs adjacent to the heteroatom to give 264, regardless of the identity of the heteroatom (Figure 6B). Along with olefin metathesis,99 this represents one of the few unified reactivity manifolds of olefins that can operate with a variety of heteroatom substituents.
Figure 6.
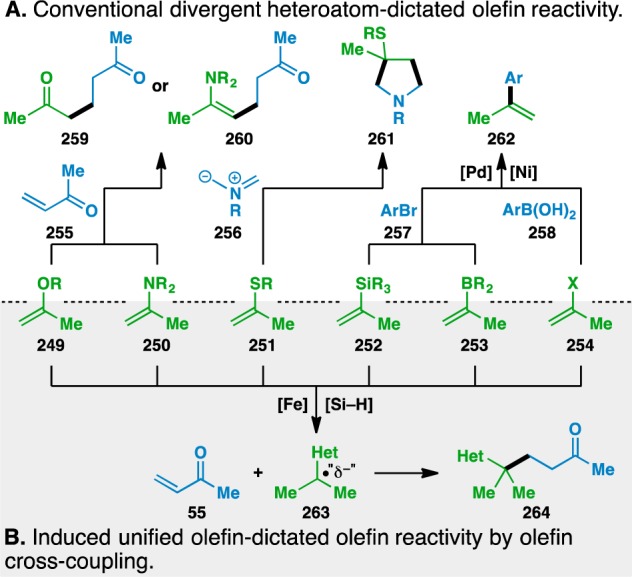
Functionalized olefin cross-coupling overrides inherent reactivity of the heteroatom functionalized olefins.
6. Vinyl Sulfone Coupling
Having explored the scope of the donor olefins employed in the olefin cross-coupling, attention shifted to the acceptor olefins used. The generation of sulfone-containing adducts 84 and 220 (Tables 4 and 10, respectively) raised the possibility of taking advantage of the versatility associated with sulfones to generate additional complexity (Figure 7A).100 In this vein, parent olefin 265 would be coupled with a vinyl sulfone 266 using the olefin cross-coupling to create a branch point adduct 267, which would then be subjected to precedented downstream operations to access a variety of daughter congeners (268–271). However, the couplings involving phenyl vinyl sulfone (83) as the acceptor olefin were among the more recalcitrant systems probed in the olefin cross-coupling. For example, the reaction of isopropenyl silane 216 and phenyl vinyl sulfone (83) required a stoichiometric loading of Fe(acac)3 and only proceeded in 35% yield using the first-generation olefin cross-coupling conditions (Figure 7B). The potential utility of such a transformation, if generalizable, encouraged further optimization.
Figure 7.
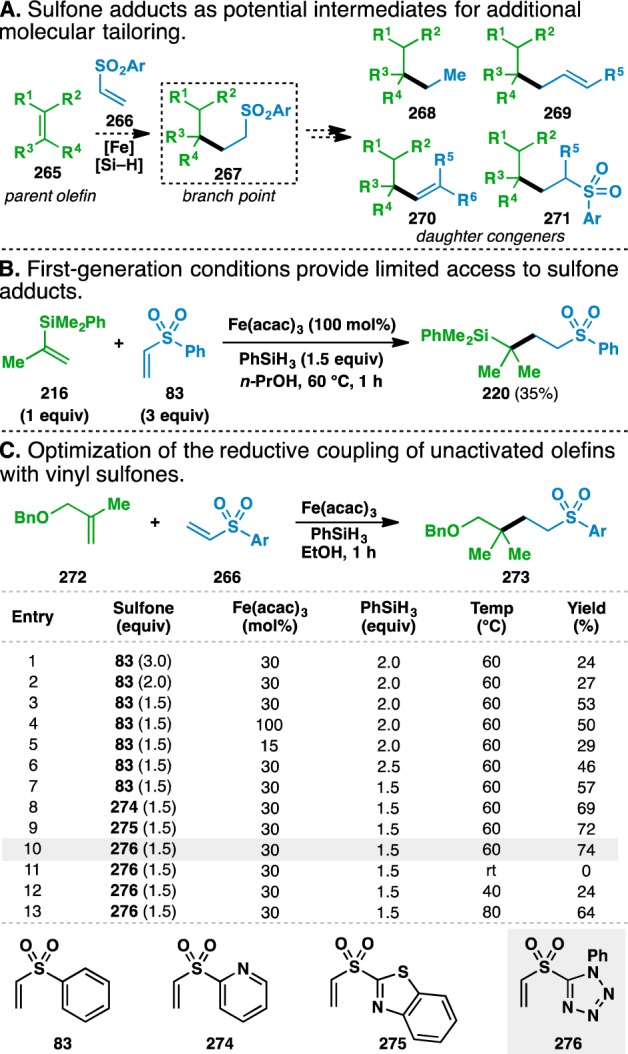
Inspiration for and development of a more efficient vinyl sulfone acceptor.
Thus, using the conditions developed in Table 1 as a starting point, the coupling of the allylic benzyl ether 272 with phenyl vinyl sulfone (83) provided the desired adduct 273 in 24% yield (entry 1). Reducing the amount of sulfone coupling partner counterintuitively increased the yield of the reaction, with 1.5 equiv providing the highest yield of 53% (entry 3). Although altering the Fe(acac)3 loading to either 100 mol% or 15 mol% led to reductions in yield (entries 4 and 5), reducing the amount of PhSiH3 used led to an increased 57% yield (entry 7). The structure of the aryl ring attached to the sulfone was next probed with the hope that altering its electronic properties would facilitate the radical conjugate addition. Switching to heteroaromatic substituents increased the yield of the coupling, with the N-phenyl tetrazole (PT) sulfone 276 providing the highest yield at 74% (entry 10). While this manuscript was in preparation, DFT calculations by Cid showed that the olefin of PT vinyl sulfone (276) to be more highly polarized than that of phenyl vinyl sulfone (83), which makes 276 a superior Michael acceptor in Giese conjugate additions.101 Running the reaction at temperatures either lower (entries 11 and 12) or higher (entry 13) than 60 °C resulted in lower yields, and thus, the system shown in entry 10 represented the optimized conditions.
The use of PT sulfone 276 in the olefin cross coupling was examined using a variety of donor olefins (Table 12). Geminally disubstituted olefins were found to participate in the reaction to give 279, 281, 286, 288, 290, and 292. Oxygen- and nitrogen-based functionality could be located at allylic positions (e.g., 272 and 280) and other polar functionalities, such as aldehydes (e.g., 282), ketones (e.g., 284), and carbamates (e.g., 61 and 287) could also be tolerated. β-Pinene (289) could be used to generate caged structure 290, which bears a quaternary carbon center. The mild nature of the reaction is demonstrated by the reaction of (+)-limonene oxide (291) to give 292, where the epoxide moiety remains untouched. Terminal olefins (e.g., 293, 295, 297, and 299) could also be used to generate the products of secondary radical conjugate additions. Unprotected hydroxyl groups and phenols were tolerated to give 294 and 296, respectively. Apronal (297) and sugar derivative 299 could also be used to fashion 298 and 300, respectively.
Table 12. Scope of the Alkyl-Substituted Donor Olefins Used in the Vinyl Sulfone Coupling.
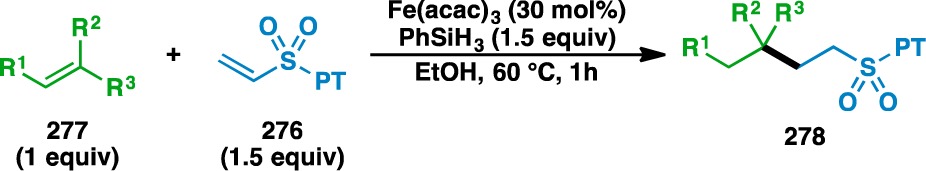
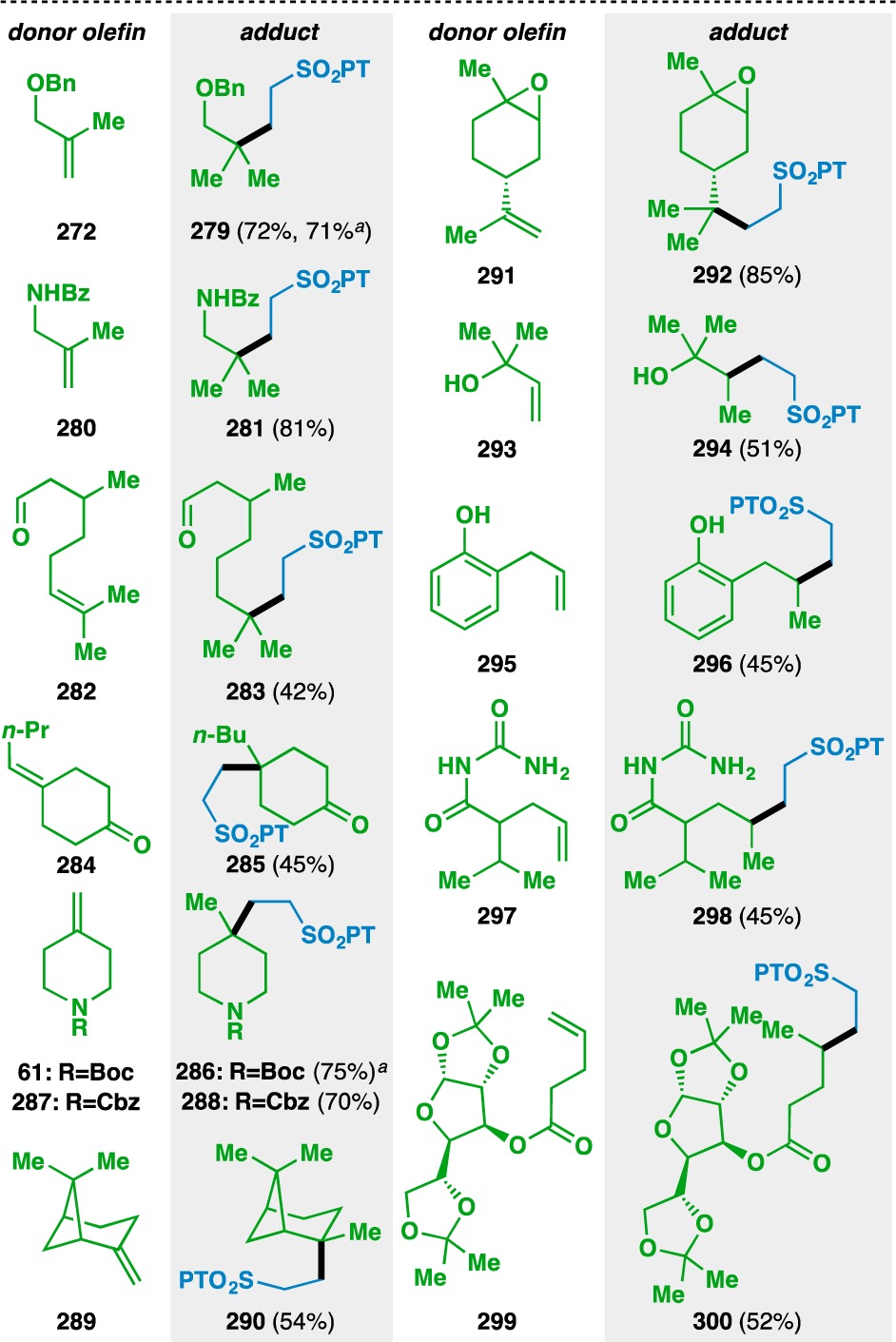
Run on gram scale.
Heteroatom-functionalized donor olefins could also be coupled with PT sulfone 276 (Table 13). Silyl enol ethers 120 and 121 could be used, as could the enecarbamate 164 and phenyl vinyl sulfide (187). Alkenyl boronic esters also gave the desired adducts, although cyclopentenyl 308 gave lower yields than isopropenyl 201, which correlates with the observation by Norton that HAT to trisubstituted olefins is less efficient than that to geminally disubstituted olefins.102 Finally, the couplings of 2-chloroallyl alcohol (237) and 2-bromoallyl alcohol (311) proceeded with conservation of the halogen atoms to give 310 and 312, respectively.
Table 13. Scope of the Heteroatom-Substituted Donor Olefins Used in the Vinyl Sulfone Coupling.
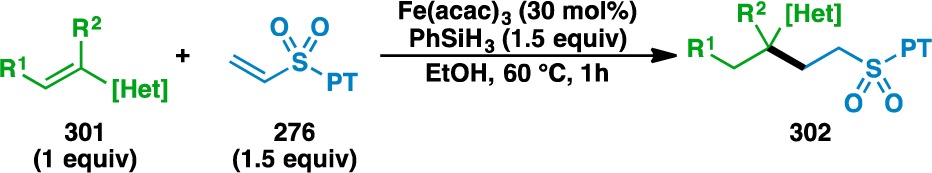
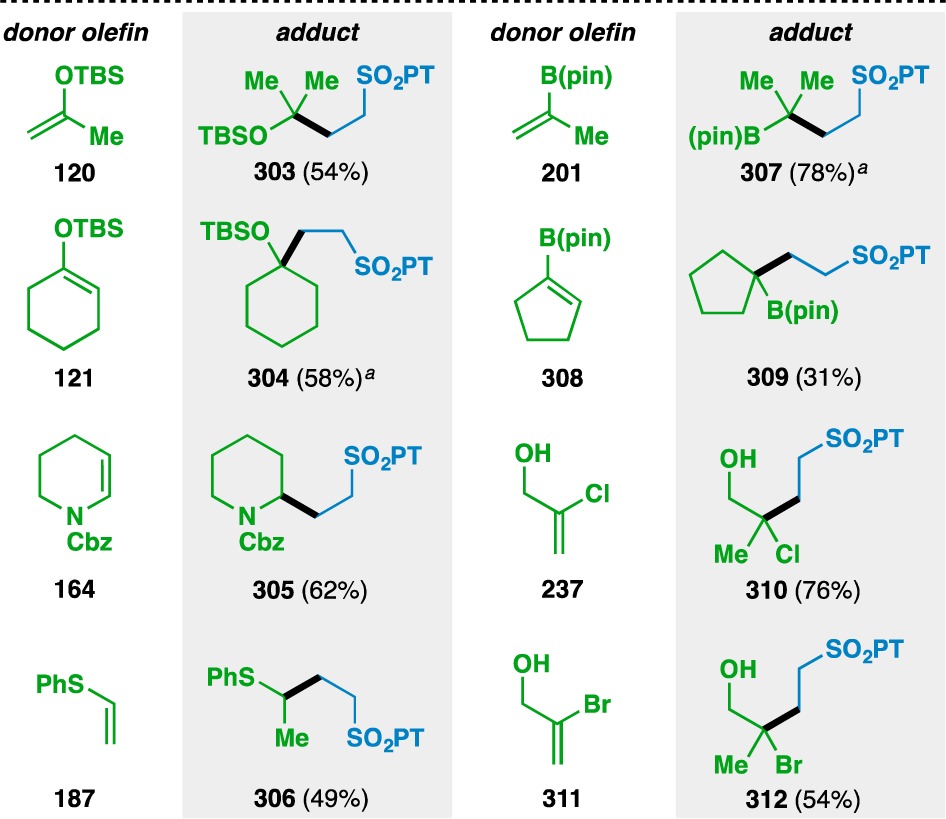
3 equiv vinyl sulfone and 3 equiv PhSiH3 used.
As expected, the PT sulfone thus incorporated was found to be a convenient handle for further reactions of adduct 286 (Figure 8A). Reductive desulfonylation of the C–S bond with SmI2103 led to the isolation of 313, the product of a net hydroethylation of an unactivated olefin. The position adjacent to the sulfone of 286 could be alkylated or acylated under basic conditions to give 314 and 315, respectively. The PT sulfone moiety of 286 could be exploited in a Julia-Kocienski olefination104 with para-bromobenzaldehyde to give 316 as a single olefin isomer. Additionally, the PT group could be cleaved via a SNAr reaction with NaSEt to give the sodium sulfinate salt 317, which itself is a radical precursor that can be used for further functionalization of heterocycles.105 α-Halogenation with NBS and NCS gave 318 and 319, respectively, and led to the development of a one-pot method, where α-difluorination is followed by net elimination of N-phenyltetrazole sulfinic acid, to give the 1,1-difluoroalkene 320.106
Figure 8.
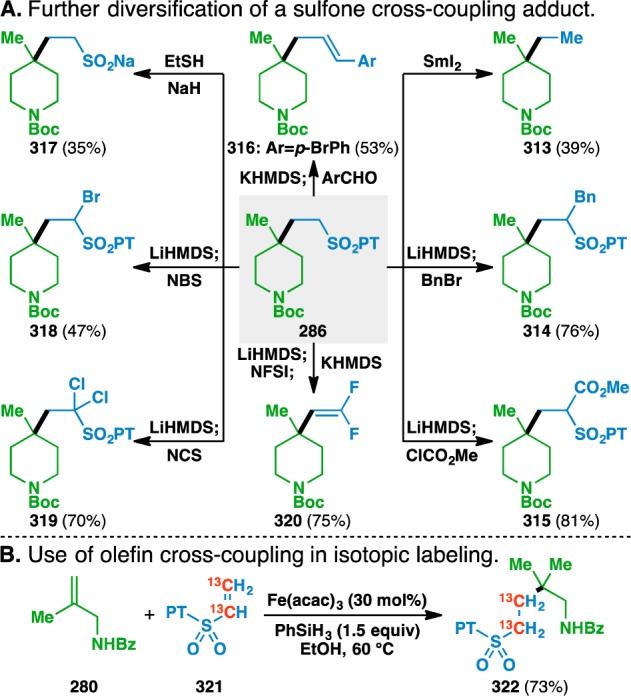
Additional applications of the vinyl sulfone coupling.
Moreover, coupling with donor olefin 280 with 13C-labeled sulfone 321 led to the isolation of 13C-labeled adduct 322 in 73% yield (Figure 8B), which is similar to the unlabeled system (Table 12, 281, 81%). Although 13C is a stable isotope, these results suggest that the olefin coupling could find use in radiolabeling settings.107
7. Olefin-Based Minisci Reaction
The pursuit of an olefin-based Minisci reaction67 was spurred by the initial finding reported in 2014 that acridine (100) could serve as an acceptor for disubstituted donor 59 to provide the reductively functionalized adduct 101 in 48% yield (Table 5 and Figure 9B). The utility of such a transformation is clear as alkylated heterocycles can be challenging to prepare and Minisci-type disconnections are an excellent retrosynthetic shortcut to access them.108 While the optimization of our initial finding was in progress,109 a pair of reports in the literature further suggested that such a process would be possible (Figure 9A). Herzon’s olefin hydropyridylation, where the nucleophilic radical intermediate 27 is captured with N-methoxypyridium salts 323, was particularly relevant.110 Although this transformation necessitates the use of 5 equiv of the pyridinium coupling partner, which itself requires 1 step to prepare from the commercial N-oxide, the ability to access the 4-alkylated pyridine motifs represented by 324 justifies the effort. These products can alternatively be obtained through an orthogonal and programmable reaction manifold developed by Shenvi that uses a Ni catalyst to enable the coupling of intermediate radical 27 to an aryl or heteroaryl iodide 325.111
Figure 9.
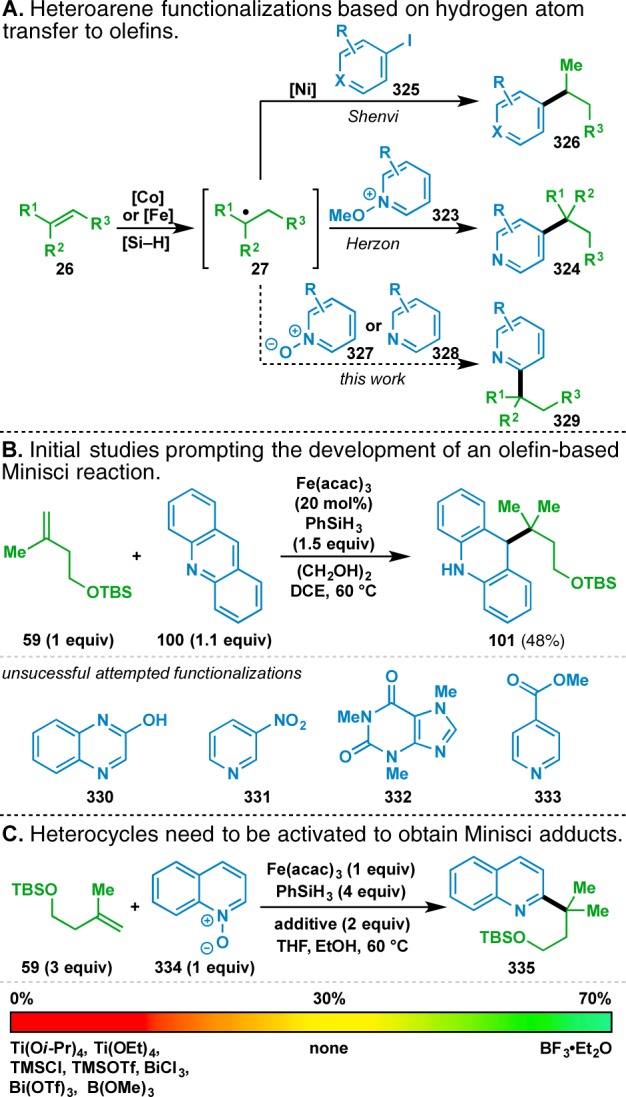
Inception and development of an olefin-based Minisci reaction.
In our studies, readily available coupling partners, such as a N-oxide 327 or the parent heterocyclic scaffold 328 were pursued as the acceptor components. Initial attempts to build on the result with acridine were disappointing, as other heterocyclic acceptors such as quinoxazoline (330), 3-nitropyridine (331), caffeine (332), and methyl isonicotinate (333) did not yield the desired adducts (Figure 9B). Presumably those heterocycles had more aromatic character than acridine and were not as activated for the addition of nucleophilic radicals. In traditional Minisci reactions, less reactive heterocycles are rendered more electrophilic by in situ protonation.112 However, the conditions depicted in Figure 9B do not ostensibly lead to protonation of the heterocycle. Additionally, the use of Brønsted acidic additives in the olefin cross-coupling led to reaction inhibition (vide supra), suggesting that alternative modes of heterocycle activation would need to be pursued.
In one such approach, Itami and Li have shown that activation of pyridines through formation of their N-oxides can facilitate radical-based C–H functionalizations of the heterocyclic cores.113 Inspired by these findings, we tested heteroarene N-oxides as coupling partners for the nucleophilic radical intermediate. Activation of quinoline as its N-oxide (i.e., 334) and its subsequent subjection to the reaction conditions led to the successful isolation of the alkylated quinoline derivative 335 (Figure 9C), albeit in 30% yield with substantial reduction of 334 to quinoline.
Although capable of accessing the desired product, N-oxide 334 was presumably still not electrophilic enough to obtain high yields of 335, and further activation would be necessary. An alternative to the N-oxide alkylation employed by Mitchell114 and Herzon110 to increase electrophilicity could conceivably involve coordination with a Lewis acid. Nucleophilic functionalizations of heteroaromatic N-oxides have previously been shown to be facilitated by Lewis acid complexation, lending credibility to this hypothesis.115 Thus, a sampling of Lewis acids was screened to identify one that could facilitate the radical addition (Figure 9C). Although most Lewis acids completely inhibited the reaction, the use of 2 equiv of BF3·Et2O increased the yield of the reaction to 70%. It is presently unclear whether the N-oxide is being activated by direct complexation with BF3 or by protonation with a BF3·EtOH complex generated in situ. In a subsequent control reaction, it was determined that N-oxide formation was not necessary when using BF3·Et2O as an activating agent; however, this did result in regioisomeric mixtures (vide infra, Figure 10B).
Figure 10.
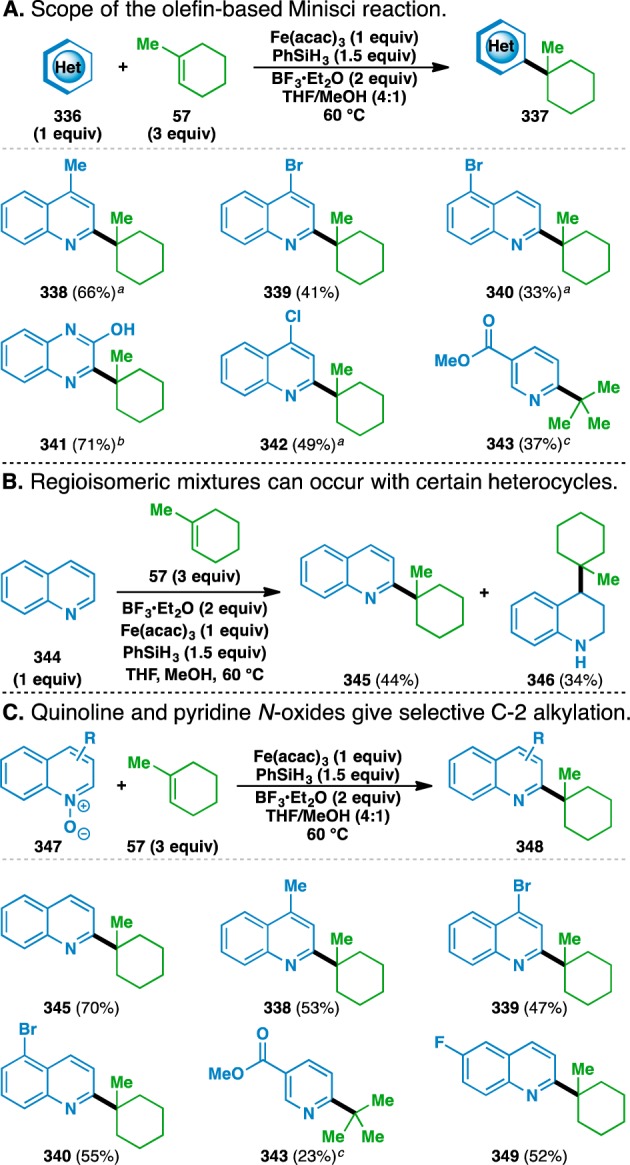
Developed olefin-based Minisci reaction functionalizes heterocycles and their N-oxides. a1 equiv PhSiH3 used. bAfter subsequent heating with chloranil (2 equiv) at 60 °C for 2 h. c10 equiv isobutylene instead of 1-methylcyclohexene, 4 equiv BF3·Et2O, and 4 equiv PhSiH3 used.
Thus, the scope of the newly developed olefin-based Minisci reaction was probed using 1-methylcyclohexene (57) as the donor olefin (Figure 10A). The use of lepidine as the acceptor gave rise to alkylated adduct 338 in 66% isolated yield. Bromine atoms could be incorporated at both the 4 and 5 positions of the quinoline ring to give 339 and 340 in yields of 41% and 33%, respectively. Similar to the case of the functionalized olefin cross-coupling, the conservation of the bromine atom in the final product demonstrates the reaction’s high degree of chemoselectivity, as such substituents are frequently prone to competitive dehalogenation in other transition-metal-mediated systems.116 Other heteroarene acceptors could also be used, such as quinoxazoline (330), which gave 341 in 71% yield after a one-pot reoxidation of the initially formed dihydro adduct with chloranil.117 Alkylated 342 could be generated from 4-chloroquinoline in 49% yield, and methyl nicotinate could be coupled with isobutylene to give methyl 6-tert-butylnicotinate (343).
The adducts formed in each of these cases depicted in Figure 10A were isolated as single regioisomers, with the remaining mass balance attributed to competitive reduction of the heteroarenes and reduction or hydration of the donor olefin. However, mixtures of regioisomers were observed in certain cases. The use of quinoline (344) gave a mixture of both 2-alkylated quinoline 345 and 4-alkylated tetrahydroquinoline 346 (Figure 10B).118 In these cases, the 2-alkylated regioisomer can be formed exclusively by using the corresponding N-oxide as the electrophilic acceptor.115 For example, 2-alkylated 345 was formed in 70% yield as the sole regioisomer (Figure 10C). Various other N-oxides were alkylated with 1-methylcyclohexene (57) to give the same adducts as those formed in Figure 10A, namely 338–340 and 343. Additionally, 6-fluoroquinoline N-oxide could be used to give 349 in 52% yield.
8. Elucidation of the Olefin Cross-Coupling Reaction Mechanism and Further Development
Although Mukaiyama-type Markovnikov olefin hydrofunctionalizations have been known for decades, early mechanistic proposals were supported by little to no experimental evidence. Only recently have more rigorous mechanistic studies been performed, with the most extensive to date by Carreira on his Co-catalyzed olefin hydrohydrazination and hydroazidation.53 A comprehensive mechanistic understanding of the olefin cross-coupling would likely have a positive impact on this field and perhaps lead to further reaction development.
8.1. Identification of the Iron Species Present
The results of numerous mechanistic studies (vide infra) have culminated in the rather complex mechanistic picture illustrated in Figure 11A.119 Solvolysis of the FeL3 precatalyst (L=acac or dibm) with 2 equiv of EtOH generates the bridged dimeric Fe species 350.120 This species has been independently synthesized121 and was found to be catalytically competent when used in place of Fe(acac)3 (Figure 11B [A]). In the proposed mechanism, reaction of 350 with 2 equiv of PhSiH3 or PhSi(OEt)H2 (351, vide infra) gives an unobserved dihydride 352, which spontaneously loses H2 to give FeL2 (353). This proposal is supported by a stoichiometric reaction, where stirring 350′ with PhSiH3 gave [Fe(acac)2]2 in 88% yield with concomitant evolution of H2 gas, as detected by chromatography (Figure 11B [B]). Additional studies suggested that the possibility of an alternative PhSiH3 oxidative addition/reductive elimination pathway to form FeL2 is unlikely (see Supporting Information).
Figure 11.

Proposed mechanism of the olefin cross-coupling with supporting evidence.
57Fe Mössbauer spectroscopy indicated that the observable majority of Fe(acac)3 is converted into Fe(acac)2 over the course of the reaction (Figure 11B [C]). This iron(II) species could even be isolated from the reaction mixture as crystals of Fe(acac)2·2EtOH, which were characterized by X-ray crystallography (see Supporting Information, including CIF file). However, running the olefin cross-coupling under anaerobic conditions with [Fe(acac)2]2 in the place of Fe(acac)3 gave no product, indicating that Fe(acac)2 is not directly involved in the conversion of the donor olefin into the radical intermediate.122 It is common for the predominant metal species observed to lie off the catalytic cycle.123 However, the cross-coupling reaction did occur under aerobic conditions when using [Fe(acac)2]2, presumably due to oxidation of the Fe(acac)2 to catalytically active Fe(acac)3 by the O2 present in air.124 This notion was supported by the observation that the exposure of a solution of [Fe(acac)2]2125 in toluene to O2 or air provided Fe(acac)3 along with a second unidentified Fe species (Figure 11B [D]).
8.2. Interplay of Fe(acac)2 and PhSiH3
Although it was found that [Fe(acac)2]2 was catalytically incompetent in the olefin cross-coupling in the absence of O2, a control reaction showed that it catalyzes the solvolysis of PhSiH3 with EtOH to provide a mixture of PhSiH2(OEt) (351), PhSiH(OEt)2 (357), and PhSi(OEt)3 (369, Figure 11B [E]). Although silane 351 was only detected in small amounts from this reaction, it could be synthesized independently (see Supporting Information). Stirring a d6-benzene solution of 351 with EtOH and Fe(acac)2 gave silanes 357 and 369,126 suggesting that 351 is a feasible precursor to the other silanes (i.e., 357 and 369) present the reaction in Figure 11B-E. The FeL2-catalyzed solvolysis of PhSiH3 to form the active PhSi(OEt)H2 is hypothesized to begin by formation of a complex between FeL2 (353) and EtOH to provide 354. This would then react with a molecule of PhSiH3, presumably through a transition state represented by 355, to give 356 upon losing H2. Decomplexation of PhSi(OEt)H2 (351) from the Fe center would regenerate FeL2 (353).
In accord with previous studies reported by Shenvi on the efficacy of PhSi(Oi-Pr)H2 in Mukaiyama-type transformations,127 PhSi(OEt)H2 (351) was found to be superior to PhSiH3, and the use of purified 351 facilitated the olefin cross-coupling at room temperature within 15 min (Figure 11B [F]).128 However, PhSi(OEt)2H (357) was not as effective in the catalytic reaction.129 These observations suggest that silane 351 is the most active terminal reductant in the catalytic cycle, where it could convert [FeL2(OEt)]2 (350) to the bridged monohydride 358 through a transmetalation. The bridged monohydride 358 would then fragment to give Fe ethoxide 359 and Fe hydride 360.
8.3. Coupling of the Donor and Acceptor Olefins
The next step of the proposed mechanism involves the transfer of a hydrogen atom from the transient Fe hydride 360 to the donor olefin 362, giving the intermediate alkyl radical 363. A deuterium labeling experiment using PhSiD3 provided d1-Me adduct 371 from donor 120 and methyl vinyl ketone (55), indicating that the hydrogen atom incorporated into the donor olefin originates from PhSiH3 (Figure 11B [G]). Furthermore, the initial rates of the olefin cross-coupling of donor 59 with benzyl acrylate (131) were measured to give estimates of the reactant orders. Although archetypal substrates (e.g., 59 and 131) were studied, reactant orders are ultimately dependent on the specific system studied. Thus, these results illustrate only one possible (and hopefully typical) scenario (vide infra). The positive orders in donor, Fe(acac)3, and PhSiH3, as well as the inverse order in acceptor are consistent with the conversion of 362 to 363 being the rate-determining step of the reaction (Figure 11A).130
Performing the olefin cross-coupling of donor 59 with acceptor 131 using PhSiD3 showed a kinetic isotope effect (KIE) of 1.5 (Figure 11B [H]) on the initial rate of the reaction, providing further evidence that the hydride from PhSiH3 is involved in the rate-determining step. However, HAT from transition metal hydrides to olefins is typically characterized by inverse KIEs [e.g., Mn(CO)5H (KIE=0.4),131 CpW(CO)3H (KIE=0.55),132 CpCr(CO)3H (KIE=0.45),132 and CpFe(CO)2H (KIE=0.86)133], and Halpern has suggested that inverse KIEs are diagnostic of a HAT mechanism.134 These inverse KIEs have been previously rationalized by the higher strength of the C–H (or C–D) bond being formed over the M–H (or M–D) bond being broken.134
Although less common, normal KIEs for HAT have been observed in the reductions of certain high-energy olefins [i.e., benzylidenefluorene (KIE=1.22) and bifluorenylidene (KIE=2.01)].135 The authors reasoned that these specific systems are characterized by early transition states (instead of the typical late transition states), resulting in ΔGD⧧ > ΔGH⧧ and thus kH > kD. However, Eisenberg and Norton argue that a more likely explanation is that the KIE is the result of a two-step process that consists of (1) HAT to form a solvent-caged radical pair and (2) solvent cage escape.61 In the olefin reductions by transition metal carbonyl hydrides (e.g., Mn(CO)5H, CpW(CO)3H, CpCr(CO)3H, and CpFe(CO)2H), the inverse KIEs can be attributed to the initial HAT being reversible (i.e., k–1 > k2, Figure 12A). The rate of the reverse process in the case of hydrogen (k–1H, 382 → 380 + 381) would be faster than that in the case of deuterium (k–1D, 386 → 385 + 381) since the C–H bond is weaker than the C–D bond. This would result in an overall larger buildup of the solvent-caged radical pair 386, as compared to 382. After solvent cage escape, this equilibrium isotope effect136 manifests itself as an inverse KIE.
Figure 12.
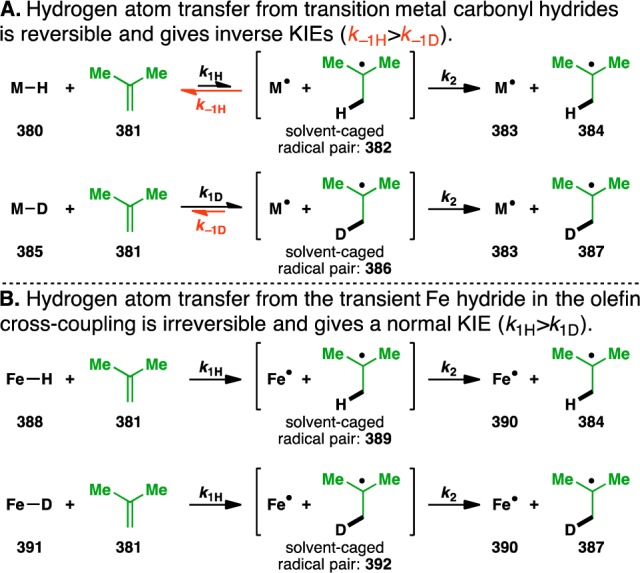
Possible explanation of the unexpected normal kinetic isotope effect observed for the olefin cross-coupling.
However, in the olefin cross-coupling (and other cases where normal KIEs are observed), the initial HAT is irreversible (Figure 12B). Now the relative amounts of the solvent-caged radical pairs are solely determined by the forward HAT from the Fe hydride or deuteride to the olefin. As the Fe–H bond is weaker than the Fe–D bond, the formation of solvent-caged radical pair 389 would be faster than that of 392 (i.e., k1H > k1D). This would lead to a normal KIE upon solvent cage escape.137 Such an argument might also explain the normal KIEs of 1.6 and 2.2 that Carreira observed for his Co-catalyzed hydroazidation and hydrohydrazination, respectively.53,138
The intermediacy of radical 363 was supported by the results of a cyclopropane ring-opening experiment (Figure 11B [I]),139 where silyl enol ether 372 was used as a donor olefin. Reaction with methyl acrylate (77) gave the diester 373, which presumably arises from the intermediacy of radical 374. Opening of the neighboring cyclopropane gives homoallyl radical 375, which adds into methyl acrylate to generate 376. The newly formed silyl enol ether then serves as a donor olefin and undergoes coupling with an additional equivalent of methyl acrylate (77), to give diester 373. Although the low yield of diester 373 is potentially indicative of other operative pathways in this system, it nevertheless provides some evidence for the formation of a carbon-centered radical upon HAT to the donor olefin.
The next step in the olefin cross-coupling mechanism is the Giese-type radical conjugate addition28,29 of alkyl radical 363 into the electron-deficient acceptor olefin 34 to form radical adduct 364. Single-electron reduction of the radical by FeL2 (361) would give the stabilized carbanion 365 with concomitant reoxidation of iron(II) 361 to iron(III) 366. The intermediacy of 365 was supported by a three-component coupling reaction, where the ester enolate intermediate formed in the coupling of donor 59 with benzyl acrylate (131) undergoes an aldol condensation with benzaldehyde (377) to deliver alcohol 378 in 20% yield (Figure 11B [J]). Furthermore, Pronin has recently realized the intramolecular version of this tandem process in his synthesis of emindole SB.140 As direct alkyl radical additions into aldehydes are quite rare,141 these results support the intermediacy of a stabilized carbanion and are inconsistent with a proton-coupled electron transfer142 terminating the olefin cross-coupling mechanism, which would instead directly deliver adduct 367 without the intermediacy of 365.
This stabilized anion 365 can be protonated with EtOH to provide the coupled adduct 367 along with the iron(III) ethoxide 359, which dimerizes to regenerate [FeL2(OEt)]2 (350). The use of EtOD as the reaction solvent in the coupling of donor 120 with methyl vinyl ketone (55) gave 379, where the deuterium atom was incorporated adjacent to the ketone (Figure 11B [K]). This is consistent with a polar protonation rather than a radical hydrogen atom abstraction ending the reaction mechanism, as the deuterium atom is the most acidic site in EtOD.143 If a radical hydrogen atom abstraction was operative, one would instead expect that the use of EtOD as a reaction solvent would give an undeuterated product since the methylene hydrogen atoms have a lower bond dissociation energy than that of the O–D bond.144
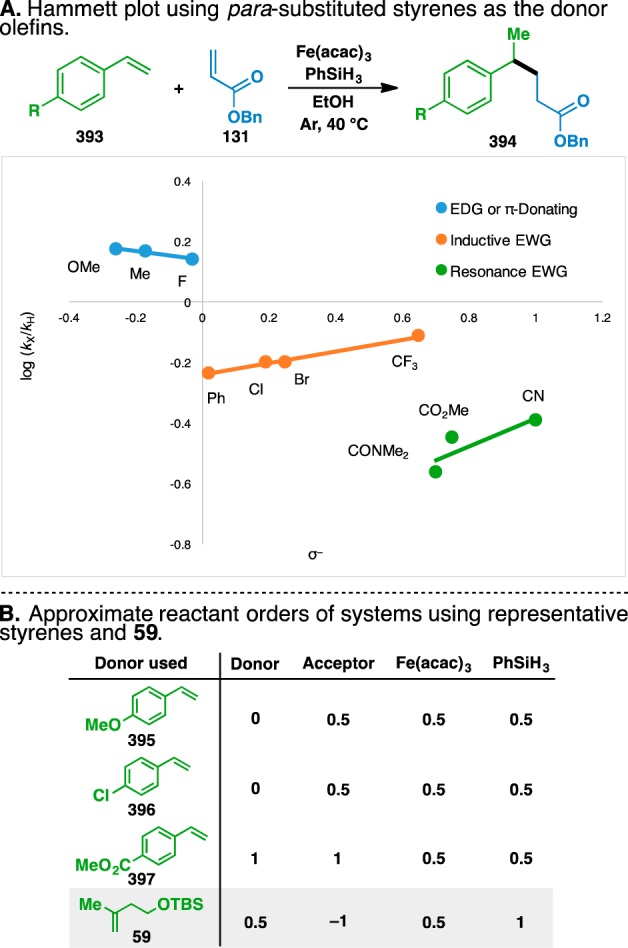
8.4. Hammett Analysis of para-Substituted Styrene Donor Olefins
A Hammett analysis was next used to probe the nature of the transition state of the rate-determining step,145 where para-substituted styrenes (393) were used as the donor component (Figure 12A). Although the transition state was presumably radical in nature, it was hoped that a Hammett plot would provide insight into the polar radical effects present.146 A survey of 10 different styrenes revealed that they could be classified into three distinct groups based on the nature of the para substituent, with inductive and π-donating groups giving a negative ρ value, inductive electron-withdrawing groups giving a positive ρ value, and resonance-delocalized electron-withdrawing groups giving a separate positive ρ value (Figure 13A). These results suggest that the rate-determining step in the olefin cross-coupling with each of the three groups may be different147 and highlight the complicated nature of this reaction.
Figure 13.
Hammett analysis of the olefin cross-coupling.
The changes in the rate-determining step were corroborated when the reactant orders of representative styrenes from each category were compared and found to differ in each case (Figure 13B). However, the negative ρ value of the electron-donating group was consistent with the proposed rate-determining step of the reaction mechanism, as the conjugate addition of the intermediate nucleophilic radical would be attenuated by the presence of electron-donating groups.146 However, the reactant orders of the representative electron-rich styrene, para-methoxystyrene (395), did not match the reactant orders of a typical alkyl-substituted donor olefin (i.e., 59), suggesting that the rate-determining steps of the two reactions are different. Similarly, the reactant orders using alkyl-substituted 59 did not match any of the other representative styrenes (i.e., 396 and 397).
8.5. Development of an Improved Set of Conditions for the (Functionalized) Olefin Cross-Coupling
Although the Hammett plot did not provide any pertinent conclusions about the rate-determining step of typical olefin cross-couplings, its construction did lead to several observations regarding the initial rate of the reaction (Figure 13; see Supporting Information for the full reaction progress curves). In the model reaction system using alkyl-substituted 59 as the donor olefin, the formation of product 368 was monitored over the first 10 min of the reaction (Figure 14). This revealed the presence of a slight induction period, as demonstrated by the upward inflection in the product formation curve (shown in blue). However, when styrene (63) was used as the donor olefin to provide 398, there was no such observed induction period (shown in green), indicating that styrene accelerated the formation of the active catalyst in the reaction. To test this hypothesis, 10 mol% styrene was added to the reaction that used alkyl-substituted 59 as a donor olefin. This led to a significant reduction in the induction period (shown in orange). Additionally, stirring the reaction overnight led to an increase in the reaction yield from 62% to 88%.148
Figure 14.

Addition of styrene removes an induction period and increases the yield of a typical system used in the olefin cross-coupling.
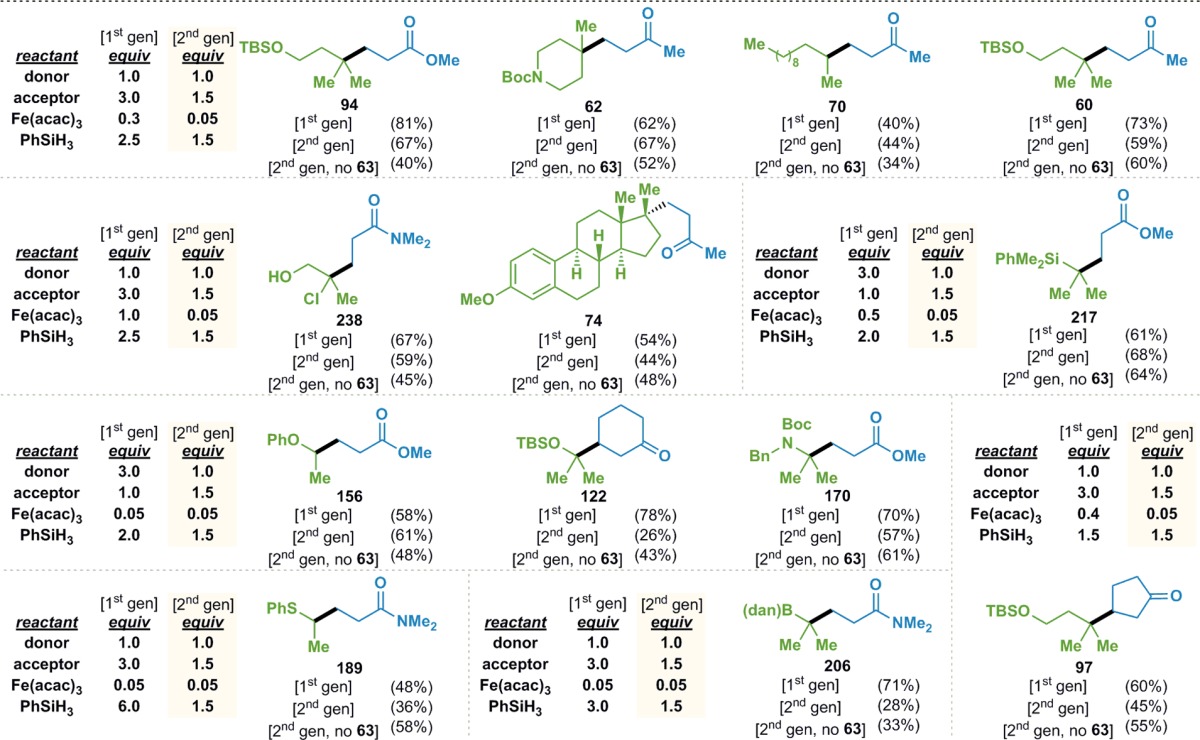
This observation led to the development of a second-generation set of conditions (Table 14), where styrene is first premixed with Fe(acac)3 and PhSiH3 at room temperature in a solvent mixture of THF and (CH2OH)2 that was sparged with Ar in a sonicator (the degassing was essential in preventing competitive Mukaiyama hydration34,35 of the donor olefin at ≤40 °C). The donor olefin, acceptor olefin, and additional PhSiH3 are then added to this solution to provide the coupled products.
Table 14. Applications of the Second-Generation Olefin Cross-Coupling Conditions.

Yields in parentheses are isolated yields.
These conditions allowed for the reductions of the Fe(acac)3 to 5 mol%, the acceptor to 1.5 equiv, and the PhSiH3 to 1.5 equiv. In the cases of 94, 62, 70, and 60, yields similar to those obtained with the less efficient first-generation conditions were realized using the second-generation conditions. The reactions to form 238 and 74 no longer required the use of stoichiometric Fe(acac)3. The formation of 217, 156, 122, and 170 each initially required the most expensive component of the reaction, the donor olefin, to be used in a threefold excess. These reactions could now be run using the donor olefin as the limiting reagent. In the case of 217, the loading of Fe(acac)3 could be decreased from 50 to 5 mol%. The formation of thioether 189 now could take place with 1.5 equiv of PhSiH3 instead of the previous 6 equiv. Gains in the efficiency of the formation of 97 could also be realized, where a similar yield was obtained using 5 mol% Fe(acac)3 instead of 40 mol%.
For currently unknown reasons, the new conditions did not provide yields of 122 and 206 comparable to those obtained with the original conditions. Furthermore, the addition of styrene was not always beneficial. In the cases of 122, 189, and 97, the inclusion of styrene inhibited product formation. The presence of the styrene additive in the formation of 60, 74, 170, and 206 had a negligible effect. It is currently unclear why some systems benefit and others suffer from the inclusion of 5 mol% styrene.
Much remains to be learned in uncovering a full mechanistic picture of these radical-based processes. Although these studies raise numerous questions, they also rule out potential pathways and have led to a tangible set of conditions that can, in many cases, improve the efficiency of this valuable reactivity.
9. Conclusion
The creation of the olefin cross-coupling described herein resulted from the fusion of two classes of influential transformations: Mukaiyama hydrofunctionalizations19 and Giese radical conjugate additions.28,29 Although these two types of reactions have been known since the 1980s, they had not been united until the initial report of the basic olefin cross-coupling in 2014.17 Subsequent studies expanded the utility of this transformation to encompass a unified reactivity for heteroatom-functionalized donor olefins18 and allowed for the creation of a strategy that uses vinyl sulfone adducts to generate additional molecular complexity. The presumed intermediacy of radicals in these transformations prompted the development of a Minisci-type reaction that allows for direct functionalization of electron-deficient heterocycles with olefins. One of the most comprehensive mechanistic interrogations of a Mukaiyama-type hydrofunctionalization to date has illuminated a detailed mechanistic picture that is consistent with all the evidence gathered thus far and led to the identification of a set of more efficient second-generation conditions. The methods developed here represent convergent approaches for small-molecule synthesis and allow for the generation of motifs, such as remote quaternary carbon centers, that are not readily accessible using other means.25 Although these reactions have only been recently disclosed, they have already enabled the syntheses of complex molecules and have served as a foundation for other advances in this area.81,140,149−156 It is hoped that the continued use of this radical chemistry27 and further extensions will simplify access to difficult structures and thus positively impact chemical synthesis.
Acknowledgments
Financial support for this work was provided by NIH/NIGMS (GM-118176 to P.S.B.), NSF (CHE-1465017 to P.L.H.), NSF and the Ellen Browning Scripps Foundation (predoctoral fellowships to J.C.L.), NDSEG (predoctoral fellowship to J.T.E.), the Shanghai Institute of Organic Chemistry, Zhejiang Medicine Co., and Pharmaron (postdoctoral fellowship for J.G.), and JSPS (postdoctoral fellowship to Y.Y.). The authors thank D.-H. Huang and L. Pasternack (TSRI) for assistance with NMR spectroscopy, A. L. Rheingold and C. E. Moore (UCSD), and the CBIC X-ray facility and Dr. Brandon Mercado (Yale) for X-ray crystallographic analysis. The authors are grateful to N. Hawbaker and M. Mower (TSRI) for helpful discussions and A. Cernijenko (TSRI) for providing the heat map used in Figure 9. The authors would like to thank M. D. Eastgate, K. Chen, and B. D. Maxwell (BMS) for additional support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b13155.
The authors declare no competing financial interest.
Supplementary Material
References
- Dewick P. M.Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons: West Sussex, 2009; pp 187–306. [Google Scholar]
- Newhouse T.; Baran P. S.; Hoffmann R. W. Chem. Soc. Rev. 2009, 38, 3010. 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns N. Z.; Baran P. S.; Hoffmann R. W. Angew. Chem., Int. Ed. 2009, 48, 2854. 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- Wu Q.-X.; Shi Y.-P.; Jia Z.-J. Nat. Prod. Rep. 2006, 23, 699. 10.1039/b606168k. [DOI] [PubMed] [Google Scholar]
- Baloglu E.; Kingston D. G. I. J. Nat. Prod. 1999, 62, 1448. 10.1021/np990176i. [DOI] [PubMed] [Google Scholar]
- Appendino G.Ingenane Diterpenoids. In Progress in the Chemistry of Organic Natural Products; Kinghorn A. D., Falk H., Gibbons S., Kobayashi J., Eds.; Springer: Cham, 2016; Vol. 102, pp 1–90. [DOI] [PubMed] [Google Scholar]
- Wang H.-B.; Wang X.-Y.; Liu L.-P.; Qin G.-W.; Kang T.-G. Chem. Rev. 2015, 115, 2975. 10.1021/cr200397n. [DOI] [PubMed] [Google Scholar]
- Liang Q.-L.; Gao Y.; Dai C.-C.; Min Z.-D. Nat. Prod. Res. Dev. 2009, 21, 545. [Google Scholar]
- Chen K.; Baran P. S. Nature 2009, 459, 824. 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- Yuan C.; Jin Y.; Wilde N. C.; Baran P. S. Angew. Chem., Int. Ed. 2016, 55, 8280. 10.1002/anie.201602235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. Science 2013, 341, 878. 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]
- Ishihara Y.; Baran P. S. Synlett 2010, 2010, 1733. 10.1055/s-0030-1258123. [DOI] [Google Scholar]
- Gaich T.; Baran P. S. J. Org. Chem. 2010, 75, 4657. 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- Christianson D. W. Chem. Rev. 2006, 106, 3412. 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- The Alkenes: Vol. 1; Patai S., Ed.; Patai’s Chemistry of Functional Groups; Wiley: London, 1964. [Google Scholar]
- The Alkenes: Vol. 2; Zabicky J., Ed.; Patai’s Chemistry of Functional Groups; Wiley: London, 1970. [Google Scholar]
- Lo J. C.; Yabe Y.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 1304. 10.1021/ja4117632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo J. C.; Gui J.; Yabe Y.; Pan C.-M.; Baran P. S. Nature 2014, 516, 343. 10.1038/nature14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley S. W. M.; Obradors C.; Martinez R. M.; Shenvi R. A. Chem. Rev. 2016, 116, 8912. 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney E. C.; Green J. C.; Baran P. S. Angew. Chem., Int. Ed. 2013, 52, 9019. 10.1002/anie.201304609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney E. C.; Lopchuk J. M.; Green J. C.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 12592. 10.1021/ja507321j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.-D.; Huang S.-X.; Han Q.-B. Nat. Prod. Rep. 2006, 23, 673. 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- See Y. Y.; Herrmann A. T.; Aihara Y.; Baran P. S. J. Am. Chem. Soc. 2015, 137, 13776. 10.1021/jacs.5b09463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasperse C. P.; Curran D. P.; Fevig T. L. Chem. Rev. 1991, 91, 1237. 10.1021/cr00006a006. [DOI] [Google Scholar]
- Alexakis A.; Bäckvall J. E.; Krause N.; Pàmies O.; Diéguez M. Chem. Rev. 2008, 108, 2796. 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]
- Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis; Christoffers J., Baro A., Eds.; Wiley: Weinheim, 2005. [Google Scholar]
- Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 12692. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. A.; Giese B.; Curran D. P. Acc. Chem. Res. 1991, 24, 296. 10.1021/ar00010a003. [DOI] [Google Scholar]
- Srikanth G. S. C.; Castle S. L. Tetrahedron 2005, 61, 10377. 10.1016/j.tet.2005.07.077. [DOI] [Google Scholar]
- Jamison C. R.; Overman L. E. Acc. Chem. Res. 2016, 49, 1578. 10.1021/acs.accounts.6b00284. [DOI] [PubMed] [Google Scholar]
- Alternative methods for homolyzing a C–O bond to generate radicals have also been developed. Although some of these more recent radical precursors are stable, they are based on the same principle of activating an alcohol, and thus do not address the potential difficulties arising from accessing the requisite alcohol (see ref (30)).
- Albini A.; Fagnoni M.. Photochemically-Generated Intermediates in Synthesis, 1st ed.; John Wiley & Sons: Hoboken, 2013; pp 43–45. [Google Scholar]
- Renaud P.; Beauseigneur A.; Brecht-Forster A.; Becattini B.; Darmency V.; Kandhasamy S.; Montermini F.; Ollivier C.; Panchaud P.; Pozzi D.; Scanlan E. M.; Schaffner A.-P.; Weber V. Pure Appl. Chem. 2007, 79, 223. 10.1351/pac200779020223. [DOI] [Google Scholar]
- Isayama S.; Mukaiyama T. Chem. Lett. 1989, 18, 569. 10.1246/cl.1989.569. [DOI] [Google Scholar]
- Isayama S.; Mukaiyama T. Chem. Lett. 1989, 18, 1071. 10.1246/cl.1989.1071. [DOI] [Google Scholar]
- Isayama S.; Mukaiyama T. Chem. Lett. 1989, 18, 573. 10.1246/cl.1989.573. [DOI] [Google Scholar]
- Kato K.; Mukaiyama T. Chem. Lett. 1992, 21, 1137. 10.1246/cl.1992.1137. [DOI] [Google Scholar]
- Shenvi R. A.; Guerrero C. A.; Shi J.; Li C.-C.; Baran P. S. J. Am. Chem. Soc. 2008, 130, 7241. 10.1021/ja8023466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Manolikakes G.; Yeh C.-H.; Guerrero C. A.; Shenvi R. A.; Shigehisa H.; Baran P. S. J. Am. Chem. Soc. 2011, 133, 8014. 10.1021/ja202103e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renata H.; Zhou Q.; Baran P. S. Science 2013, 339, 59. 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renata H.; Zhou Q.; Dünstl G.; Felding J.; Merchant R. R.; Yeh C.-H.; Baran P. S. J. Am. Chem. Soc. 2015, 137, 1330. 10.1021/ja512022r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Holte D.; Zoller J.; Umemiya S.; Simke L. R.; Baran P. S. J. Am. Chem. Soc. 2015, 137, 10160. 10.1021/jacs.5b07154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura S.; Chu H.; Felding J.; Baran P. S. Nature 2016, 532, 90. 10.1038/nature17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus P.; Waring M. J.; Scott D. A. Tetrahedron Lett. 2000, 41, 9731. 10.1016/S0040-4039(00)01728-7. [DOI] [Google Scholar]
- Iwasaki K.; Wan K. K.; Oppedisano A.; Crossley S. W. M.; Shenvi R. A. J. Am. Chem. Soc. 2014, 136, 1300. 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King S. M.; Ma X.; Herzon S. B. J. Am. Chem. Soc. 2014, 136, 6884. 10.1021/ja502885c. [DOI] [PubMed] [Google Scholar]
- Ma X.; Herzon S. B. Chem. Sci. 2015, 6, 6250. 10.1039/C5SC02476E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girijavallabhan V.; Alvarez C.; Njoroge F. G. J. Org. Chem. 2011, 76, 6442. 10.1021/jo201016z. [DOI] [PubMed] [Google Scholar]
- Waser J.; Carreira E. M. J. Am. Chem. Soc. 2004, 126, 5676. 10.1021/ja048698u. [DOI] [PubMed] [Google Scholar]
- Waser J.; Carreira E. M. Angew. Chem., Int. Ed. 2004, 43, 4099. 10.1002/anie.200460811. [DOI] [PubMed] [Google Scholar]
- Waser J.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2005, 127, 8294. 10.1021/ja052164r. [DOI] [PubMed] [Google Scholar]
- Waser J.; González-Gómez J. C.; Nambu H.; Huber P.; Carreira E. M. Org. Lett. 2005, 7, 4249. 10.1021/ol0517473. [DOI] [PubMed] [Google Scholar]
- Waser J.; Gaspar B.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2006, 128, 11693. 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]
- Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2007, 46, 4519. 10.1002/anie.200700575. [DOI] [PubMed] [Google Scholar]
- Gaspar B.; Waser J.; Carreira E. M. Synthesis 2007, 24, 3839. 10.1055/s-2007-1000817. [DOI] [Google Scholar]
- Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2008, 47, 5758. 10.1002/anie.200801760. [DOI] [PubMed] [Google Scholar]
- Gaspar B.; Carreira E. M. J. Am. Chem. Soc. 2009, 131, 13214. 10.1021/ja904856k. [DOI] [PubMed] [Google Scholar]
- Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Org. Lett. 2012, 14, 1428. 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker T. J.; Boger D. L. J. Am. Chem. Soc. 2012, 134, 13588. 10.1021/ja3063716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik T.-G.; Luis A. L.; Wang L.-C.; Krische M. J. J. Am. Chem. Soc. 2001, 123, 5112. 10.1021/ja0040971. [DOI] [PubMed] [Google Scholar]
- Eisenberg D. C.; Norton J. R. Isr. J. Chem. 1991, 31, 55. 10.1002/ijch.199100006. [DOI] [Google Scholar]
- Luche J. L. J. Am. Chem. Soc. 1978, 100, 2226. 10.1021/ja00475a040. [DOI] [Google Scholar]
- Snider B. B.; Rodini D. J.; van Straten J. J. Am. Chem. Soc. 1980, 102, 5872. 10.1021/ja00538a028. [DOI] [Google Scholar]
- Lackner G. L.; Quasdorf K. W.; Overman L. E. J. Am. Chem. Soc. 2013, 135, 15342. 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyjaszewski K.; Xia J. Chem. Rev. 2001, 101, 2921. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Giese B. Angew. Chem., Int. Ed. Engl. 1989, 28, 969. 10.1002/anie.198909693. [DOI] [Google Scholar]
- Minisci F.; Vismara E.; Fontana F. Heterocycles 1989, 28, 489. 10.3987/REV-88-SR1. [DOI] [Google Scholar]
- Chiang Y.; Kresge A. J. J. Org. Chem. 1985, 50, 5038. 10.1021/jo00225a007. [DOI] [Google Scholar]
- Brook A. G. Acc. Chem. Res. 1974, 7, 77. 10.1021/ar50075a003. [DOI] [Google Scholar]
- Simpkins L. M.; Bolton S.; Pi Z.; Sutton J. C.; Kwon C.; Zhao G.; Magnin D. R.; Augeri D. J.; Gungor T.; Rotella D. P.; Sun Z.; Liu Y.; Slusarchyk W. S.; Marcinkeviciene J.; Robertson J. G.; Wang A.; Robl J. A.; Atwal K. S.; Zahler R. L.; Parker R. Z.; Kirby M. S.; Hamann L. G. Bioorg. Med. Chem. Lett. 2007, 17, 6476. 10.1016/j.bmcl.2007.09.090. [DOI] [PubMed] [Google Scholar]
- Chu L.; Ohta C.; Zuo Z.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 10886. 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace T. J.; Gritter R. J. J. Org. Chem. 1961, 26, 5256. 10.1021/jo01070a097. [DOI] [Google Scholar]
- Urry W. H.; Juveland O. O.; Stacey F. W. J. Am. Chem. Soc. 1952, 74, 6155. 10.1021/ja01143a546. [DOI] [Google Scholar]
- Urry W. H.; Juveland O. O. J. Am. Chem. Soc. 1958, 80, 3322. 10.1021/ja01546a033. [DOI] [Google Scholar]
- The α-heteroatom-bearing radicals that 105, 108, 110, and 112–117 all target have been accessed using the developed functionalized olefin cross-coupling (vide infra).
- Similar processes have been observed using FeCl3·6H2O as a Lewis acid:Zotto C. D.; Michaux J.; Zarate-Ruiz A.; Gayon E.; Virieux D.; Campagne J.-M.; Terrasson V.; Pieters G.; Gaucher A.; Prim D. J. Organomet. Chem. 2011, 696, 296. 10.1016/j.jorganchem.2010.09.031. [DOI] [Google Scholar]
- Shigematsu T.; Matsui M.; Utsunomiya K. Bull. Inst. Chem. Res. Kyoto Univ. 1968, 46, 256. [Google Scholar]
- Hioe J.; Zipse H. Org. Biomol. Chem. 2010, 8, 3609. 10.1039/c004166a. [DOI] [PubMed] [Google Scholar]
- Stork G.; Landesman H. K. J. Am. Chem. Soc. 1956, 78, 5128. 10.1021/ja01600a087. [DOI] [Google Scholar]
- Juaristi E.; León-Romo J. L.; Reyes A.; Escalante J. Tetrahedron: Asymmetry 1999, 10, 2441. 10.1016/S0957-4166(99)00242-6. [DOI] [Google Scholar]
- Zhang H.; Li H.; Yang H.; Fu H. Org. Lett. 2016, 18, 3362. 10.1021/acs.orglett.6b01493. [DOI] [PubMed] [Google Scholar]
- Quiclet-Sire B.; Zard S. Z. J. Am. Chem. Soc. 2015, 137, 6762. 10.1021/jacs.5b03893. [DOI] [PubMed] [Google Scholar]
- Lambert J. B.; Wang G. T.; Finzel R. B.; Teramura D. H. J. Am. Chem. Soc. 1987, 109, 7838. 10.1021/ja00259a036. [DOI] [Google Scholar]
- Champagne P. A.; Desroches J.; Hamel J.-D.; Vandamme M.; Paquin J.-F. Chem. Rev. 2015, 115, 9073. 10.1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- Kropp P. J. Acc. Chem. Res. 1984, 17, 131. 10.1021/ar00100a003. [DOI] [Google Scholar]
- Giese B.; Dupuis J. Tetrahedron Lett. 1984, 25, 1349. 10.1016/S0040-4039(01)80154-4. [DOI] [Google Scholar]
- Hutchinson D. K.; Fuchs P. L. J. Am. Chem. Soc. 1987, 109, 4930. 10.1021/ja00250a029. [DOI] [Google Scholar]
- This same regioselectivity has been exploited by Herzon to achieve Mukaiyama hydroperoxidations of heteroatom-functionalized olefins. The fragmentation of the initial peroxide products forms ketones and esters. See:Ma X.; Herzon S. B. J. Org. Chem. 2016, 81, 8673. 10.1021/acs.joc.6b01709. [DOI] [PubMed] [Google Scholar]
- Seebach D. Angew. Chem., Int. Ed. Engl. 1979, 18, 239. 10.1002/anie.197902393. [DOI] [Google Scholar]
- Procter D. J. J. Chem. Soc., Perkin Trans. 1 2001, 335. 10.1039/b002081h. [DOI] [Google Scholar]
- Clayden J.; MacLellan P. Beilstein J. Org. Chem. 2011, 7, 582. 10.3762/bjoc.7.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénès F.; Pichowicz M.; Povie G.; Renaud P. Chem. Rev. 2014, 114, 2587. 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]
- Zweifel G.; Brown H. C. Org. React. 1963, 13, 1. [Google Scholar]
- Nakajima Y.; Shimada S. RSC Adv. 2015, 5, 20603. 10.1039/C4RA17281G. [DOI] [Google Scholar]
- Brown H. C.; Cole T. E. Organometallics 1983, 2, 1316. 10.1021/om50004a009. [DOI] [Google Scholar]
- Birkofer L.; Stuhl O.. General Synthetic Pathways to Organosilicon Compounds. In The Chemistry of Organic Silicon Compounds; Patai S., Rappoport Z., Eds.; Wiley: New York, 1989; pp 655–761. [Google Scholar]
- Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi E., de Meijere A., Eds.; John Wiley & Sons: New York, 2002. [Google Scholar]
- Kotali A.; Harris P. A.. Alkyl halides. In Comprehensive Organic Functional Group Transformations II; Katritzky A. R., Taylor R. J. K., Eds.; Elsevier: Oxford, 2005: Vol. 2, pp 1–21. [Google Scholar]
- Handbook of Metathesis, 2nd ed.; Grubbs R. H., O’Leary D. J., Eds.; Wiley-VCH: Weinheim, 2015: Vol. 2. [Google Scholar]
- Magnus P. D. Tetrahedron 1977, 33, 2019. 10.1016/0040-4020(77)80311-6. [DOI] [Google Scholar]
- Rodrigo E.; Alonso I.; Ruano J. L. G.; Cid M. B. J. Org. Chem. 2016, 81, 10887. 10.1021/acs.joc.6b01956. [DOI] [PubMed] [Google Scholar]
- Choi J.; Tang L.; Norton J. R. J. Am. Chem. Soc. 2007, 129, 234. 10.1021/ja066325i. [DOI] [PubMed] [Google Scholar]
- Szostak M.; Spain M.; Procter D. J. Chem. Soc. Rev. 2013, 42, 9155. 10.1039/c3cs60223k. [DOI] [PubMed] [Google Scholar]
- Blakemore P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563. 10.1039/b208078h. [DOI] [Google Scholar]
- Gianatassio R.; Kawamura S.; Eprile C. L.; Foo K.; Ge J.; Burns A. C.; Collins M. R.; Baran P. S. Angew. Chem., Int. Ed. 2014, 53, 9851. 10.1002/anie.201406622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash G. K. S.; Hu J.; Wang Y.; Olah G. A. Angew. Chem., Int. Ed. 2004, 43, 5203. 10.1002/anie.200460815. [DOI] [PubMed] [Google Scholar]
- Elmore C. S. Annu. Rep. Med. Chem. 2009, 44, 515. 10.1016/S0065-7743(09)04425-X. [DOI] [Google Scholar]
- Duncton M. A. J. MedChemComm 2011, 2, 1135. 10.1039/c1md00134e. [DOI] [Google Scholar]
- Edwards J. T; Baran P. S.. C–H Alkylation of Heterocycles. Presented at the International Chemical Congress of the Pacific Basin Societies, Honolulu, HI, Dec 15–20, 2015.
- Ma X.; Herzon S. B. J. Am. Chem. Soc. 2016, 138, 8718. 10.1021/jacs.6b05271. [DOI] [PubMed] [Google Scholar]
- Green S. A.; Matos J. L. M.; Yagi A.; Shenvi R. A. J. Am. Chem. Soc. 2016, 138, 12779. 10.1021/jacs.6b08507. [DOI] [PubMed] [Google Scholar]
- Minisci F.; Galli R.; Cecere M.; Malatesta V.; Caronna T. Tetrahedron Lett. 1968, 9, 5609. 10.1016/S0040-4039(00)70732-5. [DOI] [Google Scholar]
- Deng G.; Ueda K.; Yanagisawa S.; Itami K.; Li C.-J. Chem. - Eur. J. 2009, 15, 333. 10.1002/chem.200801893. [DOI] [PubMed] [Google Scholar]
- Katz R. B.; Mistry J.; Mitchell M. B. Synth. Commun. 1989, 19, 317. 10.1080/00397918908050984. [DOI] [Google Scholar]
- Nishida T.; Ida H.; Kuninobu Y.; Kanai M. Nat. Commun. 2014, 5, 3387. 10.1038/ncomms4387. [DOI] [PubMed] [Google Scholar]
- Alonso F.; Beletskaya I. P.; Yus M. Chem. Rev. 2002, 102, 4009. 10.1021/cr0102967. [DOI] [PubMed] [Google Scholar]
- Braude E. A.; Hannah J.; Linstead R. J. Chem. Soc. 1960, 3249. 10.1039/jr9600003249. [DOI] [Google Scholar]
- Notably, similar mixtures of regioisomers have been obtained by nucleophilic perfluoroalkylations of Lewis acid-activated heteroarenes:Nagase M.; Kuninobu Y.; Kanai M. J. Am. Chem. Soc. 2016, 138, 6103. 10.1021/jacs.6b01753. [DOI] [PubMed] [Google Scholar]
- Several of the steps depicted in Figure 11A might be reversible. This is not indicated in the mechanistic proposal since these steps have yet to be identified.
- Gumbel G.; Elias H. Inorg. Chim. Acta 2003, 342, 97. 10.1016/S0020-1693(02)01143-X. [DOI] [Google Scholar]
- Wu C.-H. S.; Rossman G. R.; Gray H. B.; Hammond G. S.; Schugar H. J. Inorg. Chem. 1972, 11, 990. 10.1021/ic50111a015. [DOI] [Google Scholar]
- These observations also imply that the Fe-based active catalyst is present only in trace amounts under these conditions, suggesting that the identification and use of the active catalyst directly in the olefin cross-coupling could result in significantly lower catalyst loadings.
- Halpern J. Inorg. Chim. Acta 1981, 50, 11. 10.1016/S0020-1693(00)83716-0. [DOI] [Google Scholar]
- Xue Z.; Daran J.-C.; Champouret Y.; Poli R. Inorg. Chem. 2011, 50, 11543. 10.1021/ic201486v. [DOI] [PubMed] [Google Scholar]
- Fe(acac)2 is dimeric in the solid state when purified by sublimation.
- Interestingly, further ethanolysis of PhSi(OEt)H2 (351) to give PhSi(OEt)2H (357) and PhSi(OEt)3 (369) did not take place upon heating in d6-benzene at 60 °C with EtOH in the absence of Fe(acac)2.
- Obradors C.; Martinez R. M.; Shenvi R. A. J. Am. Chem. Soc. 2016, 138, 4962. 10.1021/jacs.6b02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PhSi(OEt)H2 (351) disproportionates to PhSiH3 and PhSi(OEt)2H (357) at room temperature, making it unsuitable for practical use as a reductant in the olefin cross-coupling.
- Control experiments showed the PhSi(OEt)2H (357) was also able to facilitate the olefin cross-coupling. However, the yields when using 357 were approximately 25% of the yields when using PhSiH3, suggesting that 357 is a relatively inefficient terminal reductant.
- The fractional orders in donor and Fe(acac)3 are tentatively attributed to off-cycle processes that involve these two species.
- Sweany R. L.; Halpern J. J. Am. Chem. Soc. 1977, 99, 8335. 10.1021/ja00467a045. [DOI] [Google Scholar]
- Bullock R. M.; Samsel E. G. J. Am. Chem. Soc. 1990, 112, 6886. 10.1021/ja00175a024. [DOI] [Google Scholar]
- Shackleton T. A.; Baird M. C. Organometallics 1989, 8, 2225. 10.1021/om00111a020. [DOI] [Google Scholar]
- Halpern J. Pure Appl. Chem. 1986, 58, 575. 10.1351/pac198658040575. [DOI] [Google Scholar]
- Nalesnik T. E.; Freudenberger J. H.; Orchin M. J. Mol. Catal. 1982, 16, 43. 10.1016/0304-5102(82)80062-X. [DOI] [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006; pp 432–433. [Google Scholar]
- There is also the possibility that the rate-determining step of the olefin cross-coupling is entirely different from HAT to the donor olefin. An alternative scenario that is consistent with both the observed reactant orders and the normal KIE could be the formation of an Fe hydride from PhSi(OEt)H2 (351) and some Fe complex derived from the donor olefin and Fe(acac)3.
- In contrast, Shenvi has observed a very slight inverse KIE (value not determined) for his developed Mn(dpm)3-catalyzed thermodynamic reduction of olefins with PhSiH3 (see ref (127)). However, the authors do caution about overinterpreting the observed inverse KIE.
- Nonhebel D. C. Chem. Soc. Rev. 1993, 22, 347. 10.1039/cs9932200347. [DOI] [Google Scholar]
- George D. T.; Kuenstner E. J.; Pronin S. V. J. Am. Chem. Soc. 2015, 137, 15410. 10.1021/jacs.5b11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian H.; Landais Y.; Sibi M. P.. Free-Radical Additions to Carbon–Oxygen Double Bonds. In Comprehensive Organic Synthesis, 2nd ed.; Knochel P., Molander G. A., Eds.; Elsevier: Oxford, 2014; pp 724–725. [Google Scholar]
- Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Chem. Rev. 2012, 112, 4016. 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- Olmstead W. N.; Margolin Z.; Bordwell F. G. J. Org. Chem. 1980, 45, 3295. 10.1021/jo01304a032. [DOI] [Google Scholar]
- Alfassi Z. B.; Golden D. M. J. Phys. Chem. 1972, 76, 3314. 10.1021/j100667a003. [DOI] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Walling C.Free Radicals in Solution; John Wiley & Sons: New York, 1957. [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006; pp 445–453. [Google Scholar]
- The beneficial effects of styrene additives in certain transition metal-catalyzed reactions have been previously described, including a Ni-catalyzed cross-coupling (see; Jensen A. E.; Knochel P. J. Org. Chem. 2002, 67, 79. 10.1021/jo0105787. [DOI] [PubMed] [Google Scholar]; ) and a Ru-catalyzed C–H arylation (see; Hiroshima S.; Matsumura D.; Kochi T.; Kakiuchi F. Org. Lett. 2010, 12, 5318. 10.1021/ol102325f. [DOI] [PubMed] [Google Scholar]; ) However, such systems bear little resemblance to the developed olefin cross-coupling, and the role of styrene in each case is likely different.
- Dao H. T.; Li C.; Michaudel Q.; Maxwell B. D.; Baran P. S. J. Am. Chem. Soc. 2015, 137, 8046. 10.1021/jacs.5b05144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui J.; Pan C.-M.; Jin Y.; Qin T.; Lo J. C.; Lee B. J.; Spergel S. H.; Mertzman M. E.; Pitts W. J.; La Cruz T. E.; Schmidt M. A.; Darvatkar N.; Natarajan S. R.; Baran P. S. Science 2015, 348, 886. 10.1126/science.aab0245. [DOI] [PubMed] [Google Scholar]
- Ruider S. A.; Sandmeier T.; Carreira E. M. Angew. Chem., Int. Ed. 2015, 54, 2378. 10.1002/anie.201410419. [DOI] [PubMed] [Google Scholar]
- Fang X.; Yu P.; Morandi B. Science 2016, 351, 832. 10.1126/science.aae0427. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Wang D.; Cui S. Org. Lett. 2015, 17, 4572. 10.1021/acs.orglett.5b02294. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Qi J.; Cui S. Org. Lett. 2016, 18, 128. 10.1021/acs.orglett.5b03317. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Qi J.; Mao Z.; Cui S. Org. Lett. 2016, 18, 2722. 10.1021/acs.orglett.6b01173. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chen C.; Cui S. RSC Adv. 2016, 6, 93753. 10.1039/C6RA20007A. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.