


Abstract
MicroRNAs (miRNA) are short single-stranded RNA molecules derived from hairpin-forming precursors that play a crucial role as post-transcriptional regulators in eukaryotes and viruses. In the past years, many microRNA target genes (MTGs) have been identified experimentally. However, because of the high costs of experimental approaches, target genes databases remain incomplete. Although several target prediction programs have been developed in the recent years to identify MTGs in silico, their specificity and sensitivity remain low. Here, we propose a new approach called MirAncesTar, which uses ancestral genome reconstruction to boost the accuracy of existing MTGs prediction tools for human miRNAs. For each miRNA and each putative human target UTR, our algorithm makes uses of existing prediction tools to identify putative target sites in the human UTR, as well as in its mammalian orthologs and inferred ancestral sequences. It then evaluates evidence in support of selective pressure to maintain target site counts (rather than sequences), accounting for the possibility of target site turnover. It finally integrates this measure with several simpler ones using a logistic regression predictor. MirAncesTar improves the accuracy of existing MTG predictors by 26% to 157%. Source code and prediction results for human miRNAs, as well as supporting evolutionary data are available at http://cs.mcgill.ca/∼blanchem/mirancestar.
INTRODUCTION
MicroRNAs (miRNAs) form a class of evolutionarily conserved non-coding single-stranded RNA molecules involved in the regulation of gene expression by translational repression and mRNA destabilization (1–4). They are involved in the regulation of most animal and plant physiological processes (5–7), are implicated in many human diseases (8–10), and represent promising therapeutic applications (6,11).
Unlike in plants, where the gene silencing requires a near-perfect complementarity between the miRNA and its mRNA target site, the repression of mRNA expression in animals is determined in part by the complementarity of a short region of the miRNA, called the seed. The seed is usually located between positions 2 to 7 of the miRNA, but variations exist (12) and non-canonical sites are common (13). MiRNA target binding sites (MTBS) are generally located in the 3′ untranslated region (3′ UTR) of genes, but also, in a lower proportion, in their 5′ UTR and open reading frame (14). MiRNAs produced from a single locus have the potential to silence a large number of genes (henceforth called its miRNA target genes (MTG)), and silenced genes are often targeted by more than one miRNA (15).
Experimental identification of miRNA target genes involves techniques such as gene expression analysis, using expression of ectopic miRNAs followed by the quantification of remaining non-degraded target mRNA on a genome-wide scale with microarrays or RNA-seq (16), as well as approaches that directly identify interactions between mRNAs and proteins such as argonaute, including AGO2-PAR-CLIP (17). Experimentally identified miRNA target genes can be retrieved from databases such as MirTarBase (18), TarBase (19) and miRWalk (20). Importantly, these databases provide information about the strength of the experimental evidence in support of each target. Despite such databases already containing more than 300 000 human miRNA-target interactions, this is far from a complete repertoire. Indeed, the number of experiments required to identify all MTGs of all miRNAs, in all tissues, conditions and species of interest remains impractical. Therefore, computational methods to predict MTGs continue to be necessary.
Over the last few years, many MTG prediction tools have been developed and applied to various species. A first set of approaches, including miRanda (21) and PicTar (22), focused on identifying thermodynamically stable interaction sites between miRNAs and putative target genes. Later, various rule-based approaches, such as PITA (23), or machine learning approaches, such as MirTarget2 (miRDB) (24,25) and TargetMiner (26), were proposed to integrate miRNA–mRNA duplex structural information with other types of features, such as target site accessibility, A/U content or target-site abundance, in order to improve prediction accuracy (27).
Although these approaches have grown increasingly accurate over the past few years, and despite significant efforts, existing programs continue to produce high rates of false positives and false negatives (27). In an effort to alleviate this problem, several programs, including mirMark (28), Diana-microT (29) and TargetScan (30), have proposed to use inter-species sequence conservation as an indication of functional binding. MirMark considers as part of its input cross-species sequence conservation scores from PhastCons (31), and TargetScan makes direct use of UTR sequence alignments to measure conservation on each branch of a calculated phylogenetic tree.
The underlying principle of using interspecies conservation is that functional miRNA target sites are important to the appropriate regulation of a gene's expression, so mutations that would disrupt binding are generally deleterious and over time more mutations should accumulate outside target sites than within them. However, concerns about the use of site conservation criteria have been raised by Farh et al. (32) and Xu et al. (33), who observed that a large fraction of MTBS is not highly conserved among mammals. Applying strict requirements of sequence conservation thus results in an increased false-negative rate. Nevertheless, more than 60% of human protein-coding genes are under selective pressure to maintain pairing to miRNAs (34) that explains in part why many mammalian miRNA target sites are conserved above background levels (35).
The failure of conservation-based approaches to identify certain MTBS is partly due to an evolutionary process called binding site turnover (36) (Note that this concept is unrelated to that of miRNA turnover, which describes a change in miRNA expression due to its own degradation (37)). Consider a gene that was targeted by a given miRNA M at binding site A at some point in the past. Because MTBS are short, random mutations can easily create a new target site B for M in the vicinity of existing ones. Since MTBS are generally not dependant on their exact position in the UTRs of regulated genes, site B may be as potent as site A (provided B's position is in an accessible portion of the folded mRNA), thus reducing the selective pressure to maintain both. Mutations that would abrogate the function of A would thus not be deleterious. If such a mutation happens, the only functional site remaining is B and it then becomes under strong selection. This is called a turnover event, where although the target gene has continuously been targeted by M over evolutionary time, the position of the functional binding site has changed. Interspecies comparison would reveal that the sequence of neither the old nor the new site is particularly conserved, because both have been evolving neutrally for some time. This phenomenon is well characterized for transcription factor binding sites (38–40) and taking it into consideration has been shown to improve the accuracy of binding predictions (41). For miRNAs, target site turnover has been observed in cases where a target gene has multiple target sites for the same miRNA, a situation called cooperative targeting that allows MTBS to be lost and gained over time, as long as one or more remain present in the same target (42). Simkin et al. (43) have recently exhibited several cases of miRNA target site turnover within primates.
In this paper, we introduce MirAncesTar, an approach to improve the miRNA target gene predictions made by existing tools by making use of interspecies comparison while taking into account MTBS turnover. MirAncesTar uses computationally reconstructed ancestral mRNA sequences, rather than relying on pure conservation scores such as phastCons or PhyloP (31,44,45). Our approach is not a predictor in itself, but rather an accuracy booster that can be applied to any existing predictor. Applied to three of the most commonly used MTBS predictors, MirAncesTar results in a large improvement in accuracy and compares favorably with three of the recent MTG predictors making use of sequence conservation, mirMark (28), Diana-microT (29) and TargetScan (30).
MATERIALS AND METHODS
Data sets
Human miRNAs were retrieved from miRbase v20 (46,47), for a total of 2580 mature miRNAs. Experimentally validated miRNA targets (called known targets in this paper) were downloaded from miRTarBase version 6.0 (18) that contains a total of 3242 19 interactions between 2619 human miRNAs and 12 738 target genes, including 7439 strong evidence interactions. Of those, three subsets of miRNAs were considered: (i) M100 is a set of 100 miRNAs that had at least 200 known targets in the union of miRTarBase (release 5.0) and mirWalk (version 1) (Supplementary Table S1); (ii) M396 is a set of 396 miRNAs that had at least 200 known targets in the most recent version of miRTarBase 6.0 (Supplementary Table S2); (iii) M308
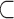 M396, a set of 308 miRNAs for which target predictions are available from both TargetScan and Diana-microT. The number of known targets (based on miRTarBase 6.0) used for the training and evaluation varies from 47 388 miRNA-target gene pairs for M100 to 150 892 pairs for M396.
M396, a set of 308 miRNAs for which target predictions are available from both TargetScan and Diana-microT. The number of known targets (based on miRTarBase 6.0) used for the training and evaluation varies from 47 388 miRNA-target gene pairs for M100 to 150 892 pairs for M396.
Human 5′ and 3′ UTRs sequences of human protein-coding genes were retrieved from the UCSC genome browser (build GRCh37/hg19, RefSeq genes annotation). PhastCons conservation scores (31) and conserved regions based on a 100-way multiple sequence alignment (48) were also retrieved from the UCSC genome browser.
Human 5′ and 3′ UTRs sequences of human protein-coding genes were retrieved from the UCSC genome browser (build GRCh37/hg19, RefSeq genes annotation). PhastCons conservation scores (31) and conserved regions based on a 100-way multiple sequence alignment (48) were also retrieved from the UCSC genome browser.
Target gene predictors
MTGs predictors were selected based on their availability and running time. We considered five target gene predictors:
MiRanda (August 2010 version; (21)), which identifies putative targets by sequence alignment and ranks them based on thermodynamic stability. Default options.
RNAhybrid (49), which determines the most stable hybridization site based on energy parameters from Mathews et al. (50), with length restrictions established for bulges and internal loops (49). Default options, except for target length option (-m 1 000 000), a P-value threshold (-P 0.1) and the appropriate species selection (-s 3utr_human).
MirMark (version 1.0; (28)), a machine learning based method using more than 700 features describing the interactions between a miRNA and a UTR, such as target site availability, structure and sequence features and PhastCons46way conservation data. Default options.
TargetScan (30), which predicts miRNA target genes by searching for the presence of 6 to 8mer sites that match the seed region of a given miRNA and make use of species alignment to locate conserved sites. We did not run this tool ourselves but instead downloaded its predictions from targetscan.org, release 7.0, august 2015. The sum of the context++ scores of conserved and non-conserved sites was considered as target score.
Diana-microT v4 (29), trained on miRbase v18, is based on binding and conservation features identified in high-throughput experimental data, and calculated for each miRNA and each miRNA recognition elements responsible for the interaction with a target gene.
For each tool, we obtained a ranked list of putative targets for each miRNA, sorted in decreasing order of the sum of confidence scores of predicted target sites.
Ancestral reconstruction
Ancestral genomes were reconstructed with an improved local version of Ancestor 1.1 (51), a tool that uses a maximum likelihood approach based on an evolutionary model that takes in account insertions, deletions and substitutions. The reconstruction is computed from whole-genome multiple alignments available from UCSC genome browser (52) for 46 vertebrate species (including 35 mammals), which were built with the blastZ/Multiz pipeline (48,53) based on a previously published phylogenetic tree (54). 5′ UTR and 3′ UTR reconstructed ancestral sequences are available as supplementary data on our site. This produced a set of up to 69 extant or ancestral orthologous mammalian sequences per human gene, although for most genes, orthologs are missing in a small number of species (average number of orthologs/ancestors per gene: 65.3; 0.1% of genes have no orthologs outside primates).
Measuring evidence of selective pressure on predicted target site count
To identify targets for a given miRNA M, target site predictions are first obtained for each human 5′ and 3′ UTRs, their orthologs and ancestral sequences, using a given Single-Sequence Target Site Predictors (SSTSP). Consider branch (p,u) of the phylogenetic tree, where p is the parent of u. We first build an evolutionary null model of the count of predicted target sites for M, which aims at describing how this number may change along branch (p,u), assuming that the sequence under consideration is not a true target of M. In other words, we model the evolution of the count of false-positive predictions in UTRs. Let Xu denote the random variable corresponding to the number of sites at node u, and let 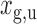 denote the observed number of target sites predicted for M in the sequence at node u for gene g. Let T(p,u)(a,b) = Pr[Xu
= b | Xp
= a] be the conditional probability of the sequence at u containing b sites given that the sequence at p contained a sites. T(p,u) is estimated on the basis that the vast majority of predicted target sites for M are false-positives, so that
denote the observed number of target sites predicted for M in the sequence at node u for gene g. Let T(p,u)(a,b) = Pr[Xu
= b | Xp
= a] be the conditional probability of the sequence at u containing b sites given that the sequence at p contained a sites. T(p,u) is estimated on the basis that the vast majority of predicted target sites for M are false-positives, so that
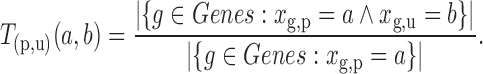 |
Figure 1 shows some of the 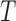 conditional distributions for branches of the tree that have different lengths. Let
conditional distributions for branches of the tree that have different lengths. Let 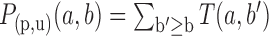 be the P-value associated to observing b sites at node u given that there were a sites at node p. The score of gene g as a putative target for miRNA M is obtained as
be the P-value associated to observing b sites at node u given that there were a sites at node p. The score of gene g as a putative target for miRNA M is obtained as
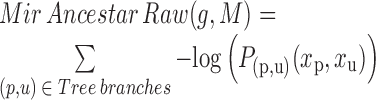 |
Figure 1.

Examples of the posterior probability of the count of predicted target sites for let7a-5p, for two different branches of the phylogenetic tree: (A) A short branch leading from the human-chimp ancestor; (B) A longer branch leading from the mouse-rate ancestor to mouse.
Normalized conservation score
To take into account the fact that longer UTRs have a higher probability to be targeted than shorter ones, we introduce a second scoring mechanism that calculates for each branch (p,u) a P-value conditioned on the (binned) length L(u) of the sequence at node u. Specifically,
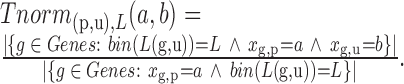 |
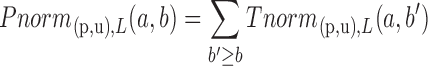 |
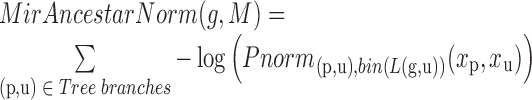 |
The length binning function bin( ) is chosen so that approximately 500 genes fall within each bin.
) is chosen so that approximately 500 genes fall within each bin.
Posterior probability normalized conservation score
While we found that the MirAncestarNorm score performed well, we realized that it over-penalizes genes with long UTRs, by intrinsically assuming that all genes are equally likely to be targets, irrespective of their UTR lengths. In reality, longer UTRs are generally more likely to be targets for any given miRNA. We thus introduced a last score called MirAncestarPost, which captures the posterior probability of a gene g being a target for M, given its length L(g) (in human) and its length-normalized score 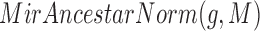 (abbreviated
(abbreviated 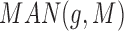 in the formula below). Let P(g,M) denote the event that g is a target of M.
in the formula below). Let P(g,M) denote the event that g is a target of M.
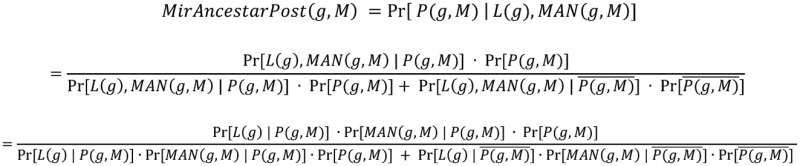
Where 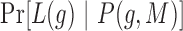 ,
, 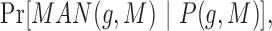
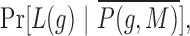 and
and 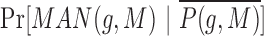 are represented using multinomial distributions and estimated from the known targets genes and non-target genes (separately in each cross-validation iteration, with binning of the MirAncestarNorm score and length).
are represented using multinomial distributions and estimated from the known targets genes and non-target genes (separately in each cross-validation iteration, with binning of the MirAncestarNorm score and length).
MirAncestar feature set and training
While the MirAncestarPost scoring approach is in itself competitive with existing SSTSPs, we are aware that it is not capturing some properties that could be useful for prediction. Thus, we elected to instead combine the three scoring schemes presented above (MirAncestarRaw, MirAncestarNorm and MirAncestarPost) with a set of seven other simpler measures:
UTRlength: The total length of the gene's UTRs in human.
TotalSitesCount: The total number of target sites predicted in the human gene, its orthologs and ancestors.
TotalSitesCountNorm: TotalSitesCount/UTRlength.
HumanTotalScore-Conserved: The sum of the SSTSP scores of all target sites predicted in the human sequence, limited to the highly conserved portions (defined by the PhastCons 46-way predictions).
HumanTotalScore-NonConserved: The sum of the SSTSP scores of all target sites predicted in the human sequence, outside of the highly conserved portions.
HumanMaxScore-Conserved: The maximum of the SSTSP scores of all target sites predicted in the human sequence, limited to the highly conserved portions.
HumanMaxScore-NonConserved: The maximum of the SSTSP scores of all target sites predicted in the human sequence, outside of the highly conserved portions.
These features were chosen because they are similar to those previously used by other tools (feature 1, 4–7 in the list above) or capture the total predicted site density across species (features 2, 3). The 10 features are combined using a logistic regression approach trained and evaluated using 10-fold cross-validation, using Weka (55). Because we work with unbalanced classes, we used a cost sensitive classifier, used to reweight training instances according to the total cost assigned to each class. This weighting method simulates stratification, avoiding downsampling the majority class and allowing taking advantage of the full available data. The cost matrix associated with the cost-sensitive classifier was set as follow: False-negatives were assigned a cost of 1, while false-positives were assigned a cost of |PositiveTrainingSet|/|NegativeTrainingSet|. The logistic regression parameters were learned based on a positive training set consisting of the set of known targets of M100 miRNAs, and the negative training set was the set of non-targets for the same miRNAs. For each SSTSP, a different set of logistic regression parameters were learned.
RESULTS
MirAncesTar is an approach that makes use of comparative genomics data to improve the accuracy of target gene predictions for a given miRNA by evaluating the conservation of the count of predicted target sites among mammalian orthologs and their ancestors. MirAncesTar exploits existing SSTSP such as miRanda (21) to identify candidate target sites in genes of the species under study (here, human), their orthologs (here, from 34 other mammals) and computationally reconstructed ancestral sequences. The method does not directly evaluate sequence conservation of target sites per se, but instead seeks evidence for selective pressure to maintain a certain number of target sites in gene's UTRs (irrespective of their position), thus allowing for target site turnover. The target site count conservation score is then combined with other simpler measures (UTR length, sum and maximum of site SSTSP scores inside and outside conserved regions, and total number of predicted sites (see Materials and Methods)), using a logistic regression predictor. Here, we report our evaluation of the accuracy of MirAncesTar compared to a variety of other existing tools, and investigate the factors that affect its performance. The complete set of target predictions for each of the 2580 human miRNAs in each isoform of the RefSeq human gene annotation is available at http://cs.mcgill.ca/∼blanchem/mirancestar.
MirAncesTar improves the accuracy of miRNA target gene prediction
For each of the 18 653 UTRs sequences of human genes annotated in RefSeq release 66 (after merging isoforms), we extracted orthologous mammalian sequences from the UCSC 46-way vertebrate whole-genome alignment (48,56), which yielded a maximum of 34 aligned mammalian orthologs. Ancestral sequences for each of the 34 internal nodes in the phylogenetic tree (Supplementary Figure S1) were inferred using a local version of Ancestors 1.1 (51,57), which was previously estimated to be able to infer ancestral mammalian sequences with an accuracy ranging from 85% to 98%, depending on the ancestral node.
We trained and tested (using 10-fold cross-validation) our various predictors on experimentally identified target sites of a set of 100 well-characterized miRNAs (see Methods). These 100 miRNAs have on average 474 known targets per miRNA. For each SSTSP P
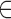 {miRanda, RNAhybrid, mirMark}, we evaluated the accuracy of MirAncesTarP, the MirAncesTar predictor based on the predictions obtained with P, and compared it to P itself when applied to the human sequences alone. For each miRNA and each predictor, we obtained the ranked list of predicted targets among RefSeq genes, sorted by the sum of confidence values (prediction score) of predicted targets. We then evaluated the proportion of all known targets captured among the top k predictions (recall), for k ranging from 1 to 1000 (Figure 2A–C). Although receiving-operator curves are a more classical way to evaluate predictors (presented in Supplementary Figure S2), we find that recall curves provide a more intuitive and practical evaluation of a predictor, by providing the answer to the question: if a researcher was to look at the top k predictions made by a given tool, what fraction of the known targets would be recovered?
{miRanda, RNAhybrid, mirMark}, we evaluated the accuracy of MirAncesTarP, the MirAncesTar predictor based on the predictions obtained with P, and compared it to P itself when applied to the human sequences alone. For each miRNA and each predictor, we obtained the ranked list of predicted targets among RefSeq genes, sorted by the sum of confidence values (prediction score) of predicted targets. We then evaluated the proportion of all known targets captured among the top k predictions (recall), for k ranging from 1 to 1000 (Figure 2A–C). Although receiving-operator curves are a more classical way to evaluate predictors (presented in Supplementary Figure S2), we find that recall curves provide a more intuitive and practical evaluation of a predictor, by providing the answer to the question: if a researcher was to look at the top k predictions made by a given tool, what fraction of the known targets would be recovered?
Figure 2.

Comparison of the recall (primary y-axis) and relative recall improvement (RRI, secondary y-axis, log-scale) of single-sequence target gene predictors and their corresponding MirAncesTar predictors. (A–C) Average (over 100 miRNAs) of the recall (percentage of known targets recovered) as a function of the number of sites being predicted (k). (A) miRanda; (B) RNAhybrid; (C) mirMark with and without PhastCons. (D–F) Recall (at k = 1000 predictions), for each of the 100 miRNAs, for each SSTSP (x-axis) and its corresponding MirAncesTar predictor (y-axis). MiRNAs for which the difference between the two recall values is statistically significant (P-value < 0.05 based on two-tailed Student's t-test) are shown in black.
Figure 2A compares the recall curves of miRanda and MirAncesTarmiRanda. The latter provides a notable improvement. For example, at k = 1000, MirAncesTarmiRanda has an average recall of 26.1%, compared to 18.4% for miRanda, a relative increase of 20.7%. The recall relative increase is actually much larger when limiting our attention to a smaller number of top predictions; e.g. at k = 100, MirAncesTarmiRanda improves the recall of miRanda by 67%. The improvements in recall are even more significant for RNAhybrid (Figure 2B) where MirAncesTar yields a 158% increase in recall (at k = 1000). MirMark is not a true single-sequence predictor because it uses as part of its input a measure of interspecies sequence conservation (PhastCons score (31)). As such, we were not able to use it directly to predict targets sites in orthologs and ancestors and instead modified it to not take sequence conservation into consideration. The resulting predictor (MirMark0) had a recall that was slightly worse than the original MirMark (Figure 2C), but MirAncesTarMirMark0 nonetheless succeeded at increasing the recall value 63% above that of MirMark (at k = 1000). (Because MirMark produces better results if we calculate the recall based on the maximum of the scores of the putative sites instead of their sum, we used the former method in this case). Overall, MirAncesTar produced significant improvements over all SSTSP we considered. The best recall curve was obtained using MirAncesTarmiRanda, which outperformed the other two MirAncesTar-based predictors, by 72–78% at k = 1000 and even more for smaller values of k.
Although MirAncesTar performs on average better than SSTSP predictors, its accuracy varies depending on the miRNA whose targets are being predicted. Figure 2D presents the recall obtained by MirAncesTarmiRanda (at k = 1000) for each miRNA, compared to that obtained with miRanda alone. MirAncesTarmiRanda improves the recall for 93 of the 100 miRNAs considered, including 39 where the improvement was statistically significant (in red in the figure; P ≤ 0.05; two-tailed Student's t-test). In one case, the recall is more than doubled. Figure 2E and F show the analogous results for RNAhybrid and MirMark. Improved recall values were obtained for 99% and 98% of miRNAs respectively, with 85% and 78% of these improvements being statistically significant.
TargetScan (30) and Diana-microT (29) are two of the most widely used miRNA target gene predictors that exploit interspecies comparisons to score putative target sites. For that reason, we could not use them as a SSTSP for MirAncesTar to be based off. Because both tools offers precomputed target predictions for a large set of miRNAs, we were able to expand our study to a larger set of 308 well-characterized miRNAs having at least 200 known targets and for which target gene predictions were available from both TargetScan and Diana-microT. To obtain recall curves, we again listed in decreasing order of scores the predicted targets provided by the tools. Figure 3 show that MirAncesTarMiranda obtains recall values that are significantly larger than those of Diana-microT (by ∼25–40%, depending on the value k). Recall values are comparable to those of TargetScan at k = 1000, but ∼10% better for k < 400. For a larger set of miR NAs (Supplementary Figure S3), MirAncesTarMiranda reports on average a higher recall rate than TargetScan for all values of k. We also evaluated each predictor on only the subset of MirTarBase targets interactions with strong experimental evidence (i.e. identified using reporter assays or Western blots), and found that all three predictors reached much higher recall values (at k = 1000, MirAncesTar: 43%; Diana-microT: 39%, TargetScan: 44%).
Figure 3.

Recall obtained by MirAncesTarMiranda, TargetScan and Diana-microT v4, averaged across 308 miRNAs.
To better understand the properties of different prediction methods, we compared the target predictions of miRanda, TargetScan, Diana-microT and MirAncesTarMiranda on the same set of 308 miRNAs. Interestingly, the set of target predictions made by the three tools have only moderate overlap (Figure 4). This suggests that the three tools are somewhat complementary. Genes predicted as targets by all four tools have large positive predictive value (PPV; fraction of positive predictions that are currently known to be correct), at 21.6%. Those predicted by three of the tools also have high PPV, ranging from 23.9% (MirAncestar+TargetScan+Diana-microT) to only 8.7% (TargetScan+Diana-microT+miRanda). Targets predicted by a single tool had lower PPV, ranging from 4.2% (miRanda alone) to 6% (TargetScan alone). This shows that significant specificity gains can be obtained by combining the three comparative genomics based predictors.
Figure 4.

Overlap among the predictions made with miRanda, MirAncesTarmiRanda, TargetScan and Diana-microT on 308 miRNAs, with k = 1000 for each tool and miRNA.
MirAncesTar exploits sequence conservation but is robust with respect to target site turnover
As seen in Figure 2D, the recall of MirAncesTarmiRanda (at k = 1000) varies quite widely between miRNAs, ranging from 7% to 57%. Two main reasons appear to explain this variability. The first is the ability of miRanda to correctly identify candidate target sites in human. Indeed, the correlation between the recall values of miRanda and MirAncesTarmiRanda is quite high (R2 = 0.84, Figure 2D); this is unsurprising, since MirAncesTarmiRanda builds off miRanda. Second, the extent to which MirAncesTarmiRanda improves the target recall (at k = 1000) compared to miRanda varies from a 2-fold increase for miR-92b-3p (from 19.2% to 38.4%) to no improvement for several miRNAs, and, in the case of let-7i-3p, miR-324-3p, 324–5p, 30b-3p, 373-3p, 30d-3p and 92a-1-5p, to a slight decrease in recall. We sought to understand the particular characteristics of a miRNA that may be associated with a gain or loss in accuracy with MirAncesTarmiRanda. We regressed the MirAncesTarmiRanda recall improvement against a number of miRNA properties (nucleotide content, average PhastCons UTR conservation scores of known targets, total predicted target sites count, etc.). The only significant interaction identified was with the average PhastCons conservation scores of known targets (P-value = 2.7 × 10−6), which suggests that, unsurprisingly, MirAncesTar is more effective for miRNAs whose target genes have a tendency to have more conserved UTRs. Those are often miRNAs that target transcription factors, especially those whose family is involved in regulation of embryonic development, such as let-7d (58), let-7e (59) and mir-124 (60), which are the three miRNAs for which MirAncesTar has the highest recall values.
One of the key innovations of MirAncesTar is its ability to tolerate MTBS turnover. This is supported by the fact that the UTRs correctly predicted as targets by MirAncesTar tend to have lower conservation levels (avg. PhastCons of 0.305) than those predicted by TargetScan, Diana-microT or MirMark (respectively avg. PhastCons score of 0.322, 0.419 and 0.507). Figure 5 illustrates the predicted target sites for hsa-let-7a-5p in the SMCR8 gene, a known target of that miRNA, which obtained a high prediction score (target ranked 39th out of 18 653 genes) from MirAncesTar but was scored poorly by other conservation-based tools (target ranking by mirMark: 4896th, TargetScan: 768th, Diana-microT: not in the top 7338 predictions available for this miRNA). Clearly, no specific target site predicted in human is conserved across all mammals. Interestingly, however, there is evidence of a turnover event in rodents (mouse, rat and kangaroo-rat), where a site that was otherwise conserved in most mammals was shifted by ∼600 bp. Overall, the number of predicted sites in extant ancestral sequences (shown on the phylogenetic tree in the figure) is remarkably constant, which is why this target is assigned a high score by MirAncesTar.
Figure 5.

Example of putative target site turnover for hsa-let-7a-5p in the SMCR8 gene. Putative target sites predicted by miRanda in each species are marked. The number of predicted target sites in each species and each computationally reconstructed ancestral sequence is shown on the nodes of the species tree. The position of sites for non-human species is converted to that of its human orthologous position through the multiple sequence alignment.
Contribution of the different features used by MirAncesTar
MirAncesTar is a logistic regression predictor where each putative target is represented using 10 features that capture in different ways the number of predicted target sites in the species of interest (human) and/or in its orthologs and ancestors (see Materials and Methods). It is instructive to consider how each of these features contributes to the overall accuracy of the predictor. Supplementary Figure S3 shows the recall curves obtained for each of the 10 features when used individually as predictor, for SSTSP = miRanda. By far the most informative feature is MirAncesTarPost, a score that captures evidence of selective pressure to maintain the number of candidate target sites during the evolution of the putative target. In itself, it is competitive with TargetScan and outperforms the three SSTSP used in this study. Interestingly, the second most predictive feature is the number of sites predicted by miRanda outside highly conserved portions of the UTR (PhastCons), which ranks better than the analogous number of target sites located within such conserved regions. This counterintuitive result is caused by the fact that most validated target UTRs contain zero conserved predicted targets.
DISCUSSION
We propose here a new algorithm that relies on ancestral sequence reconstruction to improve the accuracy of miRNA predictors in human, based on the idea that, despite the fact that UTRs are generally under negative selective pressure to maintain a given set of miRNA target sites, individual target sites are often subject to turnover. MirAncesTar builds off an evolutionary model that characterizes how the number of predicted targets in a neutrally evolving sequence changes over time, and seeks to identify UTRs that depart from that null model. It uses predictions made by existing SSTSPs, executed on UTRs of mammalian species and their ancestors, to identify genes that exhibit evidence of this type of selective pressure. It then learns how best to combine this measure of selective pressure with other simpler measures of target site content in the target species and its ancestors/orthologs. MirAncesTar significantly improved the overall accuracy of the three single-sequence target site predictors it was based off (miRanda, RNAhybrid and mirMark). For certain miRNAs, recall (at k = 1000) was more than doubled, while we found no miRNA for which recall was significantly decreased. The best overall accuracy was obtained using miRanda as SSTSP, although MirAncesTar produced its largest relative increase in accuracy for RNAhybrid (158% increase in recall at k = 1000). MirAncesTarmiRanda also outperforms existing sequence conservation based predictors Diana-microT and MirMark, and has slightly better performance than TargetScan. Notably, the accuracy gains obtained using MirAncesTarmiRanda appear to be largely due to its ability to tolerate target site turnover. Not all miRNAs benefit equally from the application of MirAncesTarmiRanda. Those for which MirAncesTar results in the largest increase in recall are those (i) whose target sites are already relatively well predicted by miRanda, and (ii) whose known targets tend to exhibit elevated levels of sequence conservation, such as miRNAs whose function is to regulate cell differentiation or organismal development.
An important benefit of MirAncesTar is that it can be used with any existing single-sequence target site predictor, and with the three predictors considered here, it results in significant gains in accuracy. By decoupling the individual sequence target site prediction task (performed by miRanda, RNAhydbrid, mirMark or other tools) from the evaluation of selective pressure on target site count (performed by MirAncesTar), we obtain an approach that will age well because it will benefit from future improvements in single sequence target site predictors (e.g. improved consideration of target site accessibility, non-canonical sites, etc.).
Although the overall recall of TargetScan and MirAncesTar are similar, the properties of predicted targets are quite different. Part of the explanation lies in how UTR length affects prediction accuracy. The recall of TargetScan is almost independent of UTR length: short targets (<500 bp) are recovered with the same recall as long ones (>5000 bp) (Supplementary Figure S4A). Conversely, the recall of MirAncesTar increases with target length, from only 3% for short UTRs to more than 50% for long ones. This is due to the fact that evidence of selective pressure on target site counts is easier to detect for target genes that contain a relatively large number of predicted sites. On the contrary, the precision (positive predictive value) of MirAncesTar is largely independent of target length: in other words, a gene that is predicted to be a target by MirAncesTar has approximately 10% probability of being a known target, irrespective of its length (Supplementary Figure S4B). Instead, the precision of TargetScan is length-dependent, ranging from only 6% for genes with very short UTRs to more than 15% for genes with relatively long UTRs. The predictions made by Diana-microT show an intermediate effect.
A similar analysis is instructive to highlight the effect of UTR sequence conservation on precision and recall. Unsurprisingly, the precision of each method improves with sequence conservation (average UTR PhastCons score) (Supplementary Figure S4C). However, large differences are observed in terms of recall (Supplementary Figure S4D): while both TargetScan and MirAncesTar recover 20–30% of known targets irrespective of their sequence conservation, Diana-microT has recall values that range from very poor (6%) for weakly conserved UTRs to very high (>40%) for highly conserved ones.
These differences have important consequences on the interpretation of the predictions made by these tools. On one hand, the length bias of MirAncesTar predictions, and the conservation bias of Diana-microT, can induce artificial functional enrichment (e.g. for a gene ontology enrichment analysis) among predicted targets. On the other hand, investigators interested in validating experimentally predicted targets should expect a length-dependent success rate if they base their study on TargetScan, but not so with MirAncesTar.
Several possible directions may prove fruitful to explore in order to further improve the accuracy of MirAncesTar. First, in its present version, the position of predicted sites is not taken into consideration; only the total count matters. While this conveniently allows for target site turnover, it could be that an approach that would be semi position-specific would have some benefits. One could, for example, consider a model where changes in target site position are allowed but penalized. Second, it may prove beneficial to perform single-sequence target site predictions in ancestral and orthologous sequences using as input the miRNA sequence from the very same species, rather than the human sequence, as was done here, in order to account for possible changes in the miRNA sequence itself. However, our initial assessment of this idea showed that miRNAs where mutations altered binding affinities were rare within mammals, probably because they would result in broad changes in the target repertoire, which would be strongly selected against. Furthermore, in the rare cases where we observed such changes, targets appeared to be under low selective pressure, thus reducing the impact of such a generalization. Third, it would be interesting to investigate the use of an approach similar to that proposed here in order to transfer target gene predictions from a species with rich experimental data (e.g. human) to less well studied species (most other mammals). Finally, improvements may be obtained by considering more sophisticated machine-learning predictors to replace our logistic regression classifier, or by considering additional sets of features. In particular, one may attempt to predict target genes based on the target site predictions of more than one SSTSP, although this would come at the expense of additional running time.
Finally, we note that although our focus here was on predicting target genes for human miRNAs, it should be equally powerful in other mammalian species (provided a sufficiently large number of known miRNA target sites are available for the training). MirAncesTar should also be applicable to other groups of species where sufficiently many closely related taxa are sequenced, such as fruit flies (61) or crucifers (62), although the accuracy of ancestral sequence reconstruction may not be as high for these lineages.
In conclusion, this paper is a striking example of a prediction task that can be achieved more accurately through a careful analysis of not only a human sequence and its orthologs, but also of computationally reconstructed ancestral sequences. Tracing the evolution of a region across the mammalian phylogeny significantly eases the detection of compensatory events such as target site turnover, by helping resolve the timing of these events. Did the loss of a particular target site precede or follow the creation of another one nearby? The answer to this question lies in the analysis of ancestral sequences, and is crucial for detecting evidence of selective pressure. We note that this concept is quite general and could quite easily be applied to other sequence-based prediction tasks. As the number of species whose genome get sequenced increases (63), so will the power of this family of approaches.
Acknowledgments
This work was funded in part by a NSERC Discovery grant to MB, and by a FRQNT scholarship to M.L. The authors would like to thank the Clumeq (Supercomputer Consortium Laval UQAM McGill and Eastern Quebec) for the access to the clusters Colosse and Guillimin, and the LICEF research center for their access to the ERASME cluster.
Footnotes
Present address: School of Computer Science and McGill Centre for Bioinformatics, McGill University, Montreal, Quebec H3A2B2, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NSERC Discovery [to M.B.]; FRQNT scholarship [to M.L.]. Funding for open access charge: NSERC Discovery grant [to M.B.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Ambros V. A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell. 1989;57:49–57. doi: 10.1016/0092-8674(89)90171-2. [DOI] [PubMed] [Google Scholar]
- 2.Ruvkun G. Molecular biology: glimpses of a tiny RNA world. Science. 2001;294:797–799. doi: 10.1126/science.1066315. [DOI] [PubMed] [Google Scholar]
- 3.Swami M. Small RNAS: An epigenetic silencing influence. Nat. Rev. Genet. 2010;11:172–173. doi: 10.1038/nrg2755. [DOI] [PubMed] [Google Scholar]
- 4.Kane N.M., Thrasher A.J., Angelini G.D., Emanueli C. Concise review: MicroRNAs as modulators of stem cells and angiogenesis. Stem Cells. 2014;32:1059–1066. doi: 10.1002/stem.1629. [DOI] [PubMed] [Google Scholar]
- 5.Osman A. MicroRNAs in health and disease–basic science and clinical applications. Clin. Lab. 2012;58:393–402. [PubMed] [Google Scholar]
- 6.Lawrie C.H. MicroRNAs in Medicine. Hoboken: John Wiley & Sons; 2013. [Google Scholar]
- 7.Teruel-Montoya R., Kong X., Abraham S., Ma L., Kunapuli S.P., Holinstat M., Shaw C.A., McKenzie S.E., Edelstein L.C., Bray P.F. MicroRNA expression differences in human hematopoietic cell lineages enable regulated transgene expression. PLoS One. 2014;9:e102259. doi: 10.1371/journal.pone.0102259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper T.A., Wan L., Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dangwal S., Bang C., Thum T. Novel techniques and targets in cardiovascular microRNA research. Cardiovasc. Res. 2012;93:545–554. doi: 10.1093/cvr/cvr297. [DOI] [PubMed] [Google Scholar]
- 10.Goodall E.F., Heath P.R., Bandmann O., Kirby J., Shaw P.J. Neuronal dark matter: the emerging role of microRNAs in neurodegeneration. Front. Cell. Neurosci. 2013;7:178. doi: 10.3389/fncel.2013.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammond S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hausser J., Zavolan M. Identification and consequences of miRNA-target interactions - beyond repression of gene expression. Nat. Rev. Genet. 2014;15:599–612. doi: 10.1038/nrg3765. [DOI] [PubMed] [Google Scholar]
- 14.Lytle J.R., Yario T.A., Steitz J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9667–9672. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gennarino V.A., D'Angelo G., Dharmalingam G., Fernandez S., Russolillo G., Sanges R., Mutarelli M., Belcastro V., Ballabio A., Verde P., et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res. 2012;22:1163–1172. doi: 10.1101/gr.130435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomson D.W., Bracken C.P., Goodall G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39:6845–6853. doi: 10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farazi T.A., Ten Hoeve J.J., Brown M., Mihailovic A., Horlings H.M., van de Vijver M.J., Tuschl T., Wessels L.F. Identification of distinct miRNA target regulation between breast cancer molecular subtypes using AGO2-PAR-CLIP and patient datasets. Genome Biol. 2014;15:R9. doi: 10.1186/gb-2014-15-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou C.-H., Chang N.-W., Shrestha S., Hsu S.-D., Lin Y.-L., Lee W.-H., Yang C.-D., Hong H.-C., Wei T.-Y., Tu S.-J., et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2015;44:D239–D247. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vlachos I.S., Paraskevopoulou M.D., Karagkouni D., Georgakilas G., Vergoulis T., Kanellos I., Anastasopoulos I.-L., Maniou S., Karathanou K., Kalfakakou D., et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43:D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dweep H., Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat. Methods. 2015;12:697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 21.Enright A.J., John B., Gaul U., Tuschl T., Sander C., Marks D.S. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krek A., Grün D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M., et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 23.Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 24.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X., El Naqa I.M. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- 26.Bandyopadhyay S., Mitra R. TargetMiner: MicroRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics. 2009;25:2625–2631. doi: 10.1093/bioinformatics/btp503. [DOI] [PubMed] [Google Scholar]
- 27.Zheng H., Fu R., Wang J.-T., Liu Q., Chen H., Jiang S.-W. Advances in the techniques for the Prediction of microRNA Targets. Int. J. Mol. Sci. 2013;14:8179–8187. doi: 10.3390/ijms14048179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menor M., Ching T., Zhu X., Garmire D., Garmire L.X. mirMark: a site-level and UTR-level classifier for miRNA target prediction. Genome Biol. 2014;15:500. doi: 10.1186/s13059-014-0500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maragkakis M., Vergoulis T., Alexiou P., Reczko M., Plomaritou K., Gousis M., Kourtis K., Koziris N., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT Web server upgrade supports Fly and Worm miRNA target prediction and bibliographic miRNA to disease association. Nucleic Acids Res. 2011;39:1–4. doi: 10.1093/nar/gkr294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwal V., Bell G.W., Nam J.-W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farh K.K.-H., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 33.Xu J., Zhang R., Shen Y., Liu G., Lu X., Wu C.-I. The evolution of evolvability in microRNA target sites in vertebrates. Genome Res. 2013;23:1810–1816. doi: 10.1101/gr.148916.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedman R.C., Farh K.K.-H.H., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie X., Lu J., Kulbokas E.J., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkataram S., Fay J.C. Is transcription factor binding site turnover a sufficient explanation for cis-regulatory sequence divergence? Genome Biol. Evol. 2010;2:851–858. doi: 10.1093/gbe/evq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogers K., Chen X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell. 2013;25:2383–2399. doi: 10.1105/tpc.113.113159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moses A.M., Pollard D.A., Nix D.A., Iyer V.N., Li X.-Y., Biggin M.D., Eisen M.B. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput. Biol. 2006;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S., et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dermitzakis E.T., Clark A.G. Evolution of transcription factor binding sites in mammalian gene regulatory regions: Conservation and turnover. Mol. Biol. Evol. 2002;19:1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- 41.Blanchette M. Exploiting ancestral mammalian genomes for the prediction of human transcription factor binding sites. BMC Bioinformatics. 2012;13(Suppl. 1):S2. doi: 10.1186/1471-2105-13-S19-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saetrom P., Heale B.S.E., Snøve O., Aagaard L., Alluin J., Rossi J.J. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 2007;35:2333–2342. doi: 10.1093/nar/gkm133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simkin A.T., Bailey J.A., Gao F.-B., Jensen J.D. Inferring the evolutionary history of primate microRNA binding sites: overcoming motif counting biases. Mol. Biol. Evol. 2014;31:1894–1901. doi: 10.1093/molbev/msu129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siepel A., Haussler D. Statistical Methods in Molecular Evolution. Springer; 2005. Phylogenetic Hidden Markov Models; pp. 325–351. [Google Scholar]
- 46.Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffiths-Jones S., Saini H.K., Van Dongen S., Enright A.J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blanchette M., Kent W.J., Riemer C., Elnitski L., Smith A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krüger J., Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–W454. doi: 10.1093/nar/gkl243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathews D.H., Sabina J., Zuker M., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 51.Diallo A.B., Makarenkov V., Blanchette M. Ancestors 1.0: a web server for ancestral sequence reconstruction. Bioinformatics. 2010;26:130–131. doi: 10.1093/bioinformatics/btp600. [DOI] [PubMed] [Google Scholar]
- 52.Miller W., Rosenbloom K., Hardison R.C., Hou M., Taylor J., Raney B., Burhans R., King D.C., Baertsch R., Blankenberg D., et al. 28-way vertebrate alignment and conservation track in the UCSC genome browser. Genome Res. 2007;17:1797–1808. doi: 10.1101/gr.6761107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy W.J., Eizirik E., O'Brien S.J., Madsen O., Scally M., Douady C.J., Teeling E., Ryder O.A., Stanhope M.J., de Jong W.W., et al. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science. 2001;294:2348–2351. doi: 10.1126/science.1067179. [DOI] [PubMed] [Google Scholar]
- 55.Hall M., Frank E., Holmes G., Pfahringer B., Reutemann P., Witten I.H. The WEKA data mining software: an update. ACM SIGKDD Explorations Newsl. 2009;11:10–18. [Google Scholar]
- 56.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blanchette M., Diallo A.B., Green E.D., Miller W., Haussler D. Computational reconstruction of ancestral DNA sequences. Methods Mol. Biol. 2008;422:171–184. doi: 10.1007/978-1-59745-581-7_11. [DOI] [PubMed] [Google Scholar]
- 58.Wong S.S.Y., Ritner C., Ramachandran S., Aurigui J., Pitt C., Chandra P., Ling V.B., Yabut O., Bernstein H.S. miR-125b promotes early germ layer specification through Lin28/let-7d and preferential differentiation of mesoderm in human embryonic stem cells. PLoS One. 2012;7:e36121. doi: 10.1371/journal.pone.0036121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colas A.R., McKeithan W.L., Cunningham T.J., Bushway P.J., Garmire L.X., Duester G., Subramaniam S., Mercola M. Whole-genome microRNA screening identifies let-7 and mir-18 as regulators of germ layer formation during early embryogenesis. Genes Dev. 2012;26:2567–2579. doi: 10.1101/gad.200758.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee M.R., Kim J.S., Kim K.S. MiR-124a is important for migratory cell fate transition during gastrulation of human embryonic stem cells. Stem Cells. 2010;28:1550–1559. doi: 10.1002/stem.490. [DOI] [PubMed] [Google Scholar]
- 61.Clark A.G., Eisen M.B., Smith D.R., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 62.Haudry A., Platts A.E., Vello E., Hoen D.R., Leclercq M., Williamson R.J., Forczek E., Joly-Lopez Z., Steffen J.G., Hazzouri K.M., et al. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet. 2013;45:891–898. doi: 10.1038/ng.2684. [DOI] [PubMed] [Google Scholar]
- 63.Koepfli K., Paten B., O'Brien S.J., Genome 10K Community of Scientists The Genome 10K Project: a way forward. Annu. Rev. Anim. Biosci. 2015;3:57–111. doi: 10.1146/annurev-animal-090414-014900. [DOI] [PMC free article] [PubMed] [Google Scholar]