

Abstract
Septic patients experience chronic immunosuppression resulting in enhanced susceptibility to infections normally controlled by T cells. Clinical research on septic patients has shown increased apoptosis and reduced total numbers of CD4 and CD8 T cells, suggesting contributing mechanism driving immunosuppression. Experimental models of sepsis, including cecal ligation and puncture, reverse translated this clinical observation to facilitate hypothesis-driven research and allow the use of an array of experimental tools to probe the impact of sepsis on T-cell immunity. In addition to numerical loss, sepsis functionally impairs the antigen-driven proliferative capacity and effector functions of CD4 and CD8 T cells. Sepsis-induced impairments in both the quantity and quality of T cells results in reduced protective capacity and increased susceptibility of mice to new or previously encountered infections. Therefore, the combined efforts of clinical and experimental sepsis research have begun to elucidate the impact of sepsis on T-cell-mediated immunity and potential T-cell-intrinsic and -extrinsic mechanisms driving chronic immunosuppression. Future work will explore the impact of sepsis on the recently appreciated tissue-resident memory (TRM) T cells, which provide robust protection against localized infections, and dendritic cells, which are needed to activate T cells and promote effective T-cell responses.
Keywords: sepsis, CD4 and CD8 T cells, apoptosis, circulatory and tissue-resident memory, cecal ligation and puncture, dendritic cells
I. INTRODUCTION
Sepsis is the result of an exaggerated immune response to a systemic infection that leads to damage or death of the host. Recent population-level studies have estimated the global burden of sepsis to be 31.5 million cases annually, with a death toll of 5.3 million individuals.1 In the United States, hospitalizations due to sepsis accounted for over $20 billion in total healthcare costs in 2007.2 A study investigating readmission rates of academic medical center-affiliated hospitals in the United States found that 20% of patients diagnosed with severe sepsis were readmitted to the hospital within 30 days of discharge, with over 60% of these readmissions due to secondary infection.3 The outcome of these studies, coupled with increases in the incidence of sepsis, underscore the need for further investigation into the possible deficits of T-cell-mediated immunity that lead to increased susceptibility to infection after a septic event.4
A variety of immunologic insults have the potential to precipitate a septic event. Pulmonary infections are the most common site of primary infection, whereas infections of the abdomen (e.g., those arising from a perforated or ischemic bowel), soft tissues, and the urinary tract are also common primary infection loci in adult septic patients.5,6 Causative microorganisms include both gram-positive (Staphylococcus aureus and Streptococcus pneumoniae) and gram-negative bacteria (Escherichia coli and Klebsiella species), with some patients experiencing polymicrobial infections.7 In addition, the number of sepsis cases caused by fungal organisms has increased substantially.8 However, as noted in a recent study, a pathogen may be unable to be isolated and identified in up to 30% of septic patients.9 The pathogen-specific biology of sepsis is an important parameter that influences host responses after a septic event, as well the efficacy of therapeutic interventions. This idea is highly relevant to the study of the immune system and to T-cell responses in particular.
After the initial septic insult, the immune system simultaneously produces both pro-inflammatory and anti-inflammatory cytokines, resulting in a “cytokine storm.”10 Although both pro-inflammatory and anti-inflammatory mediators are present, the pro-inflammatory response, hallmarked by increased levels of tumor necrosis factor-alpha (TNF-α) and interleukin 1-beta (IL-1β) in the serum of septic patients, is predominant very early after a septic event.10–12 This increase in pro-inflammatory cytokines leads to increased gene expression of inducible nitric oxide synthase (iNOS), type II phospholipase (PLA2), and cyclooxygenase-2 (COX-2), which produce NO, leukotrienes, and prostanoids.13,14 Depending on the health status of the host, the systemic effects of these pro-inflammatory cytokines and their small-molecule mediators may result in the manifestation of early clinical signs such as hypotension, shock, fever, and death.10,13,14
Septic patients that survive the initial phase dominated by pro-inflammatory mediators transition to a state of immunoparalysis and have increased susceptibility to opportunistic secondary infections.15–19 In addition to secondary infections, a high frequency of septic patients experience reactivation of latent viral infections such as cytomegalovirus (CMV), as detected by viral copy number in the plasma, or herpes simplex virus (HSV), as detected by HSV nuclear inclusions from pulmonary samples.17,20,21 Furthermore, sepsis survivors have an increased risk of death from non-septic events that extends 5 years beyond the initial septic insult, suggesting that septic patients suffer from long-term impairments.22 Despite these prolonged deficits, studies investigating the long-term consequences of a septic event in survivors are lacking.
Opportunistic secondary infections and viral reactivation indicate that septic patients may have a defect in T-cell-mediated immunity. T cells are divided into conventional CD4 and CD8 populations and provide important regulatory and effector immune functions during infection. The composition of the naive pathogen-specific CD8 T-cell repertoire is important in both the clearance of infection and the generation of memory CD8 T cells in response to infection and/or vaccination. Upon interaction with their cognate antigen (Ag) in the presence of co-stimulatory molecules and appropriate cytokines, naive Ag-specific CD8 T cells undergo vigorous proliferative expansion in numbers (Fig. 1A, model).23–25 This expanding pool gains effector functions characterized by the production of cytokines [e.g., interferon-gamma (IFN-γ) and TNF-α] and the ability to lyse infected host cells, thus providing the host with increased protection from the pathogen.25–29 Depending on the type of pathogen and pathogen biology, the peak number of Ag-specific effector CD8 T cells is achieved days to weeks after the initial infection. At this point, 95–98% of the expanded pool of Ag-specific CD8 T cells is eliminated during the programmed contraction (death) phase, with the surviving fraction encompassing a memory CD8 T-cell population with a protective capacity upon Ag re-encounter (re-infection) that depends on both the quantity and functional fitness of the CD8 T cell memory pool.25,30–34 These long-lived memory CD8 T cells undergo proliferative expansion upon pathogen re-encounter and provide increased protection after re-infection (Fig. 1B).25,35,36
FIG. 1.
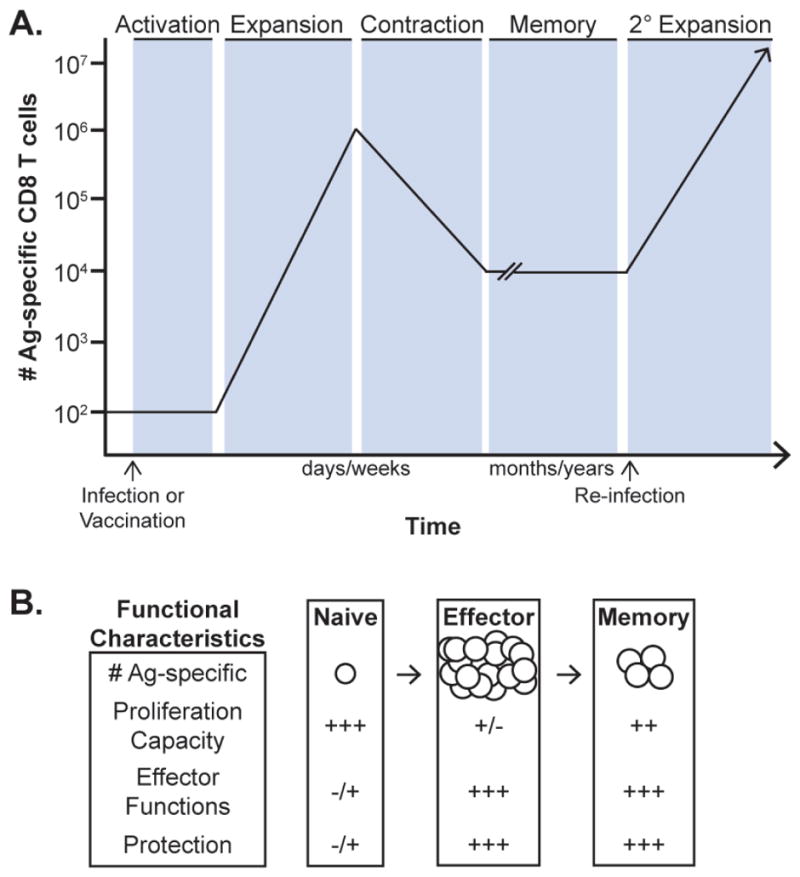
Distinct phases of the primary CD8 T-cell responses upon antigen encounter. A, Naive CD8 T cells encounter cognate Ag and appropriate signals and promote their activation and accumulation during the vigorous primary expansion phase. Upon completion of the expansion, effector CD8 T cells undergo a programmed contraction (death) phase, which leaves a stable number of memory CD8 T cells that can be maintained for the life of the host and rapidly undergo secondary expansion upon Ag re-encounter. B, Functional properties of naive, effector and memory CD8 T cells. Naive CD8 T cells do not possess effector functions, but, upon exposure to cognate Ag, this population has the capacity to undergo proliferative expansion in numbers. Effector and memory CD8 T cells possess effector functions (cytokine secretion and cytolytic capacity) that mediate host protection. Vaccination or infection-generated memory CD8 T cells exposed to cognate Ag possess the capacity to undergo proliferative expansion and gain effector functions, providing rapid protection to the host upon re-infection. (Figure adapted with permission from Badovinac et al, 2006.35).
After recognition of cognate Ag expressed by antigen-presenting cells (APCs), naive CD4 T cells are polarized to different phenotypes [T-helper 1 (Th1), Th2, Th17, regulatory T cells (Treg)] in part due to environmental cytokines.37–43 Th1 CD4 T cells serve important roles in the activation of cytotoxic CD8 T cells and the formation of memory CD8 T cells via IL-2 secretion.44–49 Th2 CD4 T cells mediate class switching of B cells via the production of IL-4 and IL-5.43,50,51 Th17 CD4 T cells, which are effector CD4 T cells that produce IL-17, IL-22, and TNF-α, are important in immunity to extracellular fungal and bacterial pathogens (especially at mucosal surfaces52) through the recruitment and activation of neutrophils.53 Treg play a vital role in the maintenance of immune homeostasis, serving to limit host immune responses after infection to prevent host tissue damage.54 Due to the importance of T cells in controlling and eradicating infection, further examination into the impact of sepsis on the number, phenotype, and function of T cells is clearly warranted.
II. CLINICAL SEPSIS INDUCES T-CELL APOPTOSIS
Apoptosis, or programmed cell death, is important in the development and homeostasis of the immune system; however, it also plays a detrimental role in disease pathology, with septic patients showing increased presence of apoptotic lymphocytes in the spleen.55–58 Post mortem samples of septic intensive care unit patients within 90 minutes of death show increased signs of apoptosis (pyknosis and karyorrhexis) compared with critically ill, non-septic controls.57 In addition, immunohistochemistry stains from septic patients demonstrate increased levels of activated caspase 3, a protease in the common apoptotic pathway, compared with non-septic patients.57–59 An increase in apoptotic markers has also been observed in the spleens of pediatric patients, suggesting that lymphocyte apoptosis during sepsis is a universal phenomenon.60
Increased frequency of apoptotic CD4 and CD8 T cells has also been detected in the peripheral blood of septic patients, which corresponds to persistent lymphopenia, compared with other non-septic, critically ill patients.61,62 Furthermore, T cells isolated from septic patients had increased levels of caspase 8 and caspase 9, suggesting that apoptosis of blood lymphocytes associated with sepsis occurs by both intrinsic and extrinsic apoptotic pathways.62 In addition to observing increased apoptosis of CD4 and CD8 T cells in septic patients, Weber et al. found that septic patients have increased expression of mRNA encoding for the pro-apoptotic proteins Bim, Bid, and Bak in the peripheral blood and down-regulate the anti-apoptotic protein Bcl-2 in CD4 T cells of the blood compared with their non-septic, critically ill counterparts.63 The apoptosis of T cells and increased expression of mRNA encoding pro-apoptotic proteins in septic patients is consistent with the state of chronic immune suppression after a septic event and suggests a possible mechanism for the observed susceptibility to opportunistic secondary infections and the reactivation of latent viral infections in sepsis survivors.
III. EXPERIMENTAL SEPSIS MODELS FACILITATE HYPOTHESIS-DRIVEN RESEARCH
Clinical observations of septic patients with enhanced T-cell apoptosis and chronic immunosuppression offers valuable insight to how sepsis impairs T-cell-mediated immunity. However, limiting factors of hypothesis-driven research in humans, including biological heterogeneity and imperfect control cohorts, has led to the necessity of an experimental sepsis model. Cecal ligation and puncture (CLP) represents a widely used model of polymicrobial sepsis that induces T-cell apoptosis. This model has many advantages, such as controllable sepsis severity by adjusting cecal ligation length and/or number of punctures,64,65 and, analogous to clinical scenarios, CLP mice receiving fluid resuscitation or broad-spectrum antibiotics have improved prognosis.66,67 With clinical observations of T-cell apoptosis during sepsis reverse translated in an experimental mouse model of sepsis, researchers are able to use additional experimental techniques to perform hypothesis-driven experiments. Ultimately, validated hypotheses using mouse models will be translated to human patients to encompass a wide array of biological and pathological heterogeneity to further test these hypotheses (Fig. 2).68
FIG. 2.

Proposed interplay of clinical and experimental research to elucidate the impact of sepsis on T-cell-mediated immunity. A, Clinical research on septic patients has shown increased apoptosis and reduced numbers of CD4 and CD8 T cells. B, Clinical observations of sepsis-induced apoptosis were successfully reverse translated in an experimental model (e.g., CLP). C, Experimental models could allow sepsis researchers to pursue hypothesis-driven research and utilize an array of experimental tools that are not feasible with human patients. The ultimate goal of experimental models is to translate hypotheses to human septic patients to further test hypotheses incorporating an array of biological and pathological heterogeneity. (Figure adapted with permission from Efron et al, 2015.68).
Mouse models are essential for the study of both general sepsis biology, as well as for dissecting immunological changes associated with the immunosuppression phase of sepsis. One key advantage of mouse models is the generation of highly reproducible conclusions due to the ability to control experimentally the severity, timing, and nature of the septic insult. In addition to these parameters, a wide array of cutting-edge experimental techniques are possible with the use of inbred mice. Unlike human patients, a priori knowledge of major histocompatibility complex (MHC) restriction and pathogen-derived T-cell epitopes in inbred mice permits precise in vivo assessment of endogenous Ag-specific T-cell responses and the role of sepsis in the development, function, and maintenance of T-cell immunity. For instance, the use of peptide: MHC I tetramer-based enrichment technology has permitted enumeration of the relatively low number of naive Ag-specific CD4 and CD8 T cells, enabling analysis of primary T-cell responses in the context of sepsis.69 One way that we have used the power of cutting-edge immunological techniques to explore specific hypotheses about sepsis-induced immune suppression is to adoptively transfer T-cell receptor-transgenic (TCR-tg) T cells into mice before or after the induction of sepsis. Adoptive transfer of such T cells, followed by infection with recombinant pathogens expressing cognate Ag, permits analyses of the impact of sepsis on a defined Ag-specific T-cell population during distinct phases of the CD8 T-cell responses. In contrast, clinical studies of septic patients typically examine the impact of sepsis on the bulk T-cell pool, which is composed of a heterogeneous population of mixed Ag specificity and unknown stimulation history. Last, an additional benefit of mouse models is the large assortment of gene knock-out mice that have proven useful in probing the contribution of individual molecules (e.g., inflammatory mediators) in the context of sepsis. In addition, gene knock-out mice were pivotal in elucidating that T-cell-sufficient hosts have higher levels of IL-6 during the acute phase of sepsis, suggesting that this subset of leukocytes plays a role in exacerbating the cytokine storm that develops during a septic event.70–72 Therefore, inbred mice generate highly reproducible results and permit an array of experimental techniques that make them valuable tools for pursuing hypothesis-driven research. Undoubtedly, a shortcoming of sepsis research utilizing inbred mice is the lack of biological heterogeneity associated with human populations. However, the use of outbred mice provides a middle ground between the biological homogeneity of inbred mice and septic patients. Some reports have recently elucidated the potential of using outbred mice (e.g. Swiss Webster) as an additional experimental tool in the context of sepsis research.73–77
However, the usefulness of some animal models of sepsis has been recently called into question with reports elucidating the discrepancies between human and mouse immune cell subset composition, TCR signaling, and surface molecule expression,78 as well as the fact that laboratory mice housed under “specific-pathogen-free” conditions have a CD8 T-cell phenotype and distribution pattern that more closely resemble human newborns.79 One critical report by Seok et al. compared the genomic responses of human and animal models of inflammatory diseases and concluded that animal models poorly mimic their human counterparts.80 However, Takao et al. re-analyzed these genomic responses of inflammatory diseases using a different analysis approach and came to the opposite conclusion: that mouse models of inflammatory diseases do highly recapitulate the human disease, which suggests the benefit of mouse models as a research tool.81 Ultimately, all models mentioned will be critical to further elucidate the impact of sepsis on T-cell-mediated immunity that leads to enhanced susceptibility of septic patients to new or previously encountered infections.
IV. ROLE OF SEPSIS IN SHAPING T-CELL RESPONSES TO NEWLY ENCOUNTERED INFECTIONS
Although a relatively small number of naive CD8 T cells specific for any particular pathogen-derived Ag exist in vivo, newly encountered infections have the capacity to trigger a substantial number of pathogen-specific naive CD8 T-cell precursors, promoting their numerical expansion, gain in effector function, and pathogen clearance.82–84 Importantly, the magnitude of the primary Ag-specific CD8 T-cell response correlates with the dose of the infection and duration of inflammation,30,85 as well as with the number of naive CD8 T-cell precursors recruited into the response.86 Therefore, sepsis-induced alterations in the number of naive T cells have the potential to compromise the ability of the host to mount effective primary T-cell responses. The first reports examining the impact of sepsis on T cells determined that sepsis-induced apoptosis of thymocytes87–89 and T-cell apoptosis was observed in nearly all tissues examined, including the spleen, lung, and colon.90 Direct examination of naive CD4 and CD8 T cells based on phenotype or using TCR-tg T cells further confirmed that T cells are highly susceptible to sepsis-induced apoptosis.69,91–93 These results show that sepsis-induced naive T-cell apoptosis and numerical loss is a phenomenon that occurs in an array of host tissues. Subsequent studies using caspase inhibitors or overexpression of the anti-apoptotic protein Bcl-2 confirmed that apoptosis was the primary mechanism driving sepsis-induced numerical loss of T cells.94,95 Interestingly, using the state-of-the-art techniques to count naive CD8 T-cell precursors specific for defined pathogen-derived antigens, we were able to show that sepsis-induced apoptosis has the capacity to change the composition of naive CD8 T-cell repertoire, leading to sustained and incomplete recovery of naive CD8 T-cell precursors and contributing to impaired CD8 T-cell responses to new infections (Fig. 3).69,74 The data also suggested that, due to the stochastic nature of the sepsis-induced changes in the composition of naive CD8 T cells, potential “holes” in the CD8 T-cell repertoire can be formed, further contributing to the reduction in primary CD8 T-cell responses to new infections in sepsis survivors.69,96 Attempts to reverse the sepsis-induced reduction in the number of naive T cells has used exogenous addition of the prosurvival cytokines IL-7 and IL-15, which enhances T-cell expression of Bcl-2 and demonstrates some efficacy in experimental models.91,97 Therefore, sepsis can result in substantial and long-lasting changes in the available T-cell repertoire, affecting the capacity of the host to respond to subsequent infections.
FIG. 3.
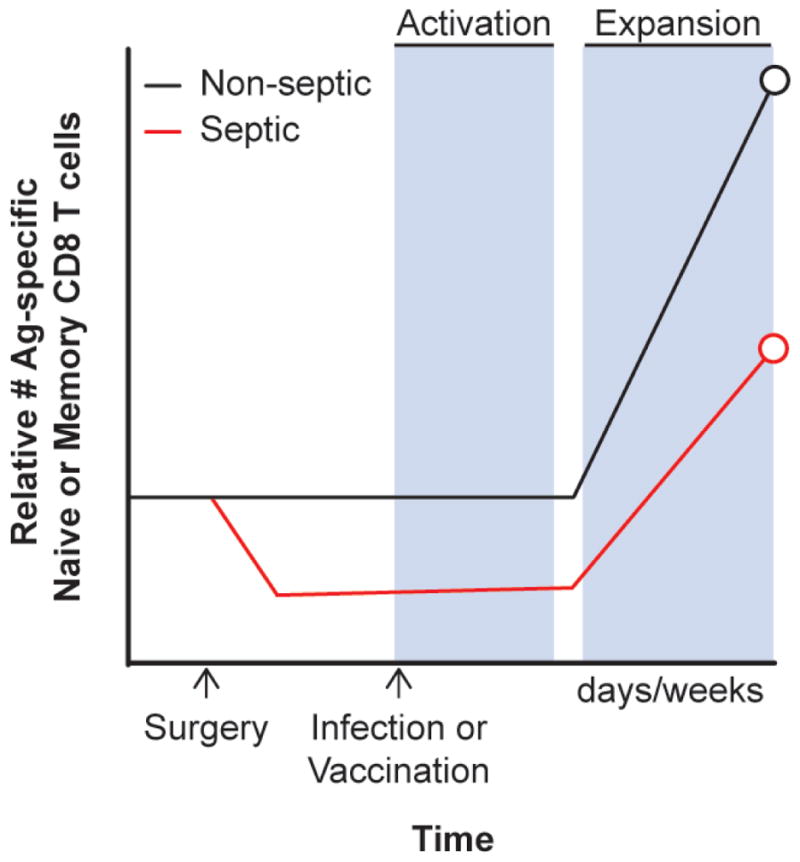
Sepsis affects the number of naive and memory CD8 T cells, influencing their ability to respond to secondary infection. Naive and primary memeory Ag-specific CD8 T cells undergo numerical loss shortly after sepsis induction. Due to their decreased numbers and/or impaired per-cell functionality, the magnitude of primary and secondary CD8 T-cell expansion is diminished in the septic host after secondary infection.
Beyond sepsis-induced numerical changes, CD8 T cells from septic patients undergo phenotypic alterations that reduce the quality (functional capacity) of CD8 T cells upon stimulation.98 Therefore, sepsis-induced changes in the quality of CD8 T cells also likely contribute to the enhanced susceptibility of septic patients to both new and previously encountered infections. The numerical loss of all lymphocytes subsets, including T cells, as a result of sepsis generates a lymphopenic environment that has the capacity to initiate homeostatic proliferation of naive T cells and their numerical recovery.99 Homeostatic proliferation, however, results in naive T cells adopting an “Ag-experienced” or “memory-like” T-cell phenotype in naive CD4 (CD11ahi CD49d+) and CD8 (CD8αlo, CD11ahi) T cells, even in the absence of cognate Ag recognition.69,93,99–101 This suggests that sepsis induces long-term phenotypic alterations of the surviving naive T-cell pool. It remains to be determined whether naive CD8 T cells express additional “memory-like” markers that could alter their migratory or proliferative capacity and how naive CD8 T-cell acquisition of this phenotype alters their response to new infections. Coinciding with this phenotypic change, sepsis-induced homeostatic proliferation alters the TCR clonotype composition of an Ag-specific CD4 T-cell population.93 Due to this finding, it is speculated sepsis-induced homeostatic proliferation of naive T cells condenses the clonal diversity of the naive T-cell pool, potentially reducing the breadth of Ag that the host can recognize.102 Importantly, over 30 days after sepsis induction in mice, not all Ag-specific naive T cells undergo complete numerical recovery as a result of homeostatic proliferation, which suggests long-term numerical impairments of the naive T-cell pool.69,93 Therefore, sepsis shapes the composition and the phenotype of the naive T-cell pool, which could impair the effectiveness of subsequent T-cell responses. Additional inquiry into the extent to which sepsis-induced alterations of the T-cell pool reduces their overall quality and affects subsequent T-cell responses is warranted. This future work will be critical to further understanding septic patients’ enhanced susceptibility to newly encountered pathogens.
The extent to which sepsis-induced alterations to the naive T-cell compartment impairs subsequent T-cell responses is important to further understanding the enhanced susceptibility of septic patients to primary infections. Specifically, septic patients may have enhanced susceptibility to either acute (short-term) or chronic (persistent) infections. Septic hosts that acquired an acute infection had impaired T-cell responses, resulting in a reduced number of effector CD8 T cells producing effector cytokines (IFN-γ, TNF-α, and IL-2).69,103 Similar impairments in the magnitude of expansion and effector functions of CD4 T cells have been seen after sepsis92,93 and be a result of sepsis impairing CD4 Th lineage commitment due to repressive histone methylation marks in the promoter regions associated with Th1 and Th2 lineages.92 Interestingly, amelioration of the deleterious effects of sepsis on the primary expansion of CD8 T cells could be achieved by administering α-TNF-related apoptosis-inducing ligand (TRAIL) antibody.104 Therefore, sepsis-induced dysfunction of memory T-cell expansion is in part TRAIL mediated.96,105 Furthermore, chronically infected septic hosts generate a primary CD8 T-cell response with impaired cytokine production and enhanced expression of inhibitory molecules (e.g. PD-1 and LAG-3), leading to increased viral burden.74 Importantly, the impairments in the CD8 T-cell response to chronic infections were seen long after the initial septic event resolved.74 Amelioration of the sepsis-enhanced CD8 T-cell exhaustion and improved control of chronic infection could be achieved in mice that received therapeutic blockade of PD-1 and LAG3.74 This report suggests that septic hosts exposed to chronic infections have an enhanced rate of CD8 T-cell exhaustion, resulting in an impaired T-cell response to chronic infections. Attempts to reduce T-cell exhaustion in clinical settings with therapeutic administration of anti-PD-1 has shown some efficacy in reducing CD4 and CD8 T-cell apoptosis and restoring effector cytokine production in septic patients.98 In short, sepsis-induced alterations in the composition of the naive T-cell pool results in increased susceptibility to new infections.
V. ROLE OF SEPSIS IN SHAPING INFECTION AND/OR VACCINE-INDUCED MEMORY T-CELL RESPONSES
The increased susceptibility to previously encountered infections and latent viral reactivation exhibited by septic patients suggests impairments in previously generated memory T-cell responses. Some of the first reports of impaired T-cell immunity in septic patients described the reduction of delayed-type hypersensitivity reactions, even to Ags known to have been encountered previously.106 Because the quantity (number) and quality (functional capacity) of memory T cells at the time of re-infection affects directly the degree of T-cell-mediated protection, the impact of sepsis on these parameters will be discussed.30,31,34
Sepsis reduces the number of memory CD4 and CD8 T cells in a variety of tissues (e.g., blood and spleen).73,91,104,107 Similar to naive T cells, numerical alterations of memory T cells are driven by sepsis-induced apoptosis, which is facilitated by cell expression of LFA-1 (CD11a).73,91,108 Reductions in the quantity of memory T cells suggest a mechanism for septic patients enhanced susceptibility to previously encountered pathogens. Upon re-infection, sepsis impairs the magnitude of secondary expansion due to a reduced number of memory T cells and impairs the proliferation capacity on a per-cell basis (Fig. 3).73,92,104 Importantly, sepsis-induced impairments in the secondary expansion of T cells results in a higher pathogen burden.73,104 However, therapeutic administration of the prosurvival cytokine IL-7 after sepsis restores the number of central and effector memory CD4 and CD8 T cells.91,109 In short, vaccination or infection-generated memory T cells are numerically susceptible to sepsis-induced apoptosis and, upon re-infection, results in a reduced magnitude of secondary expansion and impaired pathogen clearance.
Beyond numerical alterations, sepsis also reduces the functional quality of memory T cells, which contributes to the impairments in the T-cell response. Sepsis reduces the ability of memory CD8 T cells to produce IFN-γ upon encountering cognate Ag (functional avidity), suggesting that sepsis induces memory CD8 T-cell-intrinsic dysfunction on a per-cell basis.73 In addition, memory CD8 T cells do not require Ag to participate during infection because inflammatory cytokines (e.g. IL-12 and IL-18) are sufficient to drive Ag-independent activation, also known as bystander activation.110–115 Importantly, we showed recently that sepsis impairs bystander activation of memory CD8 T cells in vivo in mice.73 Experimental bystander activation was performed using lymphocytic choriomeningitis virus (LCMV)-immune mice that subsequently received Listeria monocytogenes (LM) as a heterologous infection. There is no known cross-reactivity between LCMV and LM from the CD8 T-cell perspective, so LCMV-specific memory CD8 T-cell production of effector cytokines (e.g., IFN-γ) is due to bystander activation. This impairment of memory CD8 T cells to sense and respond to heterologous infection in a bystander manner after sepsis was evident in both inbred and outbred (Swiss Webster) mice.73 However, memory CD8 T cells from septic hosts regained bystander activation capacity in vitro when cultured with the appropriate inflammatory cytokines.73 This suggests the exciting possibility that impairments in bystander activation of memory T cells was due to a T-cell-extrinsic factor, possibly due to sepsis-induced impairments in the function of innate immune cells ability to provide inflammatory cytokines (e.g., IL-12) upon heterologous infection. Together, these results suggest that sepsis-induced impairments of T-cell-mediated immunity could be the result of dysfunction in both T-cell-intrinsic and -extrinsic factors, a notion that warrants further investigation.
VI. FUTURE DIRECTIONS
A. Impact of Sepsis on Tissue-Resident Memory T-Cell-Mediated Immunity
This review, and in fact the majority of the sepsis literature, has probed the impact of sepsis on T-cell-mediated immunity by examining T cells from secondary lymphoid organs and peripheral blood. This subset of circulating memory T cells (TCIRM) migrate passively in search of their cognate Ag due to the chemotactic influence of sphingosine-1-phosphate (S1P) and/or the CCR7 ligands CCL19 and CCL21.116 Paradigmatically, it was thought the memory T-cell pool had the capacity to circulate throughout host tissues and lymphoid organs in search for cognate Ag after vaccination or infection. However, Masopust et al. discovered that memory CD8 T cells could position themselves within non-lymphoid barrier tissues (e.g., lung and small intestine) after infection, with these cells possessing enhanced cytolytic capacity compared with their splenic T-cell counterparts.117 Teologically, it was predicted that memory CD8 T cells within barrier tissues could provide protection rapidly upon homologous infection compared with their TCIRM counterparts. Subsequently, it was determined that effector CD8 T cells seed non-lymphoid tissue after vaccination or infection and generate tissue-resident memory CD8 T cells (TRM).118 Further exploring the phenotype of TRM provided insight into their tissue-resident mechanism: constitutive expression of CD69 and CD103, which interfere with S1PR1 function and tether TRM to E-cadherin expressed on epithelial cells, respectively.116,119 Much work in the memory CD8 T-cell field is now focused on elucidating the importance of TRM. The numerical appreciation of TRM was determined by enumerating P14 memory CD8 T cells generated after LCMV infection and greater than 90% of memory P14 within non-lymphoid tissue were TRM.120 In response to infection, TRM activation promotes pathogen clearance of the tissues by orchestrating innate and adaptive immune systems rapidly to provide robust protection against an array of viral and parasitic infections.121–128 As a result of TRM robust protective capacity, research effort is now focused on modifying vaccine formulations to generate site-specific TRM.129
As discussed in this review, TCIRM are susceptible to sepsis-induced apoptosis; however, it is unclear to what extent TRMs are similarly affected. We hypothesize that the distinct position of TRM within tissue parenchyma could confer some protection against the deleterious effects of sepsis. Differential survival of TCIRM and skin TRM has been observed in cutaneous T-cell lymphoma patients receiving alemtuzumab (αCD52) to deplete T cells.130,131 Further appreciation of this concept is that transplantation recipients on immunosuppressive drugs can have protracted disease-free periods that are perhaps due to the persistence of TRM.132,133 The distinction between circulation and parenchyma is seen in the experimental technique that distinguishes TCIRM from TRM. Intravenous injection of a fluorescently conjugated mAb labels TCIRM but leave TRM unlabeled due to their position outside of the circulation.134 The impact of sepsis on vascular endothelial cells, providing the biological barrier between circulation and parenchyma, will be pivotal in these pursuits. It has been speculated that severe sepsis has negative impacts on endothelial cell integrity, specifically, lipopolysaccharides and extracellular histones can induce endothelial cells apoptosis.135–139 In addition to conventional CD4 and CD8 TRM, γδ T cells and invariant natural killer tissue-resident T-cell subsets have been characterized.140 The impact of sepsis on the number, function, and protective capacity of TRM remains to be determined, as well as how the severity of sepsis dictates the susceptibility of this population. Due to the importance of TRM in providing rapid protection during localized infections and the chronic immunosuppression seen in septic patients, the impact of sepsis on TRM will be critical to further understanding sepsis-induced dysfunction of T-cell-mediated immunity.
B. Contribution of T-Cell-Extrinsic Factors Leading to Deficits in T-Cell-Mediated Immunity
As discussed previously in this review, septic survivors enter a state of chronic immunosuppression associated with increased susceptibility to secondary infections normally controlled by T-cell-mediated immunity.15–19 This immune suppression has been recapitulated in mouse models of sepsis, with septic mice showing similar immune suppression and increased susceptibility to secondary infection.74,104 Our recent work has demonstrated that septic mice have a prolonged impairment in the ability to mount primary CD8 T-cell responses (resulting from infection/vaccination) upon Ag encounter; however, the mechanism of this deficit (CD8 T-cell-intrinsic and/or -extrinsic) is not well defined.69,73,74 Primary CD8 T-cell expansion upon newly encountered infection or vaccination is reliant on CD8 T-cell-extrinsic factors (Fig. 4). These extrinsic factors, which can be provided by APCs such as dendritic cells (DCs), include the presence of Ag:MHC complex (signal 1), co-stimulatory ligands (signal 2), and appropriate cytokines such as IL-12 (signal 3).23–25,141–143 A sepsis-induced lesion in the quantity (numbers) or quality (ability to provide signals 1–3) of DCs could be an important extrinsic factor contributing to suboptimal CD8 T-cell immunity after sepsis.
FIG. 4.

T-cell-extrinsic factors mediate effective CD8 T-cell responses. The expansion of Ag-specific CD8 T cells is dependent on APCs (DCs) providing Ag:MHC (signal 1), co-stimulation (signal 2), and signal 3 cytokines (e.g. IL-12). Lack of signal 3 cytokine will lead to suboptimal CD8 T-cell expansion, reducing the effectiveness of the T-cell response. Sepsis-induced impairments of DCs’ ability to provide signals 1–3 could be an important mechanism behind the observed deficits in CD8 T-cell immunity after a septic event. (Figure adapted with permission from Haring et al., 2006.23).
Investigations into the effect of sepsis on DCs have shown that splenic DCs decline in number after a septic event in both septic patients and experimental models of sepsis.144–147 Furthermore, previous work has demonstrated that post-septic DCs produce less IL-12 and more IL-10 upon TLR stimulation than sham controls.146–148 These observed lesions in DC number and IL-12 (signal 3) production provide support for a possible T-cell-extrinsic lesion; however, further queries into the effects of sepsis on DCs are warranted. Specifically, the status of DCs in the context of impaired CD8 T-cell immunity after sepsis has not been elucidated. Future investigations will examine the manner in which DC lesions may impair CD8 T-cell responses to pathogens and probe whether recovery in the quantity and quality of DCs may help to ameliorate impairments in CD8 T-cell immunity. Moreover, there has been little investigation into the potential therapeutic benefit of DC-mobilizing cytokines such as FMS-like tyrosine kinase 3 ligand (Flt3L)149 after a septic event with the idea of restoring T-cell immunity through boosting DC numbers. The key point here is that only targeting the “T-cell side” of the equation by bolstering numbers with cytokines (e.g., IL-7) or improving function with checkpoint inhibitors may not be sufficient to fully restore T-cell immunity after a septic event if the APCs, which are required for T-cell activation, are also reduced in number/function during sepsis.
VII. CONCLUSIONS
Septic patients that survive the cytokine storm experience long-term pathological consequences, including chronic immunosuppression. Therefore, further understanding of the immunological modifications associated with sepsis-induced chronic immunosuppression is an important research goal. Clinical studies of sepsis revealed that CD4 and CD8 T cells undergo apoptosis, leading to appreciable numerical decline. Experimental models of sepsis, such as CLP, recapitulated these clinical observations. In addition, sepsis experimental models have facilitated researchers to ask hypothesis-driven questions to further probe the impact of sepsis on T-cell-mediated immunity. As discussed in this review, surviving T cells exposed to the septic environment are affected functionally in a number of ways. Sepsis-induced impairments in the quantity and quality of T cells give insight into sepsis-induced impairments of T-cell immunity. Both clinical and experimental approaches have been critical in understanding changes in T-cell-mediated immunity that help to explain septic patients’ susceptibility to infections normally controlled by T cells. However, many experimental questions still exist regarding the impact of sepsis on T-cell-mediated immunity. For example, CD4 and CD8 TRM are critical mediators of pathogen clearance but, to our knowledge, little data have been published determining the impact of sepsis on CD8 TRM. In addition, CD8 T-cell-extrinsic factors, such as DC participation in providing the appropriate signals for proper T-cell activation and function, require additional examination to further understand the changes in CD8 T-cell-mediated immunity during the immunosuppression phase of sepsis.
Acknowledgments
This work was supported by the U.S. Department of Veterans Affairs (Merit Review Award to T.S.G.) and by the National Institutes of Health (Grants GM113961, AI119160, and AI114543 to V.P.B.).
ABBREVIATIONS
- Ag
antigen
- APC
antigen-presenting cell
- CD
cluster of differentiation
- CLP
cecal ligation and puncture
- DC
dendritic cell
- IFN
interferon
- IL
interleukin
- TCR-tg
T-cell receptor-transgenic
- TNF
tumor necrosis factor
References
- 1.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K International Forum of Acute Care Trialists. Assessment of global incidence and mortality of hospital-treated sepsis: current estimates and limitations. Am J Respir Crit Care Med. 2016;193:259–72. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 2.Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med. 2012;40:754–61. doi: 10.1097/CCM.0b013e318232db65. [DOI] [PubMed] [Google Scholar]
- 3.Donnelly JP, Hohmann SF, Wang HE. Unplanned readmissions after hospitalization for severe sepsis at academic medical center-affiliated hospitals. Crit Care Med. 2015;43:1916–27. doi: 10.1097/CCM.0000000000001147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar G, Kumar N, Taneja A, Kaleekal T, Tarima S, McGinley E, Jimenez E, Mohan A, Khan RA, Whittle J, Jacobs E, Nanchal R Milwaukee Initiative in Critical Care Outcomes Research Group of Investigators. Nationwide trends of severe sepsis in the 21st century (2000–2007) Chest. 2011;140:1223–31. doi: 10.1378/chest.11-0352. [DOI] [PubMed] [Google Scholar]
- 5.Leligdowicz A, Dodek PM, Norena M, Wong H, Kumar A, Kumar A Co-operative Antimicrobial Therapy of Septic Shock Database Research G. Association between source of infection and hospital mortality in patients who have septic shock. Am J Respir Crit Care Med. 2014;189:1204–13. doi: 10.1164/rccm.201310-1875OC. [DOI] [PubMed] [Google Scholar]
- 6.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD, et al. PROWESS-SHOCK Study Group. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–64. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 7.Opal SM, Garber GE, LaRosa SP, Maki DG, Freebairn RC, Kinasewitz GT, Dhainaut JF, Yan SB, Williams MD, Graham DE, Nelson DR, Levy H, Bernard GR. Systemic host responses in severe sepsis analyzed by causative microorganism and treatment effects of drotrecogin alfa (activated) Clin Infect Dis. 2003;37:50–8. doi: 10.1086/375593. [DOI] [PubMed] [Google Scholar]
- 8.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 9.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K EPIC II Group of Investigators. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323–9. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 10.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–74. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest. 1993;103:565–75. doi: 10.1378/chest.103.2.565. [DOI] [PubMed] [Google Scholar]
- 12.Fisher CJ, Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome: results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–43. [PubMed] [Google Scholar]
- 13.Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112(6 Suppl):321–9S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- 14.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–8. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 15.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306:2594–605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Vught LA, Klein Klouwenberg PM, Spitoni C, Scicluna BP, Wiewel MA, Horn J, Schultz MJ, Nurnberg P, Bonten MJ, Cremer OL, van der Poll T MARS Consortium. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA. 2016;315:1469–79. doi: 10.1001/jama.2016.2691. [DOI] [PubMed] [Google Scholar]
- 17.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15(5):496–7. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med. 2014;20(4):224–33. doi: 10.1016/j.molmed.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adib-Conquy M, Cavaillon JM. Compensatory anti-inflammatory response syndrome. Thromb Haemost. 2009;101(1):36–47. [PubMed] [Google Scholar]
- 20.Limaye AP, Kirby KA, Rubenfeld GD, Leisenring WM, Bulger EM, Neff MJ, Gibran NS, Huang ML, Santo Hayes TK, Corey L, Boeckh M. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA. 2008;300(4):413–22. doi: 10.1001/jama.300.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luyt CE, Combes A, Deback C, Aubriot-Lorton MH, Nieszkowska A, Trouillet JL, Capron F, Agut H, Gibert C, Chastre J. Herpes simplex virus lung infection in patients undergoing prolonged mechanical ventilation. Am J Respir Crit Care Med. 2007;175(9):935–42. doi: 10.1164/rccm.200609-1322OC. [DOI] [PubMed] [Google Scholar]
- 22.Quartin AA, Schein RM, Kett DH, Peduzzi PN. Magnitude and duration of the effect of sepsis on survival: Department of Veterans Affairs Systemic Sepsis Cooperative Studies Group. JAMA. 1997;277(13):1058–63. [PubMed] [Google Scholar]
- 23.Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T-cell response. Immunity. 2006;25(1):19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 25.Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8(2):107–19. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- 26.Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31(6):859–71. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2(4):251–62. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 28.Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T-cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28(6):859–69. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 30.Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. 2002;3(7):619–26. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt NW, Podyminogin RL, Butler NS, Badovinac VP, Tucker BJ, Bahjat KS, Lauer P, Reyes-Sandoval A, Hutchings CL, Moore AC, Gilbert SC, Hill AV, Bartholomay LC, Harty JT. Memory CD8 T-cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc Natl Acad Sci U S A. 2008;105(37):14017–22. doi: 10.1073/pnas.0805452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9(9):1131–7. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- 33.Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7(8):913–9. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 34.Pope C, Kim SK, Marzo A, Masopust D, Williams K, Jiang J, Shen H, Lefrancois L. Organ-specific regulation of the CD8 T-cell response to Listeria monocytogenes infection. J Immunol. 2001;166(5):3402–9. doi: 10.4049/jimmunol.166.5.3402. [DOI] [PubMed] [Google Scholar]
- 35.Badovinac VP, Harty JT. Programming, demarcating, and manipulating CD8+ T-cell memory. Immunol Rev. 2006;211:67–80. doi: 10.1111/j.0105-2896.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 36.Martin MD, Kim MT, Shan Q, Sompallae R, Xue HH, Harty JT, Badovinac VP. Phenotypic and functional alterations in circulating memory CD8 T cells with time after primary infection. PLoS Pathog. 2015;11(10):e1005219. doi: 10.1371/journal.ppat.1005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136(7):2348–57. [PubMed] [Google Scholar]
- 38.Killar L, MacDonald G, West J, Woods A, Bottomly K. Cloned, Ia-restricted T cells that do not produce interleukin 4(IL 4)/B cell stimulatory factor 1(BSF-1) fail to help antigen-specific B cells. J Immunol. 1987;138(6):1674–9. [PubMed] [Google Scholar]
- 39.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 41.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T-cell populations (*) Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green AM, Difazio R, Flynn JL. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol. 2013;190(1):270–7. doi: 10.4049/jimmunol.1200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phares TW, Stohlman SA, Hinton DR, Bergmann CC. Enhanced CD8 T-cell anti-viral function and clinical disease in B7-H1-deficient mice requires CD4 T cells during encephalomyelitis. J Neuroinflammation. 2012;9:269. doi: 10.1186/1742-2094-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434(7029):88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 47.Badovinac VP, Messingham KA, Griffith TS, Harty JT. TRAIL deficiency delays, but does not prevent, erosion in the quality of “helpless” memory CD8 T cells. J Immunol. 2006;177(2):999–1006. doi: 10.4049/jimmunol.177.2.999. [DOI] [PubMed] [Google Scholar]
- 48.Sacks JA, Bevan MJ. TRAIL deficiency does not rescue impaired CD8+ T cell memory generated in the absence of CD4+ T cell help. J Immunol. 2008;180(7):4570–6. doi: 10.4049/jimmunol.180.7.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol. 2014;44(1):69–79. doi: 10.1002/eji.201343718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weinstein JS, Hernandez SG, Craft J. T cells that promote B-cell maturation in systemic autoimmunity. Immunol Rev. 2012;247(1):160–71. doi: 10.1111/j.1600-065X.2012.01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yates JL, Racine R, McBride KM, Winslow GM. T cell-dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J Immunol. 2013;191(3):1240–9. doi: 10.4049/jimmunol.1300062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen JM, Khandavilli S, Camberlein E, Hyams C, Baxendale HE, Brown JS. Protective contributions against invasive Streptococcus pneumoniae pneumonia of antibody and Th17-cell responses to nasopharyngeal colonisation. PLoS One. 2011;6(10):e25558. doi: 10.1371/journal.pone.0025558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev. 2013;252(1):89–103. doi: 10.1111/imr.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*) Annu Rev Immunol. 2009;27:551–89. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 55.Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(Suppl):S97–107. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 56.Parrino J, Hotchkiss RS, Bray M. Prevention of immune cell apoptosis as potential therapeutic strategy for severe infections. Emerg Infect Dis. 2007;13(2):191–8. doi: 10.3201/eid1302.060963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27(7):1230–51. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, Karl IE. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001;166(11):6952–63. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 59.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6(11):813–22. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 60.Felmet KA, Hall MW, Clark RS, Jaffe R, Carcillo JA. Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and multiple organ failure. J Immunol. 2005;174(6):3765–72. doi: 10.4049/jimmunol.174.6.3765. [DOI] [PubMed] [Google Scholar]
- 61.Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, Tattevin P, Thomas R, Fauchet R, Drenou B. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock. 2002;18(6):487–94. doi: 10.1097/00024382-200212000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol. 2005;174(8):5110–8. doi: 10.4049/jimmunol.174.8.5110. [DOI] [PubMed] [Google Scholar]
- 63.Weber SU, Schewe JC, Lehmann LE, Muller S, Book M, Klaschik S, Hoeft A, Stuber F. Induction of Bim and Bid gene expression during accelerated apoptosis in severe sepsis. Crit Care. 2008;12(5):R128. doi: 10.1186/cc7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19(4):198–208. doi: 10.1016/j.tim.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 66.Smith EF, 3rd, Slivjak MJ, Egan JW, Gagnon R, Arleth AJ, Esser KM. Fluid resuscitation improves survival of endotoxemic or septicemic rats: possible contribution of tumor necrosis factor. Pharmacology. 1993;46(5):254–67. doi: 10.1159/000139053. [DOI] [PubMed] [Google Scholar]
- 67.Baker CC, Chaudry IH, Gaines HO, Baue AE. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery. 1983;94(2):331–5. [PubMed] [Google Scholar]
- 68.Efron PA, Mohr AM, Moore FA, Moldawer LL. The future of murine sepsis and trauma research models. J Leukoc Biol. 2015;98(6):945–52. doi: 10.1189/jlb.5MR0315-127R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Condotta SA, Rai D, James BR, Griffith TS, Badovinac VP. Sustained and incomplete recovery of naive CD8+ T-cell precursors after sepsis contributes to impaired CD8+ T-cell responses to infection. J Immunol. 2013;190(5):1991–2000. doi: 10.4049/jimmunol.1202379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Schaik SM, Abbas AK. Role of T cells in a murine model of Escherichia coli sepsis. Eur J Immunol. 2007;37(11):3101–10. doi: 10.1002/eji.200737295. [DOI] [PubMed] [Google Scholar]
- 71.Sherwood ER, Enoh VT, Murphey ED, Lin CY. Mice depleted of CD8+ T and NK cells are resistant to injury caused by cecal ligation and puncture. Lab Invest. 2004;84(12):1655–65. doi: 10.1038/labinvest.3700184. [DOI] [PubMed] [Google Scholar]
- 72.Wesche-Soldato DE, Chung CS, Gregory SH, Salazar-Mather TP, Ayala CA, Ayala A. CD8+ T cells promote inflammation and apoptosis in the liver after sepsis: role of Fas-FasL. Am J Pathol. 2007;171(1):87–96. doi: 10.2353/ajpath.2007.061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duong S, Condotta SA, Rai D, Martin MD, Griffith TS, Badovinac VP. Polymicrobial sepsis alters antigen-dependent and -independent memory CD8 T cell functions. J Immunol. 2014;192(8):3618–25. doi: 10.4049/jimmunol.1303460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Condotta SA, Khan SH, Rai D, Griffith TS, Badovinac VP. Polymicrobial sepsis increases susceptibility to chronic viral infection and exacerbates CD8+ T-cell exhaustion. J Immunol. 2015;195(1):116–25. doi: 10.4049/jimmunol.1402473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doi K, Hu X, Yuen PS, Leelahavanichkul A, Yasuda H, Kim SM, Schnermann J, Jonassen TE, Frokiaer J, Nielsen S, Star RA. AP214, an analogue of alpha-melanocyte-stimulating hormone, ameliorates sepsis-induced acute kidney injury and mortality. Kidney Int. 2008;73(11):1266–74. doi: 10.1038/ki.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iannelli F, Chiavolini D, Ricci S, Oggioni MR, Pozzi G. Pneumococcal surface protein C contributes to sepsis caused by Streptococcus pneumoniae in mice. Infect Immun. 2004;72(5):3077–80. doi: 10.1128/IAI.72.5.3077-3080.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Belikoff BG, Hatfield S, Georgiev P, Ohta A, Lukashev D, Buras JA, Remick DG, Sitkovsky M. A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J Immunol. 2011;186(4):2444–53. doi: 10.4049/jimmunol.1001567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 79.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, Masopust D. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532(7600):512–6. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seok J1, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–12. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2015;112(4):1167–72. doi: 10.1073/pnas.1401965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. 2000;164(11):5782–7. doi: 10.4049/jimmunol.164.11.5782. [DOI] [PubMed] [Google Scholar]
- 83.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195(5):657–64. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27(2):203–13. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Badovinac VP, Porter BB, Harty JT. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol. 2004;5(8):809–17. doi: 10.1038/ni1098. [DOI] [PubMed] [Google Scholar]
- 86.Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T-cell response to infection. Immunity. 2007;26(6):827–41. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang SD, Huang KJ, Lin YS, Lei HY. Sepsis-induced apoptosis of the thymocytes in mice. J Immunol. 1994;152(10):5014–21. [PubMed] [Google Scholar]
- 88.Barke RA, Roy S, Chapin RB, Charboneau R. The role of programmed cell death (apoptosis) in thymic involution following sepsis. Arch Surg. 1994;129(12):1256–61. doi: 10.1001/archsurg.1994.01420360046005. discussion 61–2. [DOI] [PubMed] [Google Scholar]
- 89.Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood. 1996;87(10):4261–75. [PubMed] [Google Scholar]
- 90.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25(8):1298–307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 91.Unsinger J, McGlynn M, Kasten KR, Hoekzema AS, Watanabe E, Muenzer JT, McDonough JS, Tschoep J, Ferguson TA, McDunn JE, Morre M, Hildeman DA, Caldwell CC, Hotchkiss RS. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J Immunol. 2010;184(7):3768–79. doi: 10.4049/jimmunol.0903151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carson WFt, Cavassani KA, Ito T, Schaller M, Ishii M, Dou Y, Kunkel SL. Impaired CD4+ T-cell proliferation and effector function correlates with repressive histone methylation events in a mouse model of severe sepsis. Eur J Immunol. 2010;40(4):998–1010. doi: 10.1002/eji.200939739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cabrera-Perez J, Condotta SA, James BR, Kashem SW, Brincks EL, Rai D, Kucaba TA, Badovinac VP, Griffith TS. Alterations in antigen-specific naive CD4 T-cell precursors after sepsis impairs their responsiveness to pathogen challenge. J Immunol. 2015;194(4):1609–20. doi: 10.4049/jimmunol.1401711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96(25):14541–6. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, Zollner KM, Buchman TG, Korsmeyer SJ, Karl IE. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol. 1999;162(7):4148–56. [PubMed] [Google Scholar]
- 96.Condotta SA, Cabrera-Perez J, Badovinac VP, Griffith TS. T-cell-mediated immunity and the role of TRAIL in sepsis-induced immunosuppression. Crit Rev Immunol. 2013;33(1):23–40. doi: 10.1615/critrevimmunol.2013006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, Osborne DF, Clark AT, Coopersmith CM, McDunn JE, Hotchkiss RS. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. 2010;184(3):1401–9. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, Robbins P, Ulbrandt N, Suzich J, Green J, Patera AC, Blair W, Krishnan S, Hotchkiss R. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T-cell exhaustion in patients with sepsis. Crit Care. 2014;18(1):R3. doi: 10.1186/cc13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Unsinger J, Kazama H, McDonough JS, Hotchkiss RS, Ferguson TA. Differential lymphopenia-induced homeostatic proliferation for CD4+ and CD8+ T cells following septic injury. J Leukoc Biol. 2009;85(3):382–90. doi: 10.1189/jlb.0808491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rai D, Pham NL, Harty JT, Badovinac VP. Tracking the total CD8 T-cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J Immunol. 2009;183(12):7672–81. doi: 10.4049/jimmunol.0902874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McDermott DS, Varga SM. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T-cell responses are larger than we think. J Immunol. 2011;187(11):5568–76. doi: 10.4049/jimmunol.1102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cabrera-Perez J, Condotta SA, Badovinac VP, Griffith TS. Impact of sepsis on CD4 T-cell immunity. J Leukoc Biol. 2014;96(5):767–77. doi: 10.1189/jlb.5MR0114-067R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stieglitz D, Schmid T, Chhabra NF, Echtenacher B, Mannel DN, Mostbock S. TNF and regulatory T cells are critical for sepsis-induced suppression of T cells. Immun Inflamm Dis. 2015;3(4):374–85. doi: 10.1002/iid3.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gurung P, Rai D, Condotta SA, Babcock JC, Badovinac VP, Griffith TS. Immune unresponsiveness to secondary heterologous bacterial infection after sepsis induction is TRAIL dependent. J Immunol. 2011;187(5):2148–54. doi: 10.4049/jimmunol.1101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol. 2010;184(12):6766–72. doi: 10.4049/jimmunol.0904054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meakins JL, Pietsch JB, Bubenick O, Kelly R, Rode H, Gordon J, MacLean LD. Delayed hypersensitivity: indicator of acquired failure of host defenses in sepsis and trauma. Ann Surg. 1977;186(3):241–50. doi: 10.1097/00000658-197709000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sharma A, Yang WL, Matsuo S, Wang P. Differential alterations of tissue T-cell subsets after sepsis. Immunol Lett. 2015;168(1):41–50. doi: 10.1016/j.imlet.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Serbanescu MA, Ramonell KM, Hadley A, Margoles LM, Mittal R, Lyons JD, Liang Z, Coopersmith CM, Ford ML, McConnell KW. Attrition of memory CD8 T cells during sepsis requires LFA-1. J Leukoc Biol. doi: 10.1189/jlb.4A1215-563RR. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Unsinger J, Burnham CA, McDonough J, Morre M, Prakash PS, Caldwell CC, Dunne WM, Jr, Hotchkiss RS. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J Infect Dis. 2012;206(4):606–16. doi: 10.1093/infdis/jis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T cells to innate immunity: IFN-gamma secretion induced by IL-12 and IL-18. Eur J Immunol. 2002;32(10):2807–16. doi: 10.1002/1521-4141(2002010)32:10<2807::AID-IMMU2807>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 111.Kambayashi T, Assarsson E, Lukacher AE, Ljunggren HG, Jensen PE. Memory CD8+ T cells provide an early source of IFN-gamma. J Immunol. 2003;170(5):2399–408. doi: 10.4049/jimmunol.170.5.2399. [DOI] [PubMed] [Google Scholar]
- 112.Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med. 2003;198(10):1583–93. doi: 10.1084/jem.20031051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander activation of CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial pathogens. J Immunol. 2001;166(2):1097–105. doi: 10.4049/jimmunol.166.2.1097. [DOI] [PubMed] [Google Scholar]
- 114.Raue HP, Brien JD, Hammarlund E, Slifka MK. Activation of virus-specific CD8+ T cells by lipopolysaccharide-induced IL-12 and IL-18. J Immunol. 2004;173(11):6873–81. doi: 10.4049/jimmunol.173.11.6873. [DOI] [PubMed] [Google Scholar]
- 115.Martin MD, Badovinac VP. Antigen-dependent and -independent contributions to primary memory CD8 T cell activation and protection following infection. Sci Rep. 2015;5:18022. doi: 10.1038/srep18022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41(6):886–97. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291(5512):2413–7. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 118.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207(3):553–64. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, Carbone FR, Gebhardt T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. 2015;194(5):2059–63. doi: 10.4049/jimmunol.1402256. [DOI] [PubMed] [Google Scholar]
- 120.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, Masopust D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell. 2015;161(4):737–49. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 2012;483(7388):227–31. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346(6205):98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J Immunol. 2015;195(1):203–9. doi: 10.4049/jimmunol.1402975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, Scott P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med. 2015;212(9):1405–14. doi: 10.1084/jem.20142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10(5):524–30. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 126.Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, Cauley LS. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. 2014;95(2):215–24. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, Ritsma L, van Rheenen J, Maree AF, Zal T, de Boer RJ, Haanen JB, Schumacher TN. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci U S A. 2012;109(48):19739–44. doi: 10.1073/pnas.1208927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Schumacher TN. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346(6205):101–5. doi: 10.1126/science.1254803. [DOI] [PubMed] [Google Scholar]
- 129.Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol Rev. 2013;255(1):165–81. doi: 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, Dorosario AA, Chaney KS, Cutler CS, Leboeuf NR, Carter JB, Fisher DC, Kupper TS. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4(117):117ra7. doi: 10.1126/scitranslmed.3003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. 2015;7(279):279ra39. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thome JJ, Farber DL. Emerging concepts in tissue-resident T cells: lessons from humans. Trends Immunol. 2015;36(7):428–35. doi: 10.1016/j.it.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Turner DL, Gordon CL, Farber DL. Tissue-resident T cells, in situ immunity and transplantation. Immunol Rev. 2014;258(1):150–66. doi: 10.1111/imr.12149. [DOI] [PubMed] [Google Scholar]
- 134.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9(1):209–22. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 136.Ait-Oufella H, Maury E, Lehoux S, Guidet B, Offenstadt G. The endothelium: physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010;36(8):1286–98. doi: 10.1007/s00134-010-1893-6. [DOI] [PubMed] [Google Scholar]
- 137.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15(11):1318–21. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Munshi N, Fernandis AZ, Cherla RP, Park IW, Ganju RK. Lipopolysaccharide-induced apoptosis of endothelial cells and its inhibition by vascular endothelial growth factor. J Immunol. 2002;168(11):5860–6. doi: 10.4049/jimmunol.168.11.5860. [DOI] [PubMed] [Google Scholar]
- 139.Hotchkiss RS, Tinsley KW, Swanson PE, Karl IE. Endothelial cell apoptosis in sepsis. Crit Care Med. 2002;30(5 Suppl):S225–8. doi: 10.1097/00003246-200205001-00009. [DOI] [PubMed] [Google Scholar]
- 140.Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell. 2016;164(6):1198–211. doi: 10.1016/j.cell.2016.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lanzavecchia A, Sallusto F. Antigen decoding by T lymphocytes: from synapses to fate determination. Nat Immunol. 2001;2(6):487–92. doi: 10.1038/88678. [DOI] [PubMed] [Google Scholar]
- 142.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162(6):3256–62. [PubMed] [Google Scholar]
- 143.Schmidt CS, Mescher MF. Adjuvant effect of IL-12: conversion of peptide antigen administration from tolerizing to immunizing for CD8+ T cells in vivo. J Immunol. 1999;163(5):2561–7. [PubMed] [Google Scholar]
- 144.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, Cobb JP, Coopersmith C, Karl IE. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol. 2002;168(5):2493–500. doi: 10.4049/jimmunol.168.5.2493. [DOI] [PubMed] [Google Scholar]
- 145.Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, Hotchkiss RS. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. 2003;171(2):909–14. doi: 10.4049/jimmunol.171.2.909. [DOI] [PubMed] [Google Scholar]
- 146.Flohe SB, Agrawal H, Schmitz D, Gertz M, Flohe S, Schade FU. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol. 2006;79(3):473–81. doi: 10.1189/jlb.0705413. [DOI] [PubMed] [Google Scholar]
- 147.Pastille E, Didovic S, Brauckmann D, Rani M, Agrawal H, Schade FU, Zhang Y, Flohe SB. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J Immunol. 2011;186(2):977–86. doi: 10.4049/jimmunol.1001147. [DOI] [PubMed] [Google Scholar]
- 148.Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood. 2008;111(4):1797–804. doi: 10.1182/blood-2007-08-106443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, McKenna HJ. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184(5):1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]