

Abstract
Background
Toll-like receptors (TLRs) are the frontline actors in the innate immune response to various pathogens and are expected to be targets of natural selection in species adapted to habitats with contrasting pathogen burdens. The recent publication of genome sequences of giraffe and okapi together afforded the opportunity to examine the evolution of selected TLRs in broad range of terrestrial ungulates and cetaceans during their complex habitat diversification. Through direct sequence comparisons and standard evolutionary approaches, the extent of nucleotide and protein sequence diversity in seven Toll-like receptors (TLR2, TLR3, TLR4, TLR5, TLR7, TLR9 and TLR10) between giraffe and closely related species was determined. In addition, comparison of the patterning of key TLR motifs and domains between giraffe and related species was performed. The quantification of selection pressure and divergence on TLRs among terrestrial ungulates and cetaceans was also performed.
Results
Sequence analysis shows that giraffe has 94–99% nucleotide identity with okapi and cattle for all TLRs analyzed. Variations in the number of Leucine-rich repeats were observed in some of TLRs between giraffe, okapi and cattle. Patterning of key TLR domains did not reveal any significant differences in the domain architecture among giraffe, okapi and cattle. Molecular evolutionary analysis for selection pressure identifies positive selection on key sites for all TLRs examined suggesting that pervasive evolutionary pressure has taken place during the evolution of terrestrial ungulates and cetaceans. Analysis of positively selected sites showed some site to be part of Leucine-rich motifs suggesting functional relevance in species-specific recognition of pathogen associated molecular patterns. Notably, clade analysis reveals significant selection divergence between terrestrial ungulates and cetaceans in viral sensing TLR3. Mapping of giraffe TLR3 key substitutions to the structure of the receptor indicates that at least one of giraffe altered sites coincides with TLR3 residue known to play a critical role in receptor signaling activity.
Conclusion
There is overall structural conservation in TLRs among giraffe, okapi and cattle indicating that the mechanism for innate immune response utilizing TLR pathways may not have changed very much during the evolution of these species. However, a broader phylogenetic analysis revealed signatures of adaptive evolution among terrestrial ungulates and cetaceans, including the observed selection divergence in TLR3. This suggests that long term ecological dynamics has led to species-specific innovation and functional variation in the mechanisms mediating innate immunity in terrestrial ungulates and cetaceans.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-017-0901-7) contains supplementary material, which is available to authorized users.
Keywords: Toll-like receptors; Giraffe; Terrestrial ungulates; Cetaceans; Adaptive evolution, Functional variation
Background
Mammalian Toll-like receptors (TLRs) are membrane-bound proteins expressed in defense cells where they have evolved to mediate innate immune system through recognition of various pathogen-associated molecular patterns (PAMPs) [1, 2]. Functional classification of TLRs depends on the cellular location and the ligands they bind. For example, TLR2 is located on the outer membrane and forms dimer complex with TLR1 or TLR6 to recognize peptidoglycans, lipoproteins or lipoteichoic acid of gram positive bacteria [3, 4]. Other outer membrane TLRs include TLR4 which dimerize to recognize lipopolysaccharides (LPS) of gram negative bacteria [5], TLR5 which recognizes flagellins [6] and TLR10 which has recently been shown to have anti-inflammatory effects, and perhaps in combination with TLR2 may be associated with mycobacterial infections [7, 8]. Endosome confined TLRs includes TLR3, TLR7, TLR8 and TLR9. TLR3 recognizes double-stranded RNA (dsRNA), TLR7 and TLR8 are activated upon contact with single-stranded RNA (ssRNA) while TLR9 recognizes CpG DNA from virus, fungi and other invading pathogens [9–11].
TLRs share the same basic architecture comprising of a large extracellular domain (ECD) responsible for PAMP binding, a single-pass trans-membrane (TM) domain believed to play a role in membrane receptor stabilization and receptor-receptor oligomerization [12] and an intracellular Toll/interleukin-1 receptor (TIR) domain responsible for intracellular signal transduction and orchestration of cellular responses [1, 13]. The extracellular portion contains Leucine-rich repeat (LRR) motifs, which are arrays of 20–30 amino acid-long protein sequences enriched with the hydrophobic amino acid Leucine. Activation of TLRs by ligands initiates a cascade of events that leads the TIR domain to engage TIR domain-containing adaptor proteins. Adaptors proteins such as myeloid differentiation primary-response protein 88 (MYD88) or TIR domain-containing adaptor protein inducing IFNβ (TRIF) play a role in linking TLRs to nuclear transcription factors [2]. Such transcription factors include activator protein 1 (AP-1), Interferon regulatory factors (IRFs) or Nuclear Factor kappaB (NF-kB) which induce production of pro-inflammatory factors to mediate immune responses.
The evolution of TLRs is believed to have ancient and complex history that can be traced back to basal metazoans like sponges, Hydra (Hydra magnipapillata) and sea anemone (Nematostella vectensis) [14, 15]. Episodes of gene duplication, gene loss and gene conversion appear to have produced different TLR repertoire and functional diversification in various vertebrate species [16, 17]. The vertebrate ancestor at least possessed fifteen TLR members: TLR1-5, 7–9, 11–14, 19, 21, and 22 [18, 19]. Comprehensive phylogenetic studies of vertebrate TLRs reveal complete absence of TLR21 and TL22 and rarity of TLR12-14 and TLR 19 in land-dwelling vertebrates suggesting that pervasive TLR gene loss has taken place during transition from water to land [18, 20]. Even among mammals, TLRs are present in varying numbers; for example, primates and ungulates have ten TLRs [21–23] while some rodents (e.g. Mus musculus) possess 12 TLRs [14]. Novel TLRs combination in different animal groups reflects the need to acquire elaborate and efficient system to recognize and respond to diverse pathogens presented by different environments.
Conventionally, genes involved in immunity should exhibit an accelerated evolutionary rate indicative of adaptive struggle between host and the invading pathogens. However, various studies have found TLRs to be evolutionarily conserved within and between lineages [24–26]. Nevertheless, characterizing TLRs according to their domains provides finer resolution on the role of natural selection on TLRs. Several studies have shown purifying selection dominating in TLR regions responsible for oligomerization while considerable degree of variation was observed in TLR regions responsible for PAMP binding [27]. Other studies taking individual species, specific taxa and location of receptor into account have found evidence of adaptive substitutions in bovine TLR2 and TLR5 [28, 29] and TLR4 in primates [30, 31].
In a recently published work in our group, we identified genes associated with innate immunity to have been overrepresented among positively selected genes in the giraffe lineage when compared to okapi and cattle [32]. Giraffes are generally susceptible to viral and bacterial infections such as rinderpest [33], anthrax [34] and tuberculosis [35], that also affect all wild and livestock ruminants. Repeated exposure to infectious agents may result in extinction or adaptation of species [36], signs of the latter expected to be detected on genes especially those mediating defense against infections. Furthermore, members of the family Giraffidae have diverged over extended evolutionary periods in contrasting environments: giraffes occupy the trypanosome infested savannah while okapi is restricted to serene Congo forests suggesting that differential adaptation in response to infectious agents may be expected between the two species.
In a broader context, giraffe and okapi are members of a diverse group of terrestrial ungulates with wide zoogeographic distribution and exhibiting remarkable diversity in size, diet and habitat. The diversity of ecological specializations in ungulates seems to be attributed to habitat changes during the so-called Eocene Climatic Optimum approximately 40 million years ago. The period was also accompanied by the emergence of different habitat niches creating possibilities for variety of body forms and dietary innovations [37]. This diversification phenomenon was even more pronounced in ruminants which have displayed an extraordinary variety of body sizes and diets ranging from very small (<20 kg) and diet generalists (e.g. dik dik) to very large (>700 kg) and diet specialists (e. g. giraffe). Occupation of varied ecological niches and dynamic dietary preferences presented challenges in finding symbiotic balance between the host immune system and endemic rumen microbial population [38, 39]. Long term, this immune system–microbiota relationship may allow for the species- and/or niche-specific adaptations in the development and maintenance of regulatory homeostasis in response to pathogenic invasion.
Closely related to terrestrial ungulates are cetaceans, including whales, dolphins, and porpoises, which, by contrast, are a group of secondarily adapted marine mammals with a history of terrestrial occupation before re-colonizing aquatic habitats [20, 40]. In addition to anatomical and physiological innovations required for life in water [41], cetaceans must have been confronted with even more formidable challenges from ever-changing water-borne pathogens.
We hypothesized that adaptive evolutionary pressure mediated by infectious agents due to ecological diversity has contributed to the evolution of TLR diversity in species with complex evolutionary history as exemplified by ungulates and cetaceans. To understand the extent of functional variation in the genes modulating innate immunity in this group, we have taken advantage of the availability of giraffe and okapi genomes to identify seven TLRs and examine adaptive sequence changes in comparison with other related species. The ultimate goal is to gain insight on the adaptive pressure on the innate immune system associated with the divergence of terrestrial ungulates and cetaceans.
Methods
Species and sequences
Giraffe and okapi TLR sequences (Additional file 1) were sequenced as part of the giraffe genome project [32]. The TLR sequences of cetaceans and other artiodactyls used in the analyses were retrieved from Reference Sequence (RefSeq) database of the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov) or Ensembl at the European Bioinformatics Institute (www.ensembl.org). For sequences obtained from NCBI, identification of putative TLR orthologs for the target species was achieved using BLAST against RNA RefSeq database. BioMart was used to extract orthologs for sequences obtained from Ensembl. For each TLR included in the study, giraffe, okapi and a subset of other species in Cetartiodactyla including at least one species from an outgroup taxon (horse, rhino or both), were used in the analysis. To qualify for inclusion in the analysis the sequences had to have complete coding length in all species considered. Thus, TLR1, TLR6 and TLR8 were not considered for analysis as they were either not successfully sequenced or were of partial sequences in giraffe and okapi. Moreover, to ensure reliability of protein coding quality for each of the TLR in the target species, their sequences should have had no any internal termination codon. For example, baiji dolphin (Lipotes vexillifer) TLR5 sequence was found to have internal stop codon and was removed from subsequent analysis as it was not known whether in this species the sequence is a pseudogene or a result of a sequencing error. The final list of species used for each TLR and their Ensembl identity or accession numbers, excluding giraffe and okapi, are presented in Additional file 2. Protein translation of TLRs coding sequences were aligned using MUSCLE [42], back-translated using RevTrans [43] and phylogenetic trees constructed using PhyML [44].
TLRs sequence and motif comparison
A web based simple modular architecture research tool (SMART) utilizes Hidden Markov models to query a collection of well annotated domain families associated with wide variety of nuclear, signaling and extracellular proteins [45]. The structural organization of TLR domains in the studied TLRs was analyzed using SMART. Web based LRRfinder is derived from a large database of unique, naturally occurring LRRs (tLRRdb) allowing the identification of not only highly conserved LRR sequences, but also those which uniquely deviate from the commonly described LxxLxLxxN/CxL consensus [46]. In this study, the LRRfinder was used to detect the number of LRRs present in the deduced amino acid sequences of giraffe, okapi and cattle TLRs. To identify whether there was significant difference in the number of giraffe LRRs and closely related ruminants, comparison was performed with the corresponding numbers of LRRs in okapi and cattle (Additional file 3: Table S1). In addition, comparison was performed on total number of nucleotide and amino acid sequence differences of each TLR gene among giraffe, okapi and cattle (Additional file 3: Table S1).
Site-based analyses of positive selection
Multiple alignments of TLR sequences and corresponding phylogenetic trees were used as inputs for codon-based analysis of positive selection. We applied site-based analyses which assume that all branches in a phylogeny are evolving at the same rate but certain sites may be under differing selection pressure i.e. the individual sites may be under purifying, neutral or positive selection [47]. The analyses were implemented using CODEML program of the Phylogenetic Analysis by Maximum Likelihood (PAML) package. Different model-based tests of selection exist in PAML which generally produce equivalent results although some tests are observed to be more conservative than others [48, 49]. To increase the likelihood of detection of positively selection, we used the less conservative M7/M8 test to examine the extent of selection acting on TLRs. M7 serves as null selection model by only allowing codons to evolve neutrally or under purifying selection following a beta distribution while the alternative M8 adds an extra class of sites under positive selection. The likelihood ratio test (LRT) was applied to determine significant cases of positive selection. Significant amino acids sites under positive selection were determined using Bayes Empirical Bayes (BEB) approach with posterior probability at 95% cut-off.
Simultaneously, we applied an alternative approach based on maximum likelihood to examine the extent of evolutionary pressure occurring at every codon in all TLRs using the “site-wise likelihood ratio” method as implemented in the SLR package [50]. The SLR test consists of performing a likelihood-ratio test on site-wise basis, testing the null model (neutrality, ω = 1) against an alternative model (ω ≠ 1). The method test whether a given site has undergone selection or not, and the test statistic summarizes the strength of the evidence for selection rather than the strength of the selection itself. The sites that were predicted to undergo positive selection using M8 model were cross-checked against the sites that were predicted as significant by the SLR method. Positively selected sites that were concordantly identified by the two methods as significant were assumed to be adaptively important. These sites were mapped to human TLRs Swiss-Prot entries to determine their functional relevance based on whether they map onto key TLR domains and motifs (Additional file 4: Table S2).
Clade models analyses of selection divergence
To identify whether divergent selection would be detected between terrestrial ungulates and cetaceans clades in their combined phylogeny, we applied PAML’s clade models. Clade Model C (CmC) partitions different branches within the phylogeny as “background” and “foreground” as well as existence of three site categories, two of which experience uniform selection across the entire phylogeny (either purifying selection (0 < ω0 < 1) or neutral evolution (ω1 = 1)) while the third is allowed to vary between background (ω2 > 0) and foreground (ω3 > 0) branches [51]. We used the recently developed null model of the CmC (M2a_rel) which does not allow the third site class to vary between two or more branch types, to test for the existence of divergent selection between terrestrial ungulates and cetacean clades [52]. In the case where significance was detected between CmC and M2a_rel, we proceeded to test for existence of positive selection between the two clades using the branch-site models [53] assuming, among other things, that the divergent site class has evolved by positive selection in the cetacean branch (ω3 > 1) while the background branches has been under the influence of purifying selection or neutral evolution.
Structural analysis
For the TLR which showed significant selection divergence between terrestrial ungulates and cetacean clades, we were interested to determine the functional significance of specific changes in the TLR during giraffe evolution. To this end, we obtained and reviewed the crystal structure of the TLR to identify which residues are critical in the ligand-receptor interaction. Moreover, we reviewed site-directed mutagenesis studies to identify sites predicted to have any TLR functional impacts. We also performed a PolyPhen screen [54] to identify sites that are predicted to be probably functionally consequential if a substitution has taken place in a giraffe TLR when compared to closely related species. Finally, we identified positively selected sites based on BEB prediction on this TLR. Following the identification of all the important sites, we referenced giraffe TLR substitutions against the identified important sites of the TLR structure for correspondence.
Results
TLRs sequence and motif analysis
We successfully retrieved complete coding sequence of seven TLRs (TLR2-5, TLR7 and TLR9-10) from giraffe and okapi genome sequences. The percent nucleotide and amino acid difference of the giraffe TLR coding sequences when compared with TLRs from okapi and cattle is shown in Additional file 3: Table S1. As expected, there was a small degree of nucleotide difference with okapi sequences (<2%) and 3–5% with cattle sequences, and when comparison takes into account amino acids differences, similar pattern is observed. The receptor with the highest degree of similarity among the three species was TLR7. According to SMART predictions, comparing the patterning of the ECD, TM and TIR domains of giraffe, okapi and cattle TLRs revealed no observable differences (Fig. 1). However, for some TLRs, there were variations in the predicted numbers of LRRs among the three species despite their highly conserved sequences (Additional file 4: Table S2). Giraffe is observed to have lower number of LRRs in TLR3 (21) compared to the usual number of TLR3 LRRs in mammals (23). Okapi is observed to have lower number of LRRs (19) in TLR5 compared to 21 observed in giraffe and cattle.
Fig. 1.
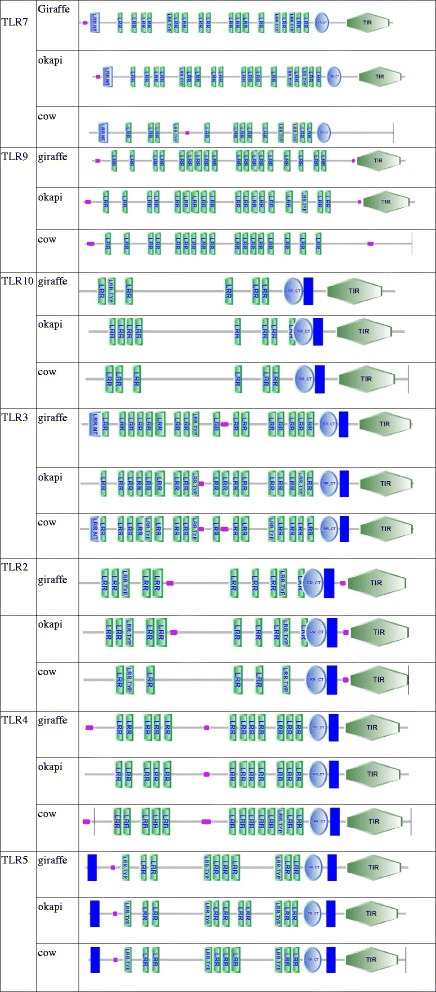
Comparison of domain architecture of TLRs in giraffe, okapi and cattle revealed no observable differences in spatial organization of major TLR domain areas (low complexity region (pink), LRRs and TIR)
Identification and distribution of selection pressure in the TLRs
The two Maximum Likelihood approaches detected evidence of positive selection in all of the TLRs studied. The results produced by M8 model indicated that the ω of all the TLR genes examined varied among codons with multiple significant codons under positive selection in five of the TLRs (Table 1). For all receptors, it was found that the proportion of sites with evidence of purifying selection (f0) is consistently larger than the proportion of sites with evidence of positive selection (f1). Thus, the majority of sites within the proteins were functionally constrained (Fig. 2). The number of positively selected codons observed for each TLR studied ranged between 23 and 113 which corresponded to 2.5–11% of the aligned codons. When significant positively selected sites based on BEB (P ≥ 95%) in specific TLRs are considered, TLR4 was the receptor with the highest proportion of codons under significant positive selection (13 sites), followed by TLR7 (11 sites) (Table 1). The TLR with the fewest number of positively selected sites appeared to be TLR2 and TLR10 with a single significant positive site in each of the TLRs.
Table 1.
Parameter estimates for PAML models across TLR genes
| Gene | Model | Parameter estimates | CODEML/SLR significant sites, giraffe residue, Bayes Empirical Bayes score |
|---|---|---|---|
| TLR5 | M7 (β) M8 (β & ω) |
p = 0.17, q = 0.26 p = 0.24, q = 0.45, f0 = 0.94 ω = 2.68, f1 = 0.06 |
31, L,96.1; 63,T, 97.4; 102,G, 96.3; 165,R, 97.1; 419,P, 97.36; 56, T,95.7 |
| LR4 | M7 (β) M8 (β & ω) |
p = 0.05, q = 0.04 p = 0.23, q = 0.38, f0 = 0.90 ω = 2.59, f1 = 0.10 |
293,D,99.3; 297,A, 99.9; 318,S, 97.9; 320,E, 98.0; 336,V, 96.1; 340,V, 99.4; 362,V, 99.3; 369,F, 97.6; 370,V, 97.5; 414,V, 99.1; 499,V, 99.2; 513,T, 95.2; 832,N, 97.1; |
| TLR2 | M7 (β) M8 (β & ω) |
p = 0.14, q = 0.24 p = 0.22, q = 0.43, f0 = 0.96 ω = 2.55, f1 = 0.04 |
765, M, 98.4 |
| TLR3 | M7 (β)M8 (β & ω) |
p = 0.19, q = 0.35 p = 0.28, q = 0.75, f0 = 0.98ω = 2.71, f1 = 0.02 |
4, H, 98.3; 277, V, 96.9; 382, F, 97.3 |
| TLR7 | M7 (β) M8 (β & ω) |
p = 0.03, q = 0.07 p = 0.15, q = 0.59, f0 = 0.95 ω = 2.23, f1 = 0.05 |
100,I,97.4; 161,L,96.6; 282,I,96.5; 393,R,97.2; 461,A,95.5; 566,H,98.5; 667,L,98.8; 693,G,97.6; 697,N,96.9; 723,H,97.5; 776,N,97.1 |
| TLR9 | M7 (β) M8 (β & ω) |
p = 0.13, q = 0.53 p = 0.17, q = 1.44, f0 = 0.93 ω = 1.72, f1 = 0.07 |
693,R,95.5; 722,K,95.7 |
| TLR10 | M7 (β) M8 (β & ω) |
p = 0.19, q = 0.24 p = 0.35, q = 0.57, f0 = 0.94 ω = 2.28, f1 = 0.06 |
611, G, 97.1 |
Fig. 2.
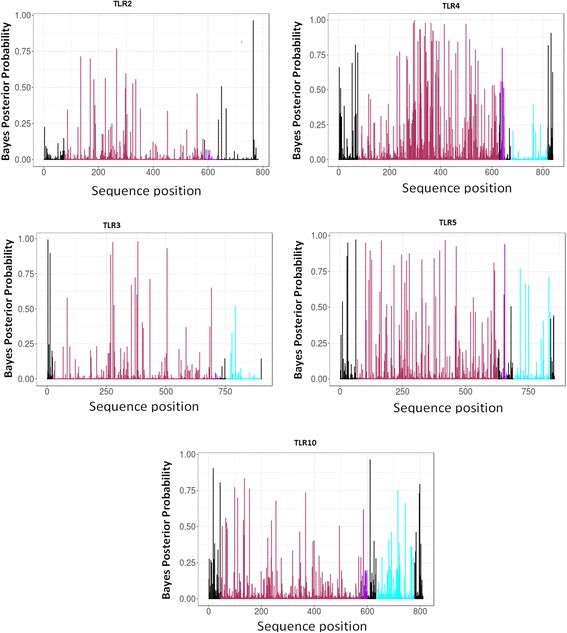
Graphical representation of distribution of selection pressure in Certatiodactyl Toll-like receptors. The majority of sites are under purifying selection. However positive selection is likely to occur in the ecto-domain (ECD) (brown-highlighted) than the transmembrane (TM) and Toll/Interleukin receptor (TIR) domain. Only TLR2-5 and TLR10 are presented for which clear positional demarcation of all three TLR domains was confidently predicted by SMART using cattle TLR as reference
The majority of significantly positively selected codons predicted by M8 method were also detected by the SLR methods suggesting high concordance between the two methods. Mapping of the positive selection concordant sites to annotated TLRs identified some positively selected sites to be located within the key domains and LRR motifs suggesting potential residues of adaptive significance in various species (Additional File 3: Table S1).
Clade-specific selection divergence
Clade model test of selection divergence revealed that the majority of TLRs did not undergo selection divergence during cetaceans’ divergence from terrestrial ungulates (Table 2). However, the null hypothesis for the clade model (M2a_rel) was significantly rejected in favor of clade model C for TLR3 (LRT = 12.2, P < 0.001). The divergent site class in TLR3 appears to evolve under stronger positive selection in cetaceans clade with an estimated ω ≥ 4, about twice that observed in terrestrial ungulates (Table 2). In order to determine if the inference of positive selection can be made on the cetaceans’ clade as a result of selection divergence, the branch-site model was applied on TLR3 to test for the presence of positive selection on cetaceans’ clade against the background of terrestrial ungulates. Branch-site analysis did not find support for positive selection in any of the divergent clades.
Table 2.
Results of Clade models testing for divergent selection among codons between Ungulate (Clade 0) and Cetacean (Clade 1)
| Clade model | Branch-site model | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| lnL | Site classes | ||||||||
| Gene | M2a_rel (np) | Clade model C (np) | LRT | 0 | 1 | 2 | P- value | LRT | P-value |
| TLR2 | −8561.3 (28) | −8561.1 (29) | 0.4 |
P
0 = 0.66 ω0 = 0.08 |
P
1 = 0.31 ω1 = 1 |
P
2 = 0.03 ωClade 0 = 4.6 ωClade 1 =2.78 |
0.5 | ||
| TLR3 | −9856.6 (42) | −9850.5 (43) | 12.2 |
P
0 = 0.77 ω0 = 0.1 |
P
1= 0.21 ω1 = 1 |
P
2 = 0.02 ωClade 0 = 2.7 ωClade 1 = 4.7 |
<0.001 | 0.000 | >0.05 |
| TLR4 | −10128.7 (34) | −10128.7 (35) | 0.0 |
P
0 = 0.6 ω0 = 0.08 |
P
1 = 0.3 ω1 = 1 |
P
2 = 0.1 ωClade 0 = 2.85 ωClade 1 =2.67 |
0.8 | ||
| TLR5 | −10467.5 (38) | −10467.3 (39) | 0.3 |
P
0 = 0.6 ω0 = 0.09 |
P
1 = 0.3 ω1 = 1 |
P
2 = 0.1 ωClade 0 = 2.98 ωClade 1 =3.71 |
0.6 | ||
| TLR7 | −9947.7 (43) | −9946.8 (42) | 1.8 |
P
0 = 0.0 ω0 = 0.0 |
P
1 = 0.2 ω1 = 1 |
P
2 = 0.8 ωClade 0 = 0.05 ωClade 1 = 0.00 |
0.2 | ||
| TLR9 | −10345.2 (30) | −10345.1 (31) | 0.2 |
P
0 = 0.57 ω0 = 0.00 |
P
1 = 0.12 ω1 = 1 |
P
2 = 0.31 ωClade 0 = 0.17 ωClade 1 = 0. 20 |
0.7 | ||
| TLR10 | −8606.2 (40) | −8605.4 (41) | 1.5 |
P
0 = 0.61 ω0 = 0.10 |
P
1 = 0.38 ω1 = 1 |
P
2 = 0.01 ωClade 0 = 4.99 ωClade 1 =0.00 |
0.2 | ||
The TLR with significant divergent selection was further subjected to branch-site analysis to determine if divergent selection corresponds to positive selection in the foreground (Cetacean) branch
Mapping of important substitutions on the TLR3 structure
We were still interested to find if giraffe possesses key substitutions within its TLR3 that localize to important sites of the receptor based on the crystal structure of the TLR3 ECD and site-directed mutagenesis experiments [55, 56]. First, we ensured that the observed sequence changes were not a result of sequencing errors by cross-checking if the sequences involved are identical in the two giraffes that were sequenced in the Giraffe Genome Project. Mapping of giraffe residues corresponding to sites of positive selection on the TLR3 ECD structure showed that two of these sites, Valine at position 278 and Phenylalanine at position 383 (Fig. 3b) are located on the concave side of the ECD. This concave surface was precluded by Choe et al. [55] as potential location for dsRNA ligand binding due to the presence of high amount of carbohydrates. Secondly, a PolyPhen screen on the TLR3 protein reported one unique giraffe substitution, T267I, as probably significant with a PolyPhen score of > 0.99 (Fig. 3b). However, the site does not correspond with any of the residues found in various experiments to be essential in dsRNA ligand binding. Finally, we examined the TLR3-ECD 11 N-glycosylation sites that are visible in the structure [55]. Interestingly, giraffe appears to have lost N-glycosylated site at position 247 where they possess Aspartate (D247) in place of conserved Asparagine (N247). A N247D mutation was shown to result in altered receptor activity in a site-directed mutagenesis experiment [56]. Therefore, similar alterations in receptor signaling may be dictated by the singular N247D change or in combination with other sequence changes in giraffe TLR3, with respect to selection divergence of TLR3 between terrestrial ungulates and cetaceans.
Fig. 3.
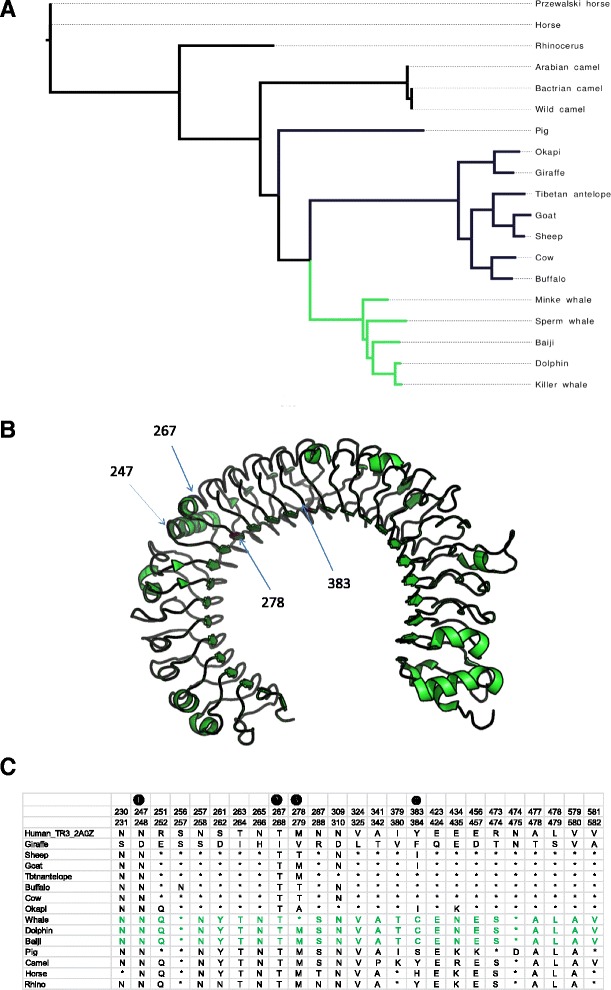
Functional prediction of important substitutions identified from giraffe TLR3. a A TLR3 cladogram (maximum likelihood) demarcating terrestrial ungulates (black) and cetacean (green) branches used in the clade analysis of functional divergence. b Crystal structure of human TLR3 ECD (PDB ID: 2A0Z) showing sites corresponding to giraffe key substitutions based on whether they are predicted to change function (PolyPhen), positive selection sites or map to empirically important site on the structure. c Partial alignment to show residues in other species at sites corresponding to giraffe important sites (❶ denotes TLR3 N-glycosylated site, ❷ show PolyPhen hit site and ❸ show positive selected sites based on PAML and SLR). Top number lane represents the residue position for human TLR and the second number lane is the residue position in the alignment. The asterisk (*) refers to residue identical to that of giraffe
Discussion
TLR sequence analysis reveals strong conservation between giraffe and related species
This is the first study presenting the sequence analysis of 7 TLR proteins from giraffe and okapi. The protein domain prediction of the TLR sequences revealed typical TLR structure with ECD, TM and TIR domains which are similar among giraffe, okapi and cattle. The results are in accordance with previous studies on TLR gene sequences from goat and buffalo which showed a high degree of sequence similarity across species [23, 57]. The high nucleotide and amino acid similarities of giraffe TLR sequences in comparison to okapi and cattle is indicative of general conservation of TLR sequences among vertebrates in general [58]. Despite the high degree of conservation, amino acid differences did exist between species, with giraffe TLR3 showing up to 25 individual amino acid differences with okapi, giraffe's closest living relative. The comparison of giraffe LRR motifs with equivalent LRR motifs in okapi and cattle TLRs indicates similar amount of LRRs in TLRs 7, 9 and 10. The remaining TLRs showed differences in the numbers of LRRs between species, although the range of differences was not remarkable (the highest observed difference in LRRs between any pair of species was 2). This supports the importance of LRRs in TLR ligand recognition. Apparently purifying selection, perhaps due to the need to maintain TLR–ligand interaction/response system resulting from similar pathogenic pressure, has kept relatively constant the number of LRRs in various vertebrates [59].
Recurrent positive selection has shaped the evolution of TLRs in ungulates and cetaceans
Various studies have comprehensively documented the importance of pathogen interaction and positive selection pressure in structuring diversity in the TLRs of mammalian species [31, 60]. The complex evolutionary history associated with divergence of cetaceans from terrestrial ungulates posed many pathogenic challenges, making members of this taxon interesting candidates of pathogen induced selection on immune genes. Results obtained in this study indicate that recurrent positive selection has shaped TLR evolution and diversity among terrestrial ungulates and cetaceans. Also, the observation that just small proportion of sites in all of the TLRs studied are affected by recurrent positive selection is consistent with the mostly accepted paradigm that purifying selection is the dominant force operating on TLRs [24]. Consistent with other studies, (e.g. [61]), our study noted the presence of more positively selected sites in bacterial-sensing TLRs than in viral-sensing counterparts. Viral PAMPs are ancient and conserved [62] while bacterial PAMPs are recognized on the cell surface and should accumulate new mutations fast at key residues to effectively evade recognition by the host [63]. Therefore viral infections are thought to exert stronger selective pressure than bacterial infections on immune genes, thus constraining the evolution of viral-sensing TLRs.
The bacterial-sensing TLR4 stood out as the gene with the strongest evidence of selection, in which more codons were found to be under recurrent positive selection at significant levels (Fig. 3). The high number of positively selected sites observed in TLR4 is also in line with previously reported results in primates and rodents [31, 61]. The malleability of TLR4 to selection pressure is often attributed to the capability of TLR4 to respond to a wide variety of ligands. The TLR4 forms a heterodimer complex with the myeloid differentiation factor 2 (MD2) to recognize a wide range of ligands ranging from Gram-negative bacteria LPS, yeast cell wall components, Trypanosoma and viruses [61, 64, 65]. The identification of numerous sites affected by positive selection in TLR4 in our study suggest that the diversity of ecological specializations among ungulates and cetaceans has combined with the TLR4 inherent factors to accelerate adaptive evolution of TLR4 in these species.
Location of strong positive selection is biased in the ECDs of TLRs
The mapping of positively selected sites to the three major TLR domains shows that 92 to 100% sites were located in the ECD, a critical domain responsible for pathogen recognition. This is consistent with several recent studies conducted on primates, birds and rodents [30, 56, 61] that have noted concentration of positively selected sites in the ECD that harbors putative sites for ligand binding. The localization of many positive selection sites in the ECD, some of which are observed to be part of the LRR motifs, implies that corresponding amino acid substitutions may exert species-specific functional significance [27, 66].
The role of terrestrial ungulates and cetaceans divergence in shaping TLRs evolution
Habitat shifts often promote adaptation and aquatic life can be considerably challenging for mammals that were originally adapted for life on land [39]. We examined patterns of TLRs in the context of terrestrial ungulates and cetaceans divergence hypothesizing that terrestrial and aquatic habitats provide contrasting environments that harbors distinct pathogenic communities. In turn, this would provide clues on specific pathogens accelerating adaptive differentiation in the immune genes operations between terrestrial-adapted ungulates and aquatic-adapted cetaceans. The data are largely in favor of functional constraint on TLRs between terrestrial ungulates and cetaceans indicating that the prevailing immune responses despite the difference in their respective habitats are a result of similar pathogenic pressure. However, we noted significant selection divergence in TLR3 suggestive of the possibility that dsRNA virus may have played a critical adaptive role in terrestrial ungulates and cetaceans divergence. In particular, divergent sites were evolving under accelerated rates in both clades but higher in cetacean clade (ω = 4.7) than in terrestrial ungulates (ω = 2.7) (Table 2). The result indicates potential adaptive response following water re-colonization and provide support for the growing appreciation of the significance of the RNA viruses in marine ecology [67, 68]. However, this result is somewhat paradoxical especially due to the fact that all RNA viruses known to infect cetaceans have thus far been single-stranded [67, 68]. Altogether, selection divergence in TLR3 and TLR7 (another viral-sensing RNA ranked second in terms of TLRs with the most number of positively selected sites), point to the increased significance of RNA viruses in the adaptations of terrestrial ungulates and cetaceans.
TLR3 divergence and species-specific functional implications
Combining clade model analysis and giraffe substitution analysis on TLR3 structure allowed us to examine possible functional significance of terrestrial ungulates versus cetaceans TLR3 divergence with respect to particular species. TLR3 has previously been identified to show disparity in species-specific adaptive functional attributes between human and mouse [69]. This difference was associated with the narrow range of TLR3 functions in humans compared to the receptor broad range of functions in mouse. Our analysis indicated that such TLR3 species-specific functional attributes may also exist in some ungulate and cetacean species. TLR3-ECD contains fifteen N-linked glycosylation sites, all of which have been experimentally mutated individually or in pairs [56, 70]. Of particular interest was the unique giraffe N247D substitution occurring at the N-glycosylated site of TLR3-ECD. The certainty of this specific sequence change on giraffe TLR3 signaling mechanism will need validation experiments given that N247D mutation in human cell lines results in reduced or complete loss of activity. Although the N-glycosylation at this site does not seem to play any role in determining the conformational stability of the ECD crystal structure, it is likely that the linked glycan moiety may be involved in important cellular function related to TLR3, such as localization of the receptor to cellular compartments [70].
Conclusions
The study has presented a molecular phylogenetic analysis of the seven TLR genes represented by giraffe, okapi, other terrestrial ungulates and cetaceans. The evidence of positive selection on the TLR genes reveals that pathogen mediated selective pressure may have shaped terrestrial ungulates and cetacean TLR evolution. The case for positive selection or selection divergence among or between terrestrial ungulates and cetaceans is supported by the correspondence of some of these sites to key TLR motifs including functionally relevant sites. The observed changes in TLRs are probably associated with different pathogenic environments that cetaceans and ungulates had to face during the course of their evolution. Sites under positive selection may have aided in their adaptation as they encountered novel environments. Further work, however, is required to ascertain the role of positively selected sites and other important substitutions identified in this study in relation to pathogen recognition. Furthermore, research is required to determine whether changes at positively selected sites and at other key sites translates to specificity in ligand recognition, signaling mechanism or differential susceptibility to pathogenic infections among ungulates and cetacean species.
Acknowledgement
We would like to thank Douglas Cavener and colleagues at the genome sequencing facility of the Department of Biology and the Huck Institute of Life Sciences, Pennsylvania State University, for making the giraffe and okapi sequences available to us. We thank Alan Orth for the computing support in accessing the HPC facility of the International Livestock Research Institute in Nairobi, Kenya. The anonymous reviewers are acknowledged for their insightful comments and suggestions.
Funding
The Tanzania government through Commission for Science and Technology (COSTECH) sponsored EI doctoral studies. MA stay at the Nelson Mandela African Institution of Science and Technology was also supported by the Tanzania government M-AIST through the Ministry of Science and Technology.
Availability of data and materials
All of the sequences used in the study are publicly accessible. TLR sequence data for giraffe and okapi were obtained from sequences generated by the Giraffe Genome Project whose genome is available in the Short Read Archive under project number SRP071593 (BioProject PRJNA313910). Sequence alignments, tree files, control files for PAML selection analyses and SLR raw data are available in TreeBASE [71] and in Dryad [72].
Authors’ contributions
EI designed the study, performed the analyses and drafted the manuscript. MA coordinated the study and edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AP-1
Activator protein 1
- BEB
Bayes Empirical Bayes
- CmC
Clade model C
- CpG
Cytosine Phosphate Guanine
- dsRNA
Double-stranded RNA
- ECD
Extracellular domain
- IRFs
Interferon regulatory factors
- LPS
Lipopolysaccharides
- LRT
Likelihood ratio test
- LRR
Leucine Rich Repeats
- MD2
Myeloid differentiation factor 2
- MYD88
Myeloid differentiation primary-response protein 88
- NF-kB
Nuclear Factor kappaB
- PAML
Phylogenetic Analysis by Maximum Likelihood
- PAMPs
Pathogen Associated Molecular Patterns
- SMART
Simple Modular Architecture Research Tool
- ssRNA
Single-stranded RNA
- TIR
Toll/Interleukin-1 Receptor
- TLRs
Toll-like receptors
- TRIF
TIR domain-containing adaptor protein inducing IFNβ
Additional files
Giraffe and okapi TLRs coding gene sequences used in the analysis. A text file containing fasta formatted raw nucleotide sequences of giraffe and okapi that were used for all TLRs analyzed in the study. (TXT 37 kb)
Selected species and accession numbers of all TLR sequences that were used in the analysis. An excel file containing the list of species and the associated RefSeq accession number of the sequence for each TLR. (XLS 13 kb)
Comparison of TLRs sequence characteristics (nucleotide, amino acids, Leucine-Rich-Repeats) between giraffe, okapi and cattle. Microsoft word document containing comparative sequence metrics between in all TLRs studied between giraffe, okapi and cattle. (DOCX 5 kb)
Mapping of significant positive selection sites that are concordant between M8 and SLR to key TLR domains and motifs. An excel file containing functionally important sites based on M8 and SLR predictions and showing correspondence of those sites with domain structure and LRR motifs of TLRs studied. (XLSX 10 kb)
Contributor Information
Edson Ishengoma, Email: ishengomae@nm-aist.ac.tz, Email: edson.ishengoma@muce.ac.tz.
Morris Agaba, Email: morris.agaba@nm-aist.ac.tz.
References
- 1.Takeda K, Kaisho T, Akira S. Toll-like Receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like Receptor Signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:1–6. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 4.Buwitt-Beckmann U, Heine H, Wiesmüller K, Jung G, Brock R, Akira S, et al. Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur J Immunol. 2005;282–9. [DOI] [PubMed]
- 5.Tsukamoto H, Fukudome K, Takao S, Tsuneyoshi N, Kimoto M. Lipopolysaccharide-binding protein-mediated Toll-like receptor 4 dimerization enables rapid signal transduction against lipopolysaccharide stimulation on membrane-associated CD14-expressing cells. Int Immunol. 2010;22:271–80. doi: 10.1093/intimm/dxq005. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 7.Oosting M, Cheng S-C, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci. 2014;111:E4478–84. doi: 10.1073/pnas.1410293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulat-Kardum LJ, Etokebe GE, Lederer P, Balen S, Dembic Z. Genetic polymorphisms in the toll-like receptor 10, interleukin (IL) 17A and IL17F genes differently affect the risk for tuberculosis in Croatian population. Scand J Immunol. 2015;82:63–9. doi: 10.1111/sji.12300. [DOI] [PubMed] [Google Scholar]
- 9.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF- k B by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 10.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 11.Takeshita F, Leifer CA, Gursel I, Ken J, Takeshita S, Gursel M, et al. Cutting edge: role of toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol. 2001;167:3555–8. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 12.Godfroy JI, Roostan M, Moroz YS, Korendovych IV, Yin H. Isolated toll-like receptor transmembrane domains Are capable of oligomerization. PLoS One. 2012;7:e48875. doi: 10.1371/journal.pone.0048875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 14.Leulier F, Lemaitre B. Toll-like receptors — taking an evolutionary approach François. Nat Rev Genet. 2008;9:165–78. doi: 10.1038/nrg2303. [DOI] [PubMed] [Google Scholar]
- 15.Bosch TCG. Rethinking the role of immunity : lessons from Hydra. Trends Immunol. 2014;35:495–502. doi: 10.1016/j.it.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Hughes AL, Piontkivska H. Functional diversification of the toll-like receptor gene family. Immunogenetics. 2008;60:249–56. doi: 10.1007/s00251-008-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roach JM, Racioppi L, Jones CD, Masci AM. Phylogeny of toll-like receptor signaling: adapting the innate response. PLoS One. 2013;8:e54156. doi: 10.1371/journal.pone.0054156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oshiumi H, Matsuo A, Matsumoto M, Seya T. Pan-vertebrate toll-like receptors during evolution. Curr Genomics. 2008;9:488–93. doi: 10.2174/138920208786241234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW. Evolution of the chicken Toll-like receptor gene family: A story of gene gain and gene loss. BMC Genom. 2008;9:62. doi: 10.1186/1471-2164-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen T, Xu S, Wang X, Yu W, Zhou K, Yang G. Adaptive evolution and functional constraint at TLR4 during the secondary aquatic adaptation and diversification of cetaceans. BMC Evol Biol. 2012;12:39. doi: 10.1186/1471-2148-12-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci. 1998;95:588–93. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hornung V, Rothenfusser S, Britsch S, Jahrsdörfer B, Giese T, Endres S, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 23.Raja A, Vignesh AR, Mary BA, Tirumurugaan KG, Raj GD, Kataria R, et al. Sequence analysis of Toll-like receptor genes 1–10 of goat (Capra hircus) Vet Immunol Immunopathol. 2011;140:252–8. doi: 10.1016/j.vetimm.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Mukherjee S, Sarkar-roy N, Wagener DK, Majumder PP. Signatures of natural selection are not uniform across genes of innate immune system, but purifying selection is the dominant signature. Proc Natl Acad Sci. 2009;106:7073–8. doi: 10.1073/pnas.0811357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quach H, Wilson D, Laval G, Patin E, Manry J, Guibert J, et al. Different selective pressures shape the evolution of Toll-like receptors in human and African great ape populations. Hum Mol Gen. 2013;22:4829–40. doi: 10.1093/hmg/ddt335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee S, Ganguli D, Majumder PP. Global footprints of purifying selection on toll-like receptor genes primarily associated with response to bacterial infections in humans. Genome Biol Evol. 2014;6:551–8. doi: 10.1093/gbe/evu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ. Variation matters: TLR structure and species-specific pathogen recognition. Trends Immunol. 2009;30:124–30. doi: 10.1016/j.it.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Jann OC, Werling D, Chang J, Haig D, Glass EJ. Molecular evolution of bovine Toll-like receptor 2 suggests substitutions of functional relevance. BMC Evol Biol. 2008;8:288. doi: 10.1186/1471-2148-8-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith SA, Jann OC, Haig D, Russell GC, Werling D, Glass EJ, et al. Adaptive evolution of Toll-like receptor 5 in domesticated mammals. BMC Evol Biol. 2012;12:122. doi: 10.1186/1471-2148-12-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakajima T, Ohtani H, Satta Y, Uno Y, Akari H, Ishida T, et al. Natural selection in the TLR-related genes in the course of primate evolution. Immunogenetics. 2008;60:727–35. doi: 10.1007/s00251-008-0332-0. [DOI] [PubMed] [Google Scholar]
- 31.Wlasiuk G, Nachman MW. Adaptation and constraint at toll-like receptors in primates. Mol Biol Evol. 2010;27:2172–86. doi: 10.1093/molbev/msq104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agaba M, Ishengoma E, Miller WC, Mcgrath BC, Hudson CN, Reina OCB, et al. Giraffe genome sequence reveals clues to its unique morphology and physiology. Nat Commun. 2016;7:1–8. doi: 10.1038/ncomms11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Normile D. Rinderpest, deadly for cattle, joins smallpox as a vanquished disease. Science. 2010;330:435. doi: 10.1126/science.330.6003.435. [DOI] [PubMed] [Google Scholar]
- 34.Ndeereh D, Obanda V, Mijele D, Gakuya F. Medicine in the wild : strategies towards healthy and breeding wildlife populations in Kenya. George Wright Forum. 2012;29:100–8. [Google Scholar]
- 35.Lewerin SS, Olsson S, Eld K, Röken B, Ghebremichael S, Koivula T, et al. Outbreak of Mycobacterium tuberculosis infection among captive Asian elephants in a Swedish zoo. Vet Rec. 2005;156:171–5. doi: 10.1136/vr.156.6.171. [DOI] [PubMed] [Google Scholar]
- 36.Smith KF, Acevedo-Whitehouse K, Pedersen AB. The role of infectious diseases in biological conservation. Anim Conserv. 2009;12:1–12. doi: 10.1111/j.1469-1795.2008.00228.x. [DOI] [Google Scholar]
- 37.Janis CM, Theodor JM. Cranial and postcranial morphological data in ruminant phylogenetics. Zitteliana B. 2014;32:15–31. [Google Scholar]
- 38.Jing L, Zhang R, Liu Y, Zhu W, Mao S. Intravenous lipopolysaccharide challenge alters ruminal bacterial microbiota and disrupts ruminal metabolism in dairy cattle. Br J Nutr. 2014;112:170–82. doi: 10.1017/S000711451400066X. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Bian G, Zhu W, Mao S. High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front Microbiol. 2015;6:167. doi: 10.3389/fmicb.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uhen MD. Evolution of marine mammals: back to the Sea after 300 million years. Anat Rec. 2007;522:514–22. doi: 10.1002/ar.20545. [DOI] [PubMed] [Google Scholar]
- 41.Reidenberg JS. Anatomical adaptations of aquatic mammals. Anat Rec. 2007;290:507–13. doi: 10.1002/ar.20541. [DOI] [PubMed] [Google Scholar]
- 42.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wernersson R, Pedersen AG. RevTrans : multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–9. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guindon S, Gascuel O. A simple, fast, and accurate method to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 45.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–4. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Offord V, Coffey TJWD. LRRfinder: a web application for the identification of leucine-rich repeats and an integrative Toll-like receptor database. Dev Comp Immunol. 2010;34:1035–41. doi: 10.1016/j.dci.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Yang Z, Nielsen R, Goldman N, Pedersen AK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–49. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen J, Kirk BD, Ma J, Wang Q. Diversifying selective pressure on influenza B virus hemagglutinin. J Med Virol. 2009;81:114–24. doi: 10.1002/jmv.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metzger KJ, Thomas MA. Evidence of positive selection at codon sites localized in extracellular domains of mammalian CC motif chemokine receptor proteins. BMC Evol Biol. 2010;10:139. doi: 10.1186/1471-2148-10-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Massingham T, Goldman N. Detecting amino acid sites under positive selection and purifying selection. Genetics. 2005;169:1753–62. doi: 10.1534/genetics.104.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bielawski JP, Yang Z. A maximum likelihood method for detecting functional divergence at individual codon sites, with application to gene family evolution. J Mol Evol. 2004;59:121–32. doi: 10.1007/s00239-004-2597-8. [DOI] [PubMed] [Google Scholar]
- 52.Weadick CJ, Chang BSW. An improved likelihood ratio test for detecting site-specific functional divergence among clades of protein-coding genes. Mol Biol Evol. 2012;29:1297–300. doi: 10.1093/molbev/msr311. [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–9. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 54.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;7(20):1–41. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309(5734):581–5. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]
- 56.De Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, et al. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem. 2005;280:38133–45. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- 57.Dubey PK, Goyal S, Periasamy K, Mishra BP, Gahlawat SK, Kataria RS. Sequence characterization of river buffalo Toll- like receptor genes 1–10 reveals distinct relationship with cattle and sheep. Int J Immunogenet. 2012;0:1–9 [DOI] [PubMed]
- 58.Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, et al. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci. 2005;102:9577–82. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genom. 2007;8:124. doi: 10.1186/1471-2164-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Areal H, Abrantes J, Esteves PJ. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol Biol. 2011;11:368. doi: 10.1186/1471-2148-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fornůsková A, Vinkler M, Pagès M, Galan M, Jousselin E, Cerqueira F, et al. Contrasted evolutionary histories of two Toll-like receptors (Tlr4 and Tlr7) in wild rodents (MURINAE) BMC Evol Biol. 2013;13:194. doi: 10.1186/1471-2148-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewis SH, Obbard DJ. Recent insights into the evolution of innate viral sensing in animals. Curr Opin Microbiol. 2014;20:170–5. doi: 10.1016/j.mib.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zipfel C, Robatzek S. Pathogen-associated molecular pattern-triggered immunity: Veni, Vidi …? Plant Physiol. 2010;154:551–4. doi: 10.1104/pp.110.161547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park BS, Song DH, Kim HM, Choi B, Lee H, Lee J. The structural basis of lipopolysaccharide recognition by the TLR4 – MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 65.Medeiros MM, Peixoto JR, Oliveira A, Cardilo-reis L, Koatz VLG, Van Kaer L, et al. Toll-like receptor 4 (TLR4)-dependent proinflammatory and immunomodulatory properties of the glycoinositolphospholipid (GIPL) from Trypanosoma cruzi. J Leukoc Biol. 2007;82:488–96. doi: 10.1189/jlb.0706478. [DOI] [PubMed] [Google Scholar]
- 66.Mucha R, Bhide MR, Chakurkar EB, Novak MMI. Toll-like receptors TLR1, TLR2 and TLR4 gene mutations and natural resistance to Mycobacterium avium subsp. paratuberculosis infection in cattle. Vet Immunol Immunopathol. 2009;128:381–8. doi: 10.1016/j.vetimm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 67.Rima BK, Collin AMJ, Earle JAP. Completion of the sequence of a cetacean morbillivirus and comparative analysis of the complete genome sequences of four morbilliviruses. Virus Genes. 2005;30:113–9. doi: 10.1007/s11262-004-4588-7. [DOI] [PubMed] [Google Scholar]
- 68.Lang AS, Rise ML, Culley AI, Steward GF. RNA viruses in the sea. FEMS Microbiol Rev. 2008;33:295–323. doi: 10.1111/j.1574-6976.2008.00132.x. [DOI] [PubMed] [Google Scholar]
- 69.Webb AE, Gerek ZN, Morgan CC, Walsh TA, Loscher CE, Edwards SV, et al. Adaptive evolution as a predictor of species-specific innate immune response. Mol Biol Evol. 2015;32:1717–29. doi: 10.1093/molbev/msv051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Botos I, Liu L, Wang Y, Segal DM, Davies DR. The toll-like receptor 3: dsRNA signaling complex. Biochim Biophys Acta-Gene Regul Mech. 2009;1789:667–74. doi: 10.1016/j.bbagrm.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishengoma E, Agaba M (2017) Data from: Evolution of toll-like receptors in the context of terrestrial ungulates and cetaceans diversification. TreeBASE. http://purl.org/phylo/treebase/phylows/study/TB2:S20429. [DOI] [PMC free article] [PubMed]
- 72.Ishengoma E, Agaba M (2017) Data from: Evolution of toll-like receptors in the context of terrestrial ungulates and cetaceans diversification. Dryad Digital Repository. http://dx.doi.org/10.5061/dryad.80p28. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the sequences used in the study are publicly accessible. TLR sequence data for giraffe and okapi were obtained from sequences generated by the Giraffe Genome Project whose genome is available in the Short Read Archive under project number SRP071593 (BioProject PRJNA313910). Sequence alignments, tree files, control files for PAML selection analyses and SLR raw data are available in TreeBASE [71] and in Dryad [72].