

Abstract
Background: Germline mutations of the REarranged during Transfection (RET) proto-oncogene cause multiple endocrine neoplasia 2 (MEN2). It is unclear whether the distribution of RET mutations varies among populations. The first nationwide study of the distribution of RET mutations was conducted, and the results were compared to those of other populations.
Methods: This retrospective cohort study included 1583 patients who underwent RET gene testing in one of three centers covering all of Denmark between September 1994 and December 2014. Primary testing method was Sanger sequencing, which included exons 8–11 and 13–16. Mutations were defined according to the ARUP database July 1, 2016.
Results: RET mutations were identified in 163 patients from 36 apparently unrelated families. Among the 36 families 13 (36.1%) carried mutations in codon 611, four (11.1%) in codon 618, three (8.3%) in codon 620, one (2.8%) in codon 631, six (16.7%) in codon 634, one (2.8%) in codon 790, one (2.8%) in codon 804, one (2.8%) in codon 852, one (2.8%) in codon 883, and five (13.9%) in codon 918. Among the 13 families with codon 611 mutations, 12 had the p.C611Y mutation.
Conclusions: The distribution of RET mutations in Denmark appears to differ from that of other populations. Mutations in codon 611 were the most prevalent, followed by more frequently reported mutations. This might be due to a possible founder effect for the p.C611Y mutation. However, further studies are needed to find possible explanations for the skewed mutational spectrum in Denmark.
Keywords: : epidemiology, genetics, molecular biology, medullary thyroid carcinoma, medullary thyroid carcinoma—genetics
Introduction
Multiple endocrine neoplasia 2 (MEN2) is an autosomal dominant inherited cancer syndrome subdivided into MEN2A and MEN2B. MEN2A associates medullary thyroid carcinoma (MTC), pheochromocytoma (PHEO), hyperparathyroidism (HPTH), cutaneous lichen amyloidosis (CLA), and Hirschsprung's disease (HD). MEN2B associates MTC, PHEO, ganglioneuromatosis of the aerodigestive tract, and facial, ophthalmologic, and skeletal abnormalities (1).
MEN2A and 2B are caused by activating missense germline mutations of the REarranged during Transfection (RET) proto-oncogene. These pivotal discoveries were made in 1993 and 1994 (2–5), and since then, >100 mutations have been identified (1).
Codon 634 mutations are reported as the most prevalent in both European (6–16) and non-European families (17–25). However, a recent Greek study of 58 RET positive families found mutations of codon 533 to be predominant (26). Smaller studies from Portugal (27), Cyprus (28), and Sardinia (29) revealed mutations prevailing in codons 611, 618, and 804, respectively. Also, a large multicenter study of 250 families from Italy showed an unusually high prevalence of codon 804 and 891 mutations (7). A high prevalence of codon 790 mutations has been noted in Germany (6). Consequently, it is unclear whether the distribution of RET mutations is comparable among populations or whether geographical differences exist.
The first nationwide study of the distribution of RET mutations was conducted, and the results were compared to those of other populations.
Patients and Methods
Patients
This retrospective cohort study included 1583 unique patients who underwent RET gene testing in one of three centers covering all of Denmark between September 1994 and December 2014. At the end of the study period on January 1, 2015, the population of Denmark was 5,659,715 (www.dst.dk).
RET testing was performed in 1056, 459, and 104 patients at (i) the Center for Genomic Medicine, Copenhagen University Hospital, (ii) the Department of Molecular Medicine, Aarhus University Hospital, and (iii) the Department of Endocrinology and Medicine, Aalborg University Hospital, respectively. If tested in more than one center, patients were considered only once. One patient was tested in all three centers, while 34 patients were tested in two centers. This yielded a total of 1583 unique patients of which 164 were positive and 1419 negative for a RET mutation (Fig. 1).
FIG. 1.
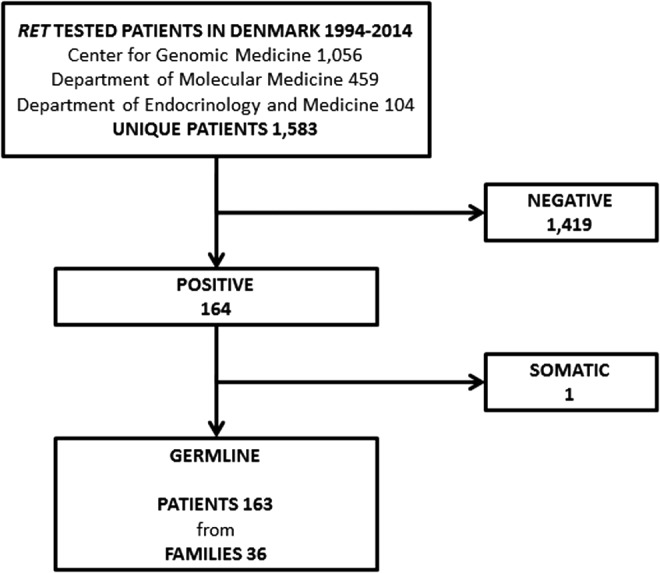
Flow chart showing patients and families tested for REarranged during Transfection (RET) mutations in Denmark 1994–2014. The Center for Genomic Medicine, Department of Molecular Medicine, and Department of Endocrinology and Medicine are part of Copenhagen University Hospital, Aarhus University Hospital, and Aalborg University Hospital, respectively.
RET testing was performed on genomic DNA extracted from whole blood in 1581 patients. One patient harbored the p.C634Y + p.Y791F tandem germline mutation. The ARUP database classifies the p.Y791F sequence change as uncertain (www.arup.utah.edu/database/) (30). However, because recent evidence has classified the sequence change as neutral, the patient was counted as a p.C634Y mutation alone (31,32).
Two patients with MTC were tested using formalin-fixed paraffin-embedded tissue. Both patients were deceased at the time of testing. One was tested using normal tissue, and one using submandibular tissue suspected of MTC metastasis. The latter proved positive for the L790F mutation, but due to suspicion of a somatic mutation in the local metastasis, the patient was excluded from our RET germline mutation cohort (Fig. 1). The patient has been described elsewhere (33).
A MEN2 family was defined by (i) clinical workup in one of the four MEN2 management centers in Denmark, (ii) a unique mutation, or (iii) a molecular proven de novo mutation (both parents of the index case tested mutation negative).
Indications for testing included MTC, PHEO, HPTH, HD, and relatives at risk of MEN2, among others.
Methods
To assess criterion (i), data were provided by collaborators from the four MEN2 management centers. For criteria (ii) and (iii), data were provided by the three RET testing centers. Sequence changes were defined as mutations if classified as pathogenic in the continually updated ARUP database on July 1, 2016 (30).
RET testing centers
Center for Genomic Medicine, Copenhagen University Hospital
From September 1994 to December 1995, Sanger sequencing of RET exons 10 and 11 was performed. Since January 1996, June 1997, February 2000, and September 2007, analysis was expanded to exons 16, 13 + 14, 15, and 8 + 9, respectively. Since October 2014, next-generation sequencing (NGS) of all RET exons has been performed. Predictive testing of relatives at risk of MEN2 was performed in the relevant exon only.
Polymerase chain reaction (PCR) was performed using the primers described in Table 1. Sanger sequencing was carried out in both directions using an ABI377 DNA sequencer as previously described (33,34) or an ABI3730 genetic analyzer, and the sequences were analyzed manually or by the use of SeqScape (Applied Biosystems).
Table 1.
Primers and Polymerase Chain Reaction Conditions for Amplification of RET Exons from the Two Major RET Testing Centers in Denmark
| RET center | Exon | Primer sequences |
|---|---|---|
| Center for Genomic Medicine, Copenhagen University Hospital | 8 | 5′-CTG TCT TTG CTG CCC TGG GTC TGT CAC-3′ |
| 5′-CGT TTC CAC CGG TGC CAT-3′ | ||
| 9 | 5′-GCT GGC AAG GCT CTG TAT ATG GT-3′ | |
| 5′-GGA GGC TCA GCT TGA TGC ATA GAA C-3′ | ||
| 10 | 5′-TCA GAA AGG CAC TGT GAC CAA GC-3′ | |
| 5′-TCC TGG GTG GAG TAA CAG AGG C-3′ | ||
| 11 | 5′-GAG CAT ACG CAG CCT GTA CCC AG-3′ | |
| 5′-GAA ATG GGG GCA GAA CAC A-3′ | ||
| 13 | 5′-CAG GAA CAT AAT GCC ACA TAC AC-3′ | |
| 5′-CCG TGG ACT CAG CTA GAC ACA-3′ | ||
| 14 | 5′-GCT CCT GGA AGA CCC AAG-3′ | |
| 5′-TGG TGG AGT CAG GGT GTG ACA-3′ | ||
| 15 | 5′-CAG GTC TCA CCA GGC CGC TAC-3′ | |
| 5′-AAG GGC CTC GGG TCA GTA TGC T-3′ | ||
| 16 | 5′-CTG TGC CCA GGA GTG TCT ACA GC-3′ | |
| 5′-CCA GCC ATT TGC CTC ACG AAC ACA T-3′ | ||
| Department of Molecular Medicine, Aarhus University Hospital | 8 | 5′-CCT TGG GCT CCA TCC GT-3′ |
| 5′-CCC CAG GAC CCC GTT T-3′ | ||
| 9 | 5′-TAT GGT GTT TCC CTA CTC A-3′ | |
| 5′-CAG GTT TCC CCT ACT ATC-3′ | ||
| 10 | 5′-GCG CCC CAG GAG GCT GAG TG-3′ | |
| 5′-TGC TGT TGA GAC CTC TGT GG-3′ | ||
| 11 | 5′-CTC GAT GGG GTG TTC TCA GG-3′ | |
| 5′-GGA GGG CAG GGG ATC TTC-3′ | ||
| 13 | 5′-GCA GGC CTC TCT GTC TGA ACT T-3′ | |
| 5′-GGA GAA CAG GGC TGT ATG GA-3′ | ||
| 14 | 5′-TCC TGG AAG ACC CAA GCT GC-3′ | |
| 5′-CAT GGT GGG CTA GAG TGT GG-3′ | ||
| 15 | 5′-CCC CCG GCC CAG GTC TCA C-3′ | |
| 5′-GCT CCA CTA ATC TTC GGT ATC TTT-3′ | ||
| 16 | 5′-TCT CCT TTA CCC CTC CTT CC-3′ | |
| 5′-CCT GGC CAA GCT GCA CAG AC-3′ |
RET, REarranged during Transfection.
NGS was carried out using biotinylated oligos (Roche NimbleGen) designed to capture all exons from the NM_020975 transcripts. Library construction was carried out using 500 ng of genomic DNA. The DNA was fragmented into an average size of 200 bp using a Covaris S2 AFA ultrasonicator, and adaptors from Illumina or Roche NimbleGen were ligated to the fragments. Illumina adaptors included in the TruSeq DNA LT Sample Preparation Kit were attached using an SPRI-works System I for the Illumina Genome Analyzer (Beckman Coulter). Adaptor sequences provided by Roche NimbleGen (SeqCap Adaptor Kit A/B) were ligated using the KAPA HTP Library Preparation Kit Illumina on a Sciclone G3 NGS Workstation (PerkinElmer). Sequence capture was performed using a double capture protocol (Roche NimbleGen), where 6–12 samples are multiplexed prior to hybridization. Finally, 2 × 76 bp paired-end sequencing was performed on the Illumina MiSeq platform to an average depth of at least 100 × .
Department of Molecular Medicine, Aarhus University Hospital
From October 1995 to January 1996, Sanger sequencing of exons 10 and 11 was performed. Since February 1996, October 1996, December 1998, September 2004, and September 2007, testing was expanded to exons 16, 13 + 14, 8 + 9, and 15, respectively. Until 2000, only the cysteine domains were tested. As testing has improved, some old blood samples initially negative of RET mutations were retested. Predictive testing of relatives at risk of MEN2 was performed in the relevant exon only.
PCR was carried out using genomic DNA extracted from EDTA-stabilized blood. The primers are shown in Table 1. Sequencing was carried out in both directions on an ABI3130XL or ABI350XL Genetic Analyzer, and sequences were analyzed using Gensearch® software (Phenosystems).
Department of Endocrinology and Medicine, Aalborg University Hospital
From November 1996 to July 2002, primer-specific PCR amplification to detect the p.C611Y mutation was performed. The method has been described in detail previously (35).
Approvals
The investigation was approved by the Danish Health Authority (3-3013-395/2) and the Danish Data Protection Agency (13/19275).
Results
RET testing of 1583 patients detected 14 different germline mutations in 163 patients from 36 apparently unrelated families. Twelve families were defined by either a unique or de novo mutation, while the remaining were defined by clinical workup (Table 2). Of the 36 families, 30 (83%) had MEN2A and six (17%) had MEN2B.
Table 2.
Families with Detected RET Germline Mutationsa in Denmark from 1994 to 2014 According to Family Number, Exon, Nucleotide and Sequence Change, RET Status in Tested Cases, and Reference of Earlier Report
| Family no. | Exon | Nucleotide change | Sequence change | RET+b/RET– | Ref. | |
|---|---|---|---|---|---|---|
| 1c | 10 | c.1833C>G | p.C611W | 6/11 | ||
| 2 | 10 | c.1832G>A | p.C611Y | 2/0 | (43) | |
| 3 | 10 | c.1832G>A | p.C611Y | 1/0 | ||
| 4 | 10 | c.1832G>A | p.C611Y | 8/3 | ||
| 5 | 10 | c.1832G>A | p.C611Y | 15/13 | ||
| 6 | 10 | c.1832G>A | p.C611Y | 2/0 | (34) | |
| 7 | 10 | c.1832G>A | p.C611Y | 9/7 | (34) | |
| 8 | 10 | c.1832G>A | p.C611Y | 2/6 | ||
| 9 | 10 | c.1832G>A | p.C611Y | 26/27 | ||
| 10 | 10 | c.1832G>A | p.C611Y | 30/30 | (44) | |
| 11 | 10 | c.1832G>A | p.C611Y | 1/3 | ||
| 12 | 10 | c.1832G>A | p.C611Y | 5/18 | (45) | |
| 13 | 10 | c.1832G>A | p.C611Y | 7/8 | ||
| 14 | 10 | c.1853G>T | p.C618F | 1/1 | ||
| 15 | 10 | c.1853G>T | p.C618F | 2/1 | (34) | |
| 16 | 10 | c.1853G>A | p.C618Y | 5/9 | ||
| 17 | 10 | c.1853G>A | p.C618Y | 3/3 | (34) | |
| 18 | 10 | c.1858T>C | p.C620R | 6/5 | ||
| 19 | 10 | c.1858T>C | p.C620R | 3/3 | (34) | |
| 20d | 10 | c.1858T>C | p.C620R | 1/3 | (34) | |
| 21c | 11 | c.1891G>T | p.D631Y | 1/0 | ||
| 22 | 11 | c.1900T>C | p.C634R | 1/1 | ||
| 23 | 11 | c.1900T>C | p.C634R | 1/2 | (34) | |
| 24 | 11 | c.1900T>C | p.C634R | 3/11 | (44) | |
| 25 | 11 | c.1900T>C | p.C634R | 1/5 | (34) | |
| 26c,e | 11 | c.1901G>A | p.C634Y | 2/0 | ||
| 27c,e | 11 + 13 | c.1901G>A+c.2372A>T | p.C634Y+Y791F | 1/2 | ||
| 28c | 13 | c.2370G>T | p.L790F | 5/2 | ||
| 29c | 14 | c.2410G>A | p.V804M | 2/1 | ||
| 30c | 14 | c.2556C>G | p.I852M | 3/4 | ||
| 31c,d | 15 | c.2647_2648GC>TT | p.A883F | 1/3 | (46) | |
| 32d | 16 | c.2753T>C | p.M918T | 1/4 | (47) | |
| 33d | 16 | c.2753T>C | p.M918T | 1/3 | ||
| 34 | 16 | c.2753T>C | p.M918T | 1/2 | ||
| 35d | 16 | c.2753T>C | p.M918T | 3/2 | (48) | |
| 36 | 16 | c.2753T>C | p.M918T | 1/1 | ||
| Total | 36 | 163/194 |
Sequence changes classified as pathogenic in the ARUP database July 1, 2016 (30).
RET+ includes index cases.
Families with unique mutations in Denmark.
Families in which both parents of the index case were tested mutation negative.
Families immigrated to Denmark. Family 26 and family 27 originate from Greece and Latvia, respectively.
Accounting for 36% of all families, RET germline mutations of codon 611 were the most frequent. Subsequently, mutations of codons 634 (17%), 918 (14%), 618 (11%), 620 (8%), 631 (3%), 790 (3%), 804 (3%), 852 (3%), and 883 (3%) followed (Table 3). No mutations of codons 292, 515, 533, 609, 630, 666, 750, 768, 891, 904, or 912 were identified.
Table 3.
| Denmark | Greece | Germany | Italy | Poland | Spain | Slovenia | Czechia | France | Spain | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | S (26) | S (6) | M (7) | S (8) | S (9) | S (10) | (11) | M (12,13) | (14) | ||
| RET mutation | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| 5 | p.V292M | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | p.C515S | 0 | 0 | 0 | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | p.G533C | 0 | 21 (36) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | p.C609R/G/F/S/Y | 0 | 0 | 1 (1) | 6 (3) | 3 (13) | 1 (4) | 0 | 1 (5) | 1 (1) | 0 |
| 10 | p.C611R/G/F/S/W/Y | 13 (36) | 0 | 6 (3) | 1 (0) | 0 | 0 | 0 | 1 (5) | 1 (1) | 1 (2) |
| 10 | p.C618R/G/F/S/Y | 4 (11) | 4 (7) | 11 (6) | 15 (6) | 2 (9) | 1 (4) | 4 (31) | 0 | 6 (6) | 3 (7) |
| 10 | p.C620R/G/F/S/W/Y | 3 (8) | 5 (9) | 14 (7) | 9 (4) | 4 (17) | 1 (4) | 0 | 0 | 12 (12) | 1 (2) |
| 11 | p.C630R/F/Y | 0 | 0 | 1 (1) | 4 (2) | 0 | 0 | 0 | 0 | 0 | 0 |
| 11 | p.D631Y | 1 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 11 | p.C634R/G/F/S/W/Y | 6 (17) | 19 (33) | 73 (38) | 85 (37) | 9 (39) | 17 (68) | 6 (46) | 11 (52) | 46 (47) | 36 (88) |
| 11 | p.K666E | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 12 | p.A750P | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 13 | p.E768D | 0 | 1 (2) | 2 (1) | 9 (4) | 0 | 1 (4) | 0 | 1 (5) | 2 (2) | 0 |
| 13 | p.L790F | 1 (3) | 0 | 26 (14) | 8 (3) | 0 | 0 | 3 (23) | 0 | 4 (4) | 0 |
| 14 | p.V804L/M | 1 (3) | 3 (5) | 19 (10) | 52 (22) | 1 (4) | 0 | 0 | 3 (14) | 15 (15) | 0 |
| 14 | p.I852M | 1 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 15 | p.A883F/T | 1 (3) | 0 | 0 | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 |
| 15 | p.S891A | 0 | 0 | 6 (3) | 23 (10) | 1 (4) | 1 (4) | 0 | 0 | 7 (7) | 0 |
| 15 | p.S904F | 0 | 0 | 0 | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 |
| 16 | p.R912P | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| 16 | p.M918T | 5 (14) | 5 (9) | 32 (17) | 17 (7) | 2 (9) | 3 (12) | 0 | 4 (19) | 3 (3) | 0 |
| Total | 36 (100) | 58 (100) | 191 (100) | 232 (100) | 23 (100) | 25 (100) | 13 (100) | 21 (100) | 97 (100) | 41 (100) |
Due to rounding up, not all sums of the numbers fit.
Sequence changes classified as pathogenic in the ARUP database July 1, 2016 (30).
Including series with a minimum of 10 European RET families ((27–29) excluded), a minimum of exons 10–11 and 13–16 tested ((16) excluded), and specifying familial prevalence.
N, nationwide; S, single center; M, multicenter; n, number of families.
Among the 13 families with codon 611 mutations, 12 had the p.C611Y mutation, while one had the p.C611W mutation.
The distribution of RET germline mutations reported in European and non-European studies is shown in Tables 3 and 4.
Table 4.
| China | China | India | Iran | China | Korea | ||
|---|---|---|---|---|---|---|---|
| S (17) | S (18) | S (19) | M (20) | (21) | M (22) | ||
| Exon | RET mutation | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) |
| 5 | p.V292M | 0 | 1 (9) | 0 | 0 | 0 | 0 |
| 8 | p.C515S | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | p.G533C | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | p.C609R/G/F/S/Y | 0 | 0 | 2 (13) | 0 | 0 | 0 |
| 10 | p.C611R/G/F/S/W/Y | 0 | 0 | 0 | 1 (9) | 0 | 0 |
| 10 | p.C618R/G/F/S/Y | 2 (20) | 1 (9) | 1 (7) | 1 (9) | 0 | 3 (23) |
| 10 | p.C620R/G/F/S/W/Y | 0 | 0 | 0 | 0 | 0 | 0 |
| 11 | p.C630R/F/Y | 0 | 0 | 0 | 1 (9) | 0 | 0 |
| 11 | p.D631Y | 0 | 0 | 0 | 0 | 0 | 0 |
| 11 | p.C634R/G/F/S/W/Y | 6 (60) | 8 (73) | 9 (60) | 6 (55) | 15 (75) | 10 (77) |
| 11 | p.K666E | 0 | 0 | 0 | 0 | 0 | 0 |
| 12 | p.A750P | 0 | 0 | 0 | 0 | 0 | 0 |
| 13 | p.E768D | 0 | 0 | 0 | 0 | 0 | 0 |
| 13 | p.L790F | 1 (10) | 1 (9) | 0 | 0 | 0 | 0 |
| 14 | p.V804L/M | 0 | 0 | 1 (7) | 1 (9) | 0 | 0 |
| 14 | p.I852M | 0 | 0 | 0 | 0 | 0 | 0 |
| 15 | p.A883F/T | 0 | 0 | 0 | 0 | 0 | 0 |
| 15 | p.S891A | 0 | 0 | 0 | 0 | 0 | 0 |
| 15 | p.S904F | 0 | 0 | 0 | 0 | 0 | 0 |
| 16 | p.R912P | 0 | 0 | 0 | 0 | 0 | 0 |
| 16 | p.M918T | 1 (10) | 0 | 2 (13) | 1 (9) | 5 (25) | 0 |
| Total | 10 (100) | 11 (100) | 15 (100) | 11 (100) | 20 (100) | 13 (100) |
Due to rounding up, not all sums of the numbers fit.
Sequence changes classified as pathogenic in the ARUP database July 1, 2016 (30).
Including series with a minimum of 10 European RET families ((25,38,39) excluded), a minimum of exons 10–11 and 13–16 tested ((23,24) excluded), and specifying familial prevalence.
S, single center; M, multicenter; n, number of families.
Discussion
In this nationwide study of 1583 patients who were RET tested from 1994 to 2014, 14 different RET germline mutations were identified in 163 patients from 36 apparently unrelated MEN2 families. Mutations of codon 611 were the most prevalent (36%), followed by mutations of codon 634 (17%).
Limitations
To estimate the true number of RET mutations in a country, the entire population needs to be tested. However, this would result in both immense socioeconomic and ethical challenges. For the best possible estimate under the given circumstances, this study included all RET tested patients in Denmark since testing became available and for more than two decades onwards.
To capture yet unrecognized MEN2 patients, the Danish MEN taskforce has recommended routine RET screening in all patients with MTC, PHEO, C-cell or parathyroid hyperplasia, familial/recurrent HPTH, and HPTH <40–50 years of age (36,37). Unfortunately, the completeness of RET screening in these patient groups is unknown and will require further studies. In most other studies, only patients with MTC, clinically diagnosed MEN2, or relatives at risk of MEN2 were submitted to mutational analysis of RET (7–29,38,39).
To establish a nationwide RET positive MEN2 cohort, data were collected in collaboration with the RET testing and MEN2 management centers in Denmark. This might cause issues of inter-variability in regards to methods and interpretation of results. However, methods and temporal expansion of RET testing were roughly similar in the two major RET testing centers, accounting for 94.7% (1499 patients) of the cohort. The remaining 84 patients were tested solely at the Aalborg University Hospital and thus only for the p.C611Y mutation. Of those, 26 were p.C611Y positive, and 45 were related to a p.C611Y patient. This leaves only 13 patients (0.7% of the total cohort) in which a RET mutation different from the p.C611Y might have been missed.
To estimate the true number of unrelated MEN2 families, a haplotype or genealogy study would be ideal. However, 24/36 apparently unrelated MEN2 families were defined by thorough clinical work-up. As in other studies, this workup is based on the index patient's recollection and knowledge of the familial history. Thus, possible relations between families carrying similar mutations might have been missed and may cause an overestimation of the number of families.
Codon 611 mutations
In the present study, mutations in codon 611 of exon 10 were the most prevalent accounting for 36% of the 36 families. This appears to be an unusually high prevalence. Numerous other studies did not detect any mutations in codon 611, despite systematic testing of RET exon 10 (8–10,16–19,21–26,28,29,39). Most of these are single-center studies, and there might be an expectation of capturing more MEN2 patients in a nationwide study such as the one presented here. However, this does not seem to be the explanation. In fact, in three large European predominantly multicenter series based on a total of 232, 191, and 97 families, the prevalence of codon 611 mutations was only 0%, 3%, and 1%, respectively (Table 3) (6,7,12,13). The highest prevalence rates reported have been from Portugal (40%), Saudi Arabia (11%), and Iran (9%). However, these reports were based on small mainly single-center studies with a total of 5, 11, and 9 families (20,27,38).
Mutations highly prevalent in other series
An unusually high prevalence of specific RET mutations has also been described in other populations (6,7,26,28,29). In Cyprus, eight families were studied, and all carried the p.C618R mutation (28). A Sardinian study of seven families found three kindreds (43%) with the p.V804M mutation (29). In a large Italian multicenter study, 22% and 10% of the families had mutations in codons 804 and 891, respectively (Table 3) (7). A large German study identified codon 790 mutations in 14% of all included families, while a recent Greek study of 58 families found that 36% harbored the p.G533C mutation in exon 8 (Table 3) (6,26).
Results from these studies indicate geographical differences in the distribution of RET mutations. On the other hand, if mutations in exon 8 are disregarded in the Greek study, the distribution of mutations would be largely similar to that of other sizeable series where exon 8 was not routinely tested (7,12,13). Also, the high prevalence of codon 804 and 891 mutations in Italy may be caused by an earlier and more extensive testing of exons 14 and 15 compared to other series. A similar explanation for the high prevalence of codon 611 mutations in Denmark seems unlikely, since the traditional RET panel included testing of exon 10 from the very beginning (40). Thus, there seems to be a genuine high prevalence of codon 611 mutations in Denmark. This is supported by the fact that 70% (114/163) of the patients studied carry a codon 611 mutation.
Possible founder effect
Among the 13 apparently unrelated families with codon 611 mutations in the present study, 12 had the p.C611Y mutation. This could indicate a possible founder effect for the p.C611Y mutation. A similar effect has been indicated in other studies (7,26–29,41,42).
In the Mediterranean islands of Cyprus and Sardinia, the authors suggested that a founder effect for the p.C618R and the p.V804M mutation could be explained by geographical isolation (28,29). However, geographical isolation alone does not seem to explain a possible founder effect in Denmark, since its southern border connects the country to Germany and the rest of Continental Europe.
In Greece, the p.G533C mutation was found especially in the southern part of Central Greece and the southeast region of the Peloponnese (26). In Italy, the p.S891A mutation was mainly present in a well-defined northern area. The Italian authors also found a high prevalence of codon 804 mutations, but excluded a founder effect by single-nucleotide polymorphism analysis (7). Only few and very small studies have proven a founder effect of RET mutations in mainland areas by haplotype analysis (27,41,42). To elaborate on this question in Denmark, further investigations are planned.
Prevalence of other mutations
Codon 634 mutations have been reported as the most predominant in several European (Table 3) (6–14) and non-European series (Table 4) (17–25). This might be explained by earlier onset of disease and a presentation frequently involving the full-blown MEN2 syndrome (MTC, PHEO, and HPTH). Mutations of codon 634 were the second most frequent alteration found in the present study, accounting for 17% (Table 3).
In the three largest studies to date, the prevalence of the p.V804L/M mutations ranged from 10% to 22% (Table 3) (6,7,12,13). In the present cohort, only one family (3%) was identified with this mutation. Despite testing for mutations in exon 14 since 1996 and 1997 in the two major RET testing centers, one center did not test the non-cysteine domains until after 2000. Retesting of patients tested before 2000 was performed but not systematically. Consequently, some patients tested for these mutations between 1996 and 2000 might have been missed, and this may result in a reduced number of families with non-cysteine codon 804 mutations. The distribution of the remaining mutations did not differ largely from that found in the literature.
Conclusion
The distribution of RET mutations in Denmark appears to differ from that of other populations. Mutations in codon 611 were by far the most prevalent alterations followed by more frequently reported mutations. This might be due to a possible founder effect for the p.C611Y mutation. However, further studies are needed to find possible explanations for the skewed mutational spectrum in Denmark.
Acknowledgments
This work was supported by the University of Southern Denmark, the Region of Southern Denmark, Odense University Hospital, Copenhagen University Hospital, the Danish Cancer Society, the Danish Cancer Research Foundation, and the A.P. Moeller Foundation. The research salary of Ulla Feldt-Rasmussen is sponsored by an unrestricted research grant from the Novo Nordic Foundation.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
- 1.Wells SA, Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG. 2015. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25:567–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L. 1993. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363:458–460 [DOI] [PubMed] [Google Scholar]
- 3.Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA., Jr 1993. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 2:851–856 [DOI] [PubMed] [Google Scholar]
- 4.Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, Pasini B, Hoppener JW, van Amstel HK, Romeo G, Lips CJ, Buys CH. 1994. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 367:375–376 [DOI] [PubMed] [Google Scholar]
- 5.Carlson KM, Dou S, Chi D, Scavarda N, Toshima K, Jackson CE, Wells SA, Jr, Goodfellow PJ, Donis-Keller H. 1994. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A 91:1579–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Machens A, Lorenz K, Sekulla C, Hoppner W, Frank-Raue K, Raue F, Dralle H. 2013. Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol 168:307–314 [DOI] [PubMed] [Google Scholar]
- 7.Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, Mian C, Castellano M, degli UE, Ceccherini I, Cremonini N, Seregni E, Orlandi F, Ferolla P, Puxeddu E, Giorgino F, Colao A, Loli P, Bondi F, Cosci B, Bottici V, Cappai A, Pinna G, Persani L, Verga U, Boscaro M, Castagna MG, Cappelli C, Zatelli MC, Faggiano A, Francia G, Brandi ML, Falchetti A, Pinchera A, Elisei R. 2010. Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol 163:301–308 [DOI] [PubMed] [Google Scholar]
- 8.Paszko Z, Sromek M, Czetwertynska M, Skasko E, Czapczak D, Wisniewska A, Prokurat A, Chrupek M, Jagielska A, Kozlowicz-Gudzinska I. 2007. The occurrence and the type of germline mutations in the RET gene in patients with medullary thyroid carcinoma and their unaffected kindred's from Central Poland. Cancer Invest 25:742–749 [DOI] [PubMed] [Google Scholar]
- 9.Fernandez RM, Navarro E, Antinolo G, Ruiz-Ferrer M, Borrego S. 2006. Evaluation of the role of RET polymorphisms/haplotypes as modifier loci for MEN 2, and analysis of the correlation with the type of RET mutation in a series of Spanish patients. Int J Mol Med 17:575–581 [PubMed] [Google Scholar]
- 10.Bergant D, Hocevar M, Besic N, Glavac D, Korosec B, Caserman S. 2006. Hereditary medullary thyroid cancer in Slovenia—genotype–phenotype correlations. Wien Klin Wochenschr 118:411–416 [DOI] [PubMed] [Google Scholar]
- 11.Jindrichova S, Vcelak J, Vlcek P, Neradilova M, Nemec J, Bendlova B. 2004. Screening of six risk exons of the RET proto-oncogene in families with medullary thyroid carcinoma in the Czech Republic. J Endocrinol 183:257–265 [DOI] [PubMed] [Google Scholar]
- 12.Niccoli-Sire P, Murat A, Rohmer V, Franc S, Chabrier G, Baldet L, Maes B, Savagner F, Giraud S, Bezieau S, Kottler ML, Morange S, Conte-Devolx B. 2001. Familial medullary thyroid carcinoma with noncysteine RET mutations: phenotype–genotype relationship in a large series of patients. J Clin Endocrinol Metab 86:3746–3753 [DOI] [PubMed] [Google Scholar]
- 13.Nguyen L, Niccoli-Sire P, Caron P, Bastie D, Maes B, Chabrier G, Chabre O, Rohmer V, Lecomte P, Henry JF, Conte-Devolx B. 2001. Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. Eur J Endocrinol 144:37–44 [DOI] [PubMed] [Google Scholar]
- 14.Sanchez B, Robledo M, Biarnes J, Saez ME, Volpini V, Benitez J, Navarro E, Ruiz A, Antinolo G, Borrego S. 1999. High prevalence of the C634Y mutation in the RET proto-oncogene in MEN 2A families in Spain. J Med Genet 36:68–70 [PMC free article] [PubMed] [Google Scholar]
- 15.Romei C, Tacito A, Molinaro E, Agate L, Bottici V, Viola D, Matrone A, Biagini A, Casella F, Ciampi R, Materazzi G, Miccoli P, Torregrossa L, Ugolini C, Basolo F, Vitti P, Elisei R. 2015. Twenty years of lesson learning: how does the RET genetic screening test impact the clinical management of medullary thyroid cancer? Clin Endocrinol 82:892–899 [DOI] [PubMed] [Google Scholar]
- 16.Klein I, Esik O, Homolya V, Szeri F, Varadi A. 2001. Molecular genetic diagnostic program of multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma syndromes in Hungary. J Endocrinol 170:661–666 [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Zhang B, Liu W, Zhang Y, Di X, Yang Y, Yan D. 2016. Screening of RET gene mutations in Chinese patients with medullary thyroid carcinoma and their relatives. Fam Cancer 15:99–104 [DOI] [PubMed] [Google Scholar]
- 18.Qi XP, Chen XL, Ma JM, Du ZF, Fei J, Yang CP, Cheng J, Song QZ, Han JS, Jin HY, Chen ZG, Wang JQ, Yang YP, Ying RB, Liu WT, Zhao Y, Chen CY, Jiang HL, Ke HP, Zhang XN. 2012. RET proto-oncogene genetic screening of families with multiple endocrine neoplasia type 2 optimizes diagnostic and clinical management in China. Thyroid 22:1257–1265 [DOI] [PubMed] [Google Scholar]
- 19.Sharma BP, Saranath D. 2011. RET gene mutations and polymorphisms in medullary thyroid carcinomas in Indian patients. J Biosci 36:603–611 [DOI] [PubMed] [Google Scholar]
- 20.Alvandi E, Akrami SM, Chiani M, Hedayati M, Nayer BN, Tehrani MR, Nakhjavani M, Pedram M. 2011. Molecular analysis of the RET proto-oncogene key exons in patients with medullary thyroid carcinoma: a comprehensive study of the Iranian population. Thyroid 21:373–382 [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Zhao Y, Cui B, Gu L, Zhu S, Li J, Liu J, Yin M, Zhao T, Yin Z, Yu C, Chen C, Wang L, Xiao B, Hong J, Zhang Y, Tang Z, Wang S, Li X, Ning G. 2007. RET proto-oncogene mutations are restricted to codons 634 and 918 in mainland Chinese families with MEN2A and MEN2B. Clin Endocrinol 67:570–576 [DOI] [PubMed] [Google Scholar]
- 22.Chung YJ, Kim HH, Kim HJ, Min YK, Lee MS, Lee MK, Kim KW, Ki CS, Kim JW, Chung JH. 2004. RET proto-oncogene mutations are restricted to codon 634 and 618 in Korean families with multiple endocrine neoplasia 2A. Thyroid 14:813–818 [DOI] [PubMed] [Google Scholar]
- 23.Sanso GE, Domene HM, Garcia R, Pusiol E, de M, Roque M, Ring A, Perinetti H, Elsner B, Iorcansky S, Barontini M. 2002. Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer 94:323–330 [DOI] [PubMed] [Google Scholar]
- 24.Hedayati M, Zarif Yeganeh M, Sheikhol Eslami S, Rezghi Barez S, Hoghooghi Rad L, Azizi F. 2011. Predominant RET germline mutations in exons 10, 11, and 16 in Iranian patients with hereditary medullary thyroid carcinoma. J Thyroid Res 2011:264248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CF, Yang WS, Su YN, Wu IL, Chang TC. 2009. Mutational spectrum of multiple endocrine neoplasia type 2 and sporadic medullary thyroid carcinoma in Taiwan. J Formos Med Assoc 108:402–408 [DOI] [PubMed] [Google Scholar]
- 26.Sarika HL, Papathoma A, Garofalaki M, Saltiki K, Pappa T, Pazaitou-Panayiotou K, Anastasiou E, Alevizaki M. 2015. Genetic screening of patients with medullary thyroid cancer in a referral center in Greece during the past two decades. Eur J Endocrinol 172:501–509 [DOI] [PubMed] [Google Scholar]
- 27.Prazeres HJ, Rodrigues F, Figueiredo P, Naidenov P, Soares P, Bugalho MJ, Lacerda M, Campos B, Martins TC. 2006. Occurrence of the Cys611Tyr mutation and a novel Arg886Trp substitution in the RET proto-oncogene in multiple endocrine neoplasia type 2 families and sporadic medullary thyroid carcinoma cases originating from the central region of Portugal. Clin Endocrinol 64:659–666 [DOI] [PubMed] [Google Scholar]
- 28.Neocleous V, Skordis N, Portides G, Efstathiou E, Costi C, Ioannou N, Pantzaris M, Anastasiadou V, Deltas C, Phylactou LA. 2011. RET proto-oncogene mutations are restricted to codon 618 in Cypriot families with multiple endocrine neoplasia 2. J Endocrinol Invest 34:764–769 [DOI] [PubMed] [Google Scholar]
- 29.Pinna G, Orgiana G, Riola A, Ghiani M, Lai ML, Carcassi C, Mariotti S. 2007. RET proto-oncogene in Sardinia: V804M is the most frequent mutation and may be associated with FMTC/MEN-2A phenotype. Thyroid 17:101–104 [DOI] [PubMed] [Google Scholar]
- 30.Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, Mao R. 2009. Multiple endocrine neoplasia type 2 RET proto-oncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30:548–556 [DOI] [PubMed] [Google Scholar]
- 31.Toledo RA, Hatakana R, Lourenco DM, Jr, Lindsey SC, Camacho CP, Almeida M, Lima JV, Jr, Sekiya T, Garralda E, Naslavsky MS, Yamamoto GL, Lazar M, Meirelles O, Sobreira TJ, Lebrao ML, Duarte YA, Blangero J, Zatz M, Cerutti JM, Maciel RM, Toledo SP. 2015. Comprehensive assessment of the disputed RET Y791F variant shows no association with medullary thyroid carcinoma susceptibility. Endocr Relat Cancer 22:65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vestergaard P, Vestergaard EM, Brockstedt H, Christiansen P. 2007. Codon Y791F mutations in a large kindred: is prophylactic thyroidectomy always indicated? World J Surg 31:996–1001 [DOI] [PubMed] [Google Scholar]
- 33.Godballe C, Jorgensen G, Gerdes AM, Krogdahl AS, Tybjaerg-Hansen A, Nielsen FC. 2010. Medullary thyroid cancer: RET testing of an archival material. Eur Arch Otorhinolaryngol 267:613–617 [DOI] [PubMed] [Google Scholar]
- 34.Hansen HS, Torring H, Godballe C, Jager AC, Nielsen FC. 2000. Is thyroidectomy necessary in RET mutations carriers of the familial medullary thyroid carcinoma syndrome? Cancer 89:863–867 [PubMed] [Google Scholar]
- 35.Kroustrup JP, Laurberg P, Madsen PH. 1999. Rapid MEN 2A gene carrier identification using primer-specific PCR amplification. Scand J Clin Lab Invest 59:643–647 [DOI] [PubMed] [Google Scholar]
- 36.Stochholm K, Sunde L, Frederiksen AL, Lihn A, Vestergaard EM, Poulsen PL, Vestergaard P, Brixen K, Eskildsen PC, Rasmussen AK, Feldt-Rasmussen U, Friis-Hansen L. 2011. [Multipel Endokrin Neoplasi. Klaringsrapport 2011]. Ugeskrift for laeger 1–50
- 37.Andersen PH, Kroustrup JP, Feldt-Rasmussen U, Hangaard J, Brixen K. 2002. [MEN Klaringsrapport], 2–12 [Google Scholar]
- 38.Qari F. 2013. RET codon 618 mutations in Saudi families with multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Ann Saudi Med 33:155–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez B, Salcedo M, Medrano ME, Mantilla A, Quinonez G, Benitez-Bribiesca L, Rodriguez-Cuevas S, Cabrera L, de Leon B, Altamirano N, Tapia J, Dawson B. 2003. RET oncogene mutations in medullary thyroid carcinoma in Mexican families. Arch Med Res 34:41–49 [DOI] [PubMed] [Google Scholar]
- 40.Frank-Raue K, Rondot S, Schulze E, Raue F. 2007. Change in the spectrum of RET mutations diagnosed between 1994 and 2006. Clin Lab 53:273–282 [PubMed] [Google Scholar]
- 41.Bugalho MJ, Domingues R, Sobrinho L. 2003. MEN 2A families: from hot spots to hot regions. Int J Mol Med 11:71–74 [DOI] [PubMed] [Google Scholar]
- 42.Peretz H, Luboshitsky R, Baron E, Biton A, Gershoni R, Usher S, Grynberg E, Yakobson E, Graff E, Lapidot M. 1997. Cys 618 Arg mutation in the RET proto-oncogene associated with familial medullary thyroid carcinoma and maternally transmitted Hirschsprung's disease suggesting a role for imprinting. Hum Mutat 10:155–159 [DOI] [PubMed] [Google Scholar]
- 43.Kjaer A, Petersen CL. 2002. Primary diagnosis of multiple pheochromocytomas in the brother of a MEN-2 patient by simultaneous MIBG scintigraphy and low-dose computed tomography. Clin Nucl Med 27:868–870 [DOI] [PubMed] [Google Scholar]
- 44.Emmertsen K. 1984. Screening for hereditary medullary cancer in Denmark. Henry Ford Hosp Med J 32:238–243 [PubMed] [Google Scholar]
- 45.Vestergaard P, Kroustrup JP, Ronne H, Eng C, Laurberg P. 1999. Neuromas in multiple endocrine neoplasia type 2A with a RET codon 611 mutation. J Endocr Genet 1:33–37 [Google Scholar]
- 46.Mathiesen JS, Stochholm K, Poulsen PL, Vestergaard EM, Christiansen P, Vestergaard P. 2015. Aggressive medullary thyroid carcinoma in a ten-year-old patient with multiple endocrine neoplasia 2B due to the A883F mutation. Thyroid 25:139–140 [DOI] [PubMed] [Google Scholar]
- 47.Sondergaard Pedersen JH, Schaffalitzky De Muckadell O. 2007. Choroidal metastases in multiple endocrine neoplasia type 2B. Acta Ophthalmol Scand 85:120–121 [DOI] [PubMed] [Google Scholar]
- 48.Mathiesen JS, Døssing H, Bender L, Godballe C. 2014. [Medullary thyroid carcinoma in a 10-month-old child with multiple endocrine neoplasia 2B]. Ugeskrift for laeger 176:V07130456. [PubMed] [Google Scholar]