


Abstract
The accumulation of somatic genomic alterations that enables cells to gradually acquire growth advantage contributes to tumor development. This has the important implication of the widespread existence of cooperative genomic alterations in the accumulation process. Here, we proposed a computational method HCOC that simultaneously consider genetic context and downstream functional effects on cancer hallmarks to uncover somatic cooperative events in human cancers. Applying our method to 12 TCGA cancer types, we totally identified 1199 cooperative events with high heterogeneity across human cancers, and then constructed a pan-cancer cooperative alteration network. These cooperative events are associated with genomic alterations of some high-confident cancer drivers, and can trigger the dysfunction of hallmark associated pathways in a co-defect way rather than single alterations. We found that these cooperative events can be used to produce a prognostic classification that can provide complementary information with tissue-of-origin. In a further case study of glioblastoma, using 23 cooperative events identified, we stratified patients into molecularly relevant subtypes with a prognostic significance independent of the Glioma-CpG Island Methylator Phenotype (GCIMP). In summary, our method can be effectively used to discover cancer-driving cooperative events that can be valuable clinical markers for patient stratification.
INTRODUCTION
Cancer is a complex and multistep disease derived from a process analogous to Darwinian evolution (1). During the process, a series of successive genetic alterations provide cells a significant fitness advantage and in turn disrupt the homeostasis of cells. Such an accumulation process can promote clonal expansion of cancer cells, resulting in multiple tumor entities linked with distinctive histological patterns and different clinical behaviors (2). Importantly, these accumulating genetic alterations do not occur at random, but mutually depend on each other (3,4). A number of co-occurring events were frequently observed in different types of cancers by using high-throughput microarray and sequencing data (5,6). And combinational alterations between cancer driver genes have already been confirmed in vitro and vivo. For example, for mice with oncogenic Kras mutation, further mutation of Scrib was more likely to develop higher grade lesions (7). What's more, some cooperative events have been reported to be clinically relevant. The 1p/19q co-deletion was associated with histological type, improved prognosis and chemosensitivity in gliomas (8,9). Thus, it is necessary to comprehensively identify and analyze cooperative events of genetic alterations so as to enhance our understanding of tumorigenesis and improve treatment strategies for precision medicine.
Many previous studies identified co-occurring pairs of somatic events in cancer genomes through classical statistical tests (6,10–12). Using Fisher's exact test, Cui et al. constructed a network of co-occurring and anti-co-occurring cancer gene mutations (13). Based hypergeometric test and Gaussian mixture model, Yeang exploited combinations of mutations from Catalog of Somatic Mutations in Cancer (COSMIC), and revealed co-occurrence and mutual exclusivity across and within pathways (5). Wang et al. proposed a stratified FDR control approach which combined with the traditional statistical test for identifying co-mutated gene pairs in cancer genome (14). Furthermore, by considering biological pathways, some studies focused on co-occurring pairs between/within pathways (15,16).
However, statistical co-occurrence is insufficient to indicate key ‘driver’ roles of these cooperation of somatic events in cancer. Several evidences support that genetic alterations are highly associated with each other. For example, a set of genes exhibit a mutually exclusive pattern because of functional redundancy or synthetic lethality (17,18). While some genes tend to co-occur, due to complementary function or epistasis interaction (15,19). Moreover, simulation trials based on the life history trade-off theory demonstrated that fitness provided by genetic alteration depended on prior mutational history (3,4). These high associations reflect that clonal evolution in cancer favors specific combinations of genetic alterations and the series of genetic alterations driving the development of tumors are context-dependent. The selective sweep process of cancer genome evolution indicated that co-occurring events are highly dependent on specific genetic contexts (20). In addition, gene cooperation in tumorigenesis usually contributes to various cancer hallmark-related functions. For example, cooperation of Myc and Kras in transgenic mouse model of breast cancer could increase proliferation and prevent cell apoptosis (21). Medulloblastomas with combined TP53 and MYC defects were enriched in relapse cases and showed locally aggressive behavior (22). It is thus reasonable to suppose that effective cooperative events should be associated with specific genomic patterns and have the functional effects on cancer hallmarks.
At present, large-scale genomic projects such as The Cancer Genome Atlas (TCGA) (23) are providing unprecedented cancer genomic data, which allows us to comprehensively explore and investigate somatic cooperative events in cancer. Here, we proposed a new and integrated method, HCOC (hallmark associated cooperation under specific genetic context), to identify cooperative events in human cancers. HCOC used ‘omics’ data sets to identify cooperative events that are associated with specific genetic contexts and contribute to cancer hallmark-associated pathways. The method consists of two components: (i) using the idea of motif matching from mapping of transcription factor binding sites to characterize the recurrent gene-dependent genomic patterns of candidate cooperative pairs (24); (ii) characterizing downstream functional effects of candidate pairs on cancer hallmarks.
Applying HCOC to somatic mutation and copy number alteration (CNA) data sets from 12 TCGA cancer types referring to >3000 samples, we totally identified 1199 cooperative events and constructed cooperative networks within and across cancers. The gene cooperative events displayed significant associations with some key drivers and could severely disturb cancer hallmark pathways. Besides, the pan-cancer cooperative network provided complementary information with tissue-of-origin to predict clinical outcome. As a case study, cooperative events in glioblastoma (GBM) were useful to characterize a clinically effective prognostic stratification of GBM patients, suggesting the clinical importance of cooperative events.
MATERIALS AND METHODS
Data source
We obtained copy number data (level 3), mutation data (level 2) and gene expression data (level 3) as well as clinical data of 3753 patients from 12 cancer types in TCGA. The detail information was shown in Supplementary Table S1. By assessing batch effects for the expression data and copy number data of 12 cancers, we observed that no data showed major effects (see Supplementary Methods for details).
Binary matrix of somatic genetic alteration
For log-transformed expression data from RNAseqV2 and RNAseq, genes with no expression in >20% of samples were filtered. For DNA copy number, we only retained high-level amplification and homozygous deletion discretized by GISTICv2. For mutations, silent mutations were discarded. Then, we removed hyper-altered patients as mentioned in (25), that is, the patients with >1000 genetic alterations were excluded, and 3049 patients were remained. For each cancer type, we integrated somatic copy number and mutation profiles to build a binary genetic alteration matrix.
HCOC (hallmark associated cooperation under specific genetic context)
Tumorigenesis is accompanied by a series of genetic alteration events. A single alteration is insufficient to make normal cells cancerous (26,27). These driver alterations are generally not independent. They can cooperate with each other and thus form a cooperative network, ultimately impairing normal cellular functions and allowing cells to acquire important cancer hallmarks (21,22). To identify cooperative events and build cooperative networks in human cancers, we proposed a computational method based on associations with specific genetic contexts and effects on cancer hallmarks by integrating multiple ‘omics’ data.
Identification of candidate cooperative gene pairs
To identifying candidate cooperative gene pairs, the genes satisfying the following requirements were remained: (i) genes with copy number alteration (CNA) have a dominant alteration type (amplification or deletion). In detail, for a given gene, let N be the number of patients with CNA. A and D represent the numbers of amplification and deletion, respectively (N = A + D). We modeled the number of the dominant alteration type as a binomial distribution with N trials with success probability p, and then inferred whether the dominant alteration type was over-presented by testing the null hypothesis H0: P = 0.5 against the alternative hypothesis H1: P > 0.5. Let L = |A – D|, then we sought to determine the rejection region L ≥ l where l is a given threshold. Since L = A – (N – A) = 2*A – N, we have
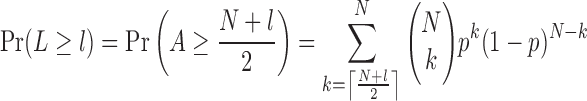 |
At a significant level α = 0.05, we calculated the least integer l under the null hypothesis. Only genes above the threshold l were kept and their dominant alteration types were recorded. For each of these genes, patients with the dominant alteration type were used, and those with the other type were regarded as noise. (ii) Genes with CNA should have concordant mRNA expression. We used a one-tailed Wilcoxon signed rank test. For a given gene with CNA, the gene expression in the amplification (or deletion) samples should be significantly higher (or lower) than wild-type samples. (iii) Genes with amplification, deletion or mutation in <3% of samples were discarded.
We enumerated all possible gene pairs from the gene set, and then selected those pairs with co-occurrence frequency >3%. Since a copy number event can refer to numerous genes (28), we filtered the gene pairs located within the same chromosome arm.
Identification of cooperative pairs with specific genetic contexts
Some important driver genes, at present, were found to contribute to the development and progression of cancer. They generally harbor high alteration frequencies and are crucial for decision making in the neoplastic transformation of various cell types. Different combinations of defects of these driver genes represent different ways of malignant transformation. We assumed that cooperative alterations are highly associated with specific alteration patterns of these driver genes when compared to wild-type and single-altered cases. To characterize the genomic patterns in each cancer type, we selected the high-confident cancer drivers according to the following criteria: (i) known cancer genes recorded in at least one public database (including OMIM (29), GAD (30) and CGC (31)); (ii) located in the wide peak regions identified by GISTICv2 (32) (for genes with CNA); (iii) recurrently mutated genes downloaded from http://www.tumorportal.org/ and identified by MutSigCV/MutSigCL/MutSigFN (33) (for genes with mutations); (iv) genes with alteration frequency >10% (5% for LAML). These driver genes were termed marker genes (Supplementary Table S2). If the overall alteration status of marker genes were similar within co-altered patients but significantly different from the others, the genomic pattern was regarded as a specific genetic context.
Inspired by the motif enrichment analysis of transcription factors (24), we utilized marker Position Frequency Matrix (mPFM) to characterize the genetic context. Let k denoted the total number of marker genes identified as described above, and {m1, m2, … mk} represents the marker gene sequence representing the alteration status of these marker genes in a specific cancer sample. For each candidate cooperative pair, to evaluate whether the gene pair is highly associated with a specific genetic context, we first extracted all co-altered patients. Then using the idea of leave-one-out, we left one patient in this co-altered group and used the others to build a mPFM. The mPFM model was constructed from a matrix, where each row represented the alteration status (alteration or wild-type) in a given sample and each column represented a marker gene. The final mPFM was a matrix which consists of frequencies of two possible alteration status of each marker gene (Supplementary Figure S1). Then a Log-Likelihood Ratio (LLR) scoring function was used to quantify the matching degree between the marker sequence in a particular sample and the mPFM model (Supplementary Figure S1).
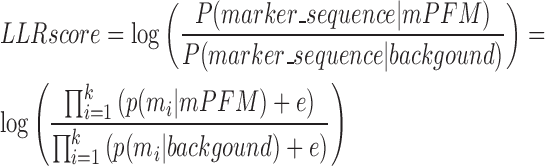 |
where marker_sequence was the alteration status of marker genes in a given sample, e was a constant (0.00001), p(mi|mPFM) represented the frequency of the alteration status of marker i in the mPFM model and p(mi|background) was the frequency in background PFM model. A high LLRscore indicated the given sample had a similar genomic alteration pattern of marker genes with the co-altered samples. Notably, when calculating the LLR score, we excluded the genes that (i) was included in the cooperative pair, or (ii) affected by CNA events and shared the same chromosome arm locations with one of the genes in the pair, from the corresponding marker sequence. We calculated the LLR scores for the left co-altered patient and the other wild-type and single-altered patients (Supplementary Figure S1). By employing the area under the ROC curve (AUC), we could assess the difference of the LLR scores between the left one co-altered patient and the other patients. After leaving the different co-altered patient out, an average AUC for each candidate cooperative pair was computed. By choosing a high cutoff of 0.75, cooperative pairs that were associated with specific genetic contexts were identified.
Identification of cooperative pairs with differential activities of hallmark pathways
To characterize whether cooperative pairs can affect cancer hallmarks, we developed a new strategy to identify pathways associated with cooperative alterations by combining expression and genomic alteration status. First, we identified biological pathways that could reflect each cancer hallmark based on sematic similarity of GO terms (Supplementary Figure S2 and Table S3). Second, activities of hallmark-associated pathways in each patient (Zsj) were assessed according to the function (34):
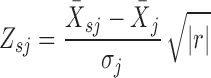 |
where |r| is the number of genes in the pathway s, and 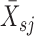 represents the mean expression level over the member genes in the pathway s in sample j.
represents the mean expression level over the member genes in the pathway s in sample j. 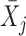 represents the mean expression level of all genes detected in sample j, and σj is the standard deviation of over all the genes in sample j.
represents the mean expression level of all genes detected in sample j, and σj is the standard deviation of over all the genes in sample j.
Next, we constructed a logistic regression model that related activities of each pathway to cooperative alteration status for determining whether activities of hallmark-associated pathway are significantly different between co-altered patients and the others. The significance of each regression model was calculated. We retained candidate cooperative pairs that markedly influenced at least one hallmark-associated pathway at a false discovery rate (FDR) of 0.25.
Ultimately, we considered candidate pairs to be cooperative if they were associated with specific genetic contexts and affected at least one cancer hallmark-associated pathway. More detailed information on the methods can be found in the Supplementary Methods.
Construction of cooperative network
The genes affected by a shared copy number event across cancer patients were combined into a ‘meta’ gene (35) (see Supplementary Methods, Supplementary Table S4). Then, for each cancer, a cooperative network was constructed by connecting all cooperative gene pairs identified above. The cooperative networks can reflect all cooperative events occurred in the corresponding cancer types.
RESULTS
The landscape of somatic cooperative alteration networks in human cancers
We developed a systematic method, HCOC, based on associations with specific genetic contexts and effects on cancer hallmarks to identify somatic cooperative events in human cancers (Figure 1). Briefly, given a pair of somatic alterations, we distinguished patients into two groups according to co-occurrence of these two aberrations, and assessed the difference in the genomic alteration patterns of some highly recurrent genes and the activities of cancer hallmark-associated pathways between the two groups. The pairs associated with specific genomic alteration patterns and significant effects on cancer hallmarks were identified as cooperative alterations (for details, see Methods and Supplementary Methods). Also, we developed an online webserver (available at http://biocc.hrbmu.edu.cn/HCOC/) for our method. It provides a user-friendly interface, allowing researchers to identify somatic cooperative events.
Figure 1.

The overview of HCOC.
Applying our method to 12 tumor types in TCGA referring to more than 3,000 samples, we identified numerous cooperative somatic alteration events (Supplementary Figure S3). The average number of cooperative events per cancer type was 108 (ranging from 1 to 622) (Supplementary Figure S4). Only two and one cooperative events were identified in LAML and BRCA, respectively. The LAML patients had a relatively stable genome (25), which results in much fewer candidate cooperative pairs (co-alteration > 3%) than other cancers. For BRCA, high heterogeneity may be a potential reason why only a few cooperative pairs were identified (36,37). There are few common cooperative events were shared by any two cancer types (Figure 2G). The cooperative alterations identified in each type of cancer were listed in Supplementary Table S5. We compared the results of HCOC with another three methods—Classical hypergeometric test, Gaussian mixture model (GMM) used in (5), CDCOCA proposed by Kumar et al. (38). Using the same pre-processing steps and thresholds as HCOC, we applied the three methods to identify recurrently co-occurring somatic events in 12 cancer types. As a result, we found that a total of 344 cooperative events that were missed by all of the three methods were uniquely identified by HCOC (Supplementary Table S12). Notably, among these cooperative events, some have been reported to have oncogenic roles in the malignant transformation of cells, such as TP53-MYC in UCEC and CDK4-TP53 in GBM (39,40). The comparison results suggested that our method HCOC provided new insights for into current research of somatic alteration combination.
Figure 2.

Networks of somatic cooperative alterations across 12 cancer types. (A-D) The distribution of the fraction of cancer genes (A), the average shortest distance in PPI network (B), the average expression correlation (C), average grail similarity rank (D) for random gene pairs in 1000 permutations. The red point indicates the average value of all cooperative events for each cancer, an empirical P value is calculated by permutation test. (E) Pan-cancer cooperative network merged from individual cancers. The different colors of edges represent different cancers. (F) Circos plot for all cooperative events in pan-cancer. The different colors of links represent different cancers. (G) The cooperative events in each cancer show few overlap. (H) The fraction of top genes ranked by the degree in pan-cancer network that are included in the CGC (red) and CGC/GAD/OMIM (blue). (I) The degree distribution of top 10 genes in pan-cancer network for each cancer and pan-cancer. (J) The cooperative partners of TP53 across 9 cancer types.
All cooperative events identified in each cancer type formed a cooperative alteration network, which could reflect the landscape of cooperative events in that cancer (Supplementary Figure S5). Some cooperative events have been confirmed in previous works. For example, in mouse model of high-grade astrocytoma (HGA) combined mutation of Pten, p53 and Rb1 significantly reduced the latency of tumor development and time to morbidity, compared to HGA induced by p53 and Pten (41), which was consistent with RB1-TP53 and RB1-PTEN identified in GBM. De novo AML patients with concurrent DNMT3A and FLT3 had a higher percentage of bone marrow blasts and extremely poor prognosis compared with single alterations (42,43). Cooperative events including TP53-ERBB2 (44) and PIK3CA-MYC (45) were also validated to be associated with tumorigenesis and clinical prognosis. Through assessing the overlap with known cancer genes recorded in CGC, we found that genes in cooperative events were enriched for cancer genes in almost all cancers (Figure 2A). Moreover, gene pairs in cooperative events were highly connected in protein-protein interaction (PPI) network (Figure 2B), more likely to be co-expressed (Figure 2C) and co-occurred in PubMed abstracts (Figure 2D), suggesting functional links of cooperative alterations (see Supplementary Methods).
In addition, we constructed a pan-cancer cooperative alteration network by merging the networks built in individual cancers (Figure 2E and F). The pan-cancer network following a power law distribution was composed of 535 genes, whose alterations covered 76% (2303/3049) of patients in 12 cancers. Genes with high degree showed a significant enrichment for known cancer-related genes (Figure 2H). In particular, all of the top ten genes with the highest degree were key genes in tumorigenesis (Figure 2I). TP53, for example, that has been widely reported to be dysregulated in various cancers (25) was identified in nine cancer types and showed the highest degree in the network, indicating a wide variety of cooperative partners. Interestingly, TP53 cooperated with completely different genes in different cancer types (Figure 2J). One reason for such substantial but heterogeneous cooperative partners was the extremely high alteration frequency of TP53 across cancers. Another possible explanation, from the aspect of cancer evolution, could be that TP53 mutation was an early clonal event in many cancers, with subsequently subclonal genomic changes selected by different tissue-specific environmental factors (46,47).
Dissecting specific genomic patterns and cancer hallmarks associated with cooperative events
It should be pointed out that the cooperative events identified using our method were associated with specific genomic patterns of some recurrent genes, thus allowing to characterize the links of cooperative pairs with these highly altered genes. Taking PTEN-KRAS identified in UCEC as an example, the patients with PTEN-KRAS co-alterations displayed a specific genomic pattern—increased mutation rates in ARID1A, RPL22 and MUC5B and decreased in FGFR2—relative to all patients (Figure 3A, P < 0.05, one-sided binomial test). PTEN and KRAS together with these recurrent genes having increased alteration frequencies appeared to form a cooperative module (Supplementary Figure S6). Indeed, PTEN and KRAS have been found to result in transformation of ovarian surface epithelial cells to low-grade adenocarcinomas (48), and PTEN could induce autocrine FGF signaling to promote tumorigenesis (49), thus, further alteration of FGFR2 was unnecessary, which was consistent with the mutual exclusive between PTEN-KRAS and FGFR2. Such specific genomic contexts thus suggested the high dependence of cooperative events on some highly recurrent genes. Subsequently, we examined all genomic patterns in different cancer types (Supplementary Figures S7 and S8) and found majority of the cooperative events significantly linking with at least one recurrent gene (Figure 3B and Supplementary Table S6). Notably, many recurrent genes, such as CDKN2A and PIK3CA, were associated with multiple cooperative events (Figure 3C) across cancers, implying that they were subjected to multiple selective pressures in tumor evolution.
Figure 3.

Characterization of genomic patterns and functional effects for gene cooperative events across 12 cancers. (A) The specific genomic pattern for PTEN-KRAS in UCEC. P value for frequency change of a marker gene is indicated by concentric circles (one concentric circle: P < 0.1, two: P < 0.05, three: P < 0.01, one-sided binomial test). (B) The summary of the number of marker genes associated with cooperative pairs (P < 0.1). (C) Marker genes associated with multiple gene cooperative events in each cancer. The number in each rectangle represents the count of associated cooperative pairs for corresponding marker gene. (D) The distribution of the average scaled activity of pathways affected by cooperative pairs in each cancer. The scaled activity of pathway s in sample j is calculated as (Zsj – Zs,min)/(Zs,max – Zs,min), where Zs,min and Zs,max represent the minimum and maximum activity scores of pathway s across all cancer samples, respectively. (E) The hallmark associated pathway activities of different sample groups for MLL-TP53 in LUAD. P value is calculated by one-sided Wilcoxon rank sum test. (F) CircleMap display of hallmark associated pathways, each track shows the fraction of cooperative events which affect a specific pathway in one cancer type. The outlayer Arabic and Roman numerals correspond to pathways and cancer hallmarks in (G), respectively. (G) The hallmark associated pathways affected by cooperative events. (H) Cooperation of PIK3CA-EIF3H in BRCA, RSRC1-CDKN2B in ESCA, TP53-TM2D2 in LUAD and TP53-MYC in UCEC are associated with elevated activity of nucleotide metabolism pathway.
In parallel, our method required that the cooperative alterations had remarkable effects on the activities of cancer hallmarks, thus enabling us to characterize cancer hallmark-associated pathways affected by cooperative events. We identified many pathways whose activities were significantly affected by the cooperative events in each cancer type (Figure 3G and Supplementary Table S7, and see Methods section for details). These hallmark-associated pathways in co-altered patients presented extreme activity relative to those in single-altered patients (Figure 3D), suggesting possible changes in phenotype. In LUAD, for example, cooperation of TP53 mutation and MLL amplification resulted in the dramatic increase of activities of hallmark-associated pathways including unwinding of DNA (Figure 3E) and regulation of cell cycle progression by PLK3 pathway, consistent with a recent report that p53 mutation binds to and upregulates the chromatin regulatory gene MLL, which promotes cancer proliferation and growth (50). Importantly, these cooperative events identified could account for >5 cancer hallmarks per cancer type (Figure 3F and G). In LAML, for example, eight of the ten cancer hallmarks, such as self-sufficiency in growth signals, were associated with the cooperative events.
Genomic instability, one of the most pervasive hallmarks of tumorigenesis (51), correlated well with seven pathways based on functional associations (see Supplementary Table S3). Nucleotide metabolism pathway harboring the strongest correlation with this hallmark was influenced by several cooperative events in multiple cancers (Figure 3h). PIK3CA-EIF3H, RSRC1-CDKN2B, TP53-TM2D2 and TP53-MYC represent the most significant cooperative events increasing the activity of this pathway in BRCA, ESCA, LUAD and UCEC, respectively (P < 0.05, Kolmogorov–Smirnoff test, Supplementary Figure S9A). To examine whether these cooperative events can affect genome instability, we assumed that patients with these cooperative events could have an increased rate of genetic alterations (including mutation and CNA). By using the sample set enrichment analysis (SSEA) method, we found that co-altered patients harbored more genetic alterations (FDR < 0.05, Supplementary Figure S9b, see Supplementary Methods). Furthermore, we analyzed the types of DNA lesion events. Interestingly, more amplification events, rather than deletions or mutations, were observed in co-altered patients relative to the other patients (Supplementary Figure S9C). A plausible explanation is that these cooperative events can lead to the increased activity of nucleotide metabolism pathway, which in turn facilitates DNA synthesis and produces more DNA for gene amplification during replication (52).
Cooperative networks contribute to prognosis in human cancers
Next, we wondered whether the cooperative events identified had clinical implications. For each cancer type, we classified patients based on the occurrence of cooperative events by using consensus clustering (see Supplementary Methods), and then performed Kaplan–Meier survival analysis. As a result, in nine of the eleven cancers, the cooperative events significantly correlated with overall survival (P < 0.05, log-rank test, Figure 4). To assess whether or not genes in the cooperative events were enough to predict prognosis, we conducted a similar classification and survival analysis, but using genes involved in cooperative events. In this case, no significant effect on overall survival was observed in any cancer type (Supplementary Figure S10). These findings suggested the importance of cooperative events in tumorigenesis and their prognostic value in clinical practice.
Figure 4.

Kaplan-Meier plot of cancer-specific survival for subtypes in each cancer. Co-clustering matrices (top) and Kaplan-Meier survival plots (bottom) were shown for ten cancer patients. For BRCA, only one cooperative event was identified, so samples were classified based on co-occurrence of this gene pair. Kaplan-Meier plot for BRCA is shown in Supplementary Figure S12D.
Furthermore, we performed k-means clustering based on all cooperative events in the pan-cancer cooperative network referring to 2303 samples (see Supplementary Methods). Seventeen clusters were identified by maximizing the average silhouette coefficient (Figure 5A). We found that most of these clusters correlated well with the cancer tissue of origin (P < 0.0001, Chi-square test) (Supplementary Table S8). For example, C3, C6, C9 and C17 were significantly enriched for patients in HNSC, UCEC, CR and LAML, respectively (P < 0.0001, hypergeometric test). Some clusters, in contrast, contained a mixture of different cancer types, such as C15, which was highly associated with the cooperative alteration of TP53-MYC (P < 0.0001, hypergeometric test) that has been reported in multiple cancer types (22,53). Importantly, the 17 clusters exhibited significantly different clinical outcomes through Kaplan–Meier analysis (P < 0.0001, log-rank test, Figure 5B). However, it is not yet clear whether they can provide additional prognostic power beyond cancer types and clinical features. Thus, we performed a multivariate Cox proportional hazards analysis. In the model, we included age at diagnosis, sex and race as clinical covariates, as well as cancer types. A likelihood ratio test conditioning first on the clinical features was performed; after the addition of either cancer types or clusters to the model, we observed a large increase in the predictive fit (P < 0.0001 and P = 0.0082, Chi-square test, Figure 5C), further supporting the clinical value of cooperative events in prognosis of human cancers.
Figure 5.

Pan-cancer cooperative network contributes to prognosis in human cancers. (A) Clustering heatmap based on all the cooperative events in 12 cancers reveals 17 groups. The groups are identified by number and color in the second bar, while the tissue of origin specified in the top bar. The cooperative event that are most significantly enriched in each cluster, are shown on the right of the heatmap. (B) Overall survival of 17 groups by Kaplan-Meier plot. (C) The estimated log-likelihood ratio statistic of a Cox proportional hazards model. The change of LR statistic as features were added to the model was assessed for significance by chi-square analysis. (D and E) Kaplan-Meier curve estimates of overall survival for PTEN-PIK3CA and DNMT3A-FLT3, which are the most significantly enriched in group C10 and group C17, respectively.
Notably, among the 17 clusters, C10 exhibited the best prognosis. PTEN-PIK3CA was found to be enriched in this cluster (P < 0.0001, hypergeometric test). It has been reported that PTEN mutation could promote phosphorylation of Akt in the context of PIK3CA mutation, and in turn provide additive effect on the activation of PI3K pathway (54). We thus wondered whether the cooperation of PTEN-PIK3CA was associated with better survival in pan-cancer. Our results showed that patients with this cooperative event had better outcomes (P = 0.0043, log-rank test, Figure 5D) compared to those without this event (i.e. single-altered or wild-type). Interestingly, in another group C6 with the second-best prognosis, the cooperative alteration of PTEN-PIK3R1 was enriched (P < 0.0001, hypergeometric test). PIK3CA and PIK3R1 encode the subunits of PI3K complex, corresponding a catalytically active protein (p110α) and a regulatory protein (p85α) respectively (55), suggesting that the combined defect of PTEN and PI3K complex is a favorable prognostic factor across multiple cancers. Opposite to C10, C17 was associated with the poorest prognosis. DNMT3A-FLT3 was significantly enriched in this group (P < 0.0001, hypergeometric test). Patients harboring the cooperative event showed significantly poorer prognosis when compared with single alterations (P < 0.0001, log-rank test, Figure 5E). Among the identified cooperative pairs, we found some key cooperative events with potential diagnostic values in seven cancer types (Supplementary Table S9) by adopting the EdgeBiomarker method proposed by Zhang et al. (56).
Case study: cooperative events in glioblastoma
Diverse genomic patterns underlie glioblastoma
Glioblastoma multiforme (GBM) is the most common and deadly primary brain tumor with extremely poor prognosis (57–59). A total of 23 cooperative events referring to many known GBM-related genes (such as IDH1, TP53, PDGFRA and EGFR) were identified. These cooperative events could be classified into five groups based on their corresponding genomic patterns (Figure 6A, see Supplementary Methods). For example, TP53-RB1 and PTEN-RB1 were grouped together, consistently showing significantly decreased alteration frequencies of EGFR, CDKN2A and CDK4 (Figure 6A). TP53, RB1 and PTEN were core members of P53 signaling, RB signaling and RTK/RAS/PI3K signaling pathways, respectively, the co-disruption of which has been demonstrated to be a general trait for GBM pathogenesis (55). These five groups with distinct genomic patterns seemed to destroy different cancer hallmark-associated pathways (Figure 6B and Supplementary Figure S11). Especially, one of the groups was composed of only one cooperative event IDH1-TP53. IDH1 mutation is highly associated with the glioma-CpG island methylator phenotype (GCIMP) subtype, which presents a hypermethylator phenotype and improved outcome (60). The IDH-mutant GCIMP subtype has been found to frequently carry TP53 mutation (61). Our analysis showed a unique genomic pattern of IDH1-TP53, consistent with previous finding that the GCIMP subtype has a significantly different biological behaviors relative to non-GCIMP (62). Notably, both IDH1 and RB1 could cooperate with TP53, while these two cooperative events showed obviously different genomic patterns (Figure 6C). PTEN mutation was absolutely mutually exclusive with IDH1-TP53, yet appeared to co-occur with TP53-RB1 (Figure 6D). The same situation could be seen for CDK4 and PIK3R1. In fact, strong mutual exclusivity between IDH1-TP53 and TP53-RB1 was observed (P < 0.0001, fisher exact test, Figure 6D). Moreover, IDH1-TP53 and TP53-RB1 affected different hallmarks, such as sustained angiogenesis of IDH1-TP53 and genome instability of TP53-RB1 (Figure 6B and E). Taken together, these cooperative events could characterize diverse genomic contexts in GBM and reflect different molecular mechanisms underlie GBM, consistent with the high heterogeneity of GBM (63).
Figure 6.

The specific genomic patterns and hallmark-associated pathways of gene cooperative events in GBM. (A) Five groups of cooperative events based on genomic pattern similarity (top). Changes of alteration frequencies of marker genes were shown (bottom). (B) The hallmark-associated pathways affected by gene cooperative events corresponding with above five groups. Dashed box denotes the pathways affected by TP53-IDH1 and TP53-RB1, respectively. (C) The genomic patterns of TP53-IDH1 and TP53-RB1 are quite different, especially for PTEN (dashed box). (D) Samples carrying TP53-IDH1 and TP53-RB1 are mutually exclusive. (E) Activity difference for representative hallmark pathways affected by TP53-IDH1 and TP53-RB1 cooperation in different sample groups.
Cooperative events revealed molecular classification of GBM
To investigate whether the cooperative events could divide a heterogeneous population of GBM tumors into clinically meaningful molecular subtypes, we classified GBM patients into six clusters (C1–C6) on the basis of the presence or absence of these 23 cooperative events using consensus clustering (Figure 7A, see Supplementary Methods). Kaplan-Meier analysis of overall survival showed that these subgroups had distinct clinical outcomes (P < 0.0001, log-rank test, Figure 7B). Meanwhile, we utilized the five best-characterized GBM subtypes (including classical, proneural, mesenchymal, neural, and GCIMP) determined by expression and DNA methylation (60,64), which were also associated with overall survival (P = 0.0052, Figure 7C). As a comparison, C1 and C2 were primarily composed of classical and mesenchymal, respectively; C4 was significantly enriched for GCIMP, and, as expected, both of them had the best prognosis. Intriguingly, when all GCIMP patients were removed, our subgroups still had a significant association with survival (P = 0.0049, log-rank test, Figure 7d), whereas the pre-defined subtypes were not predictive of survival (P = 0.5236, log-rank test, Figure 7E). Furthermore, multivariate Cox proportional hazards analysis was performed by considering age, gender, race, and Karnofsky performance score as clinical covariates. The five best-characterized subtypes and our subgroups were added to the Cox proportional hazards model in turn. We found that the predictive fits of the model showed significant improvements (P = 0.0002 and P = 0.0002, Chi-square test, Figure 7F), which revealed additional survival benefit of our subgroups. All these results suggested that the cooperative events identified could provide useful insights for cancer classification and had GCIMP-independent prognostic effect in GBM.
Figure 7.

Gene cooperation could predict prognosis in GBM. (A) Classification based on all the cooperative pairs defines six subgroups using consensus clustering. Our subgroups are identified by color in the top bar, with the pre-defined subtype specified in the bottom bar. Co-clustering matrices (top) and alteration profiles of cooperative events (bottom) were shown. (B and C) Both our subgroups (B) and the pre-defined subtypes (C) could predict prognosis. (D and E) When removing all GCIMP samples, our subgroups (D) still have a significantly different survival but the pre-defined subtypes (E) lose its significance. (F) The estimated log-likelihood ratio statistic of a Cox proportional hazards model. The change of LR statistic as features were added to the model was assessed for significance by Chi-square analysis.
In particular, our clustering result revealed a very high-risk subgroup C5 (Figure 7B and D). SPTA1-METTL1 was the most significantly enriched cooperative event in this subgroup (P < 0.0001, hypergeometric test). To our knowledge, few studies reported the roles of SPTA1 and METTL1 in GBM (65). We observed that GBM patients with SPTA1-METTL1 were associated with a poor survival (P = 0.0103, log-rank test, Figure 8A), while individual alterations of either SPTA1 or METTL1 did not correlate with poor prognosis (P = 0.7142, and P = 0.9589, log-rank test, Figure 8B). Moreover, the copy number of METTL1 had a strong influence on its expression (Pearson correlation coefficient = 0.815, Figure 8C). As for SPTA1, we found >60% (19/30) mutations of SPTA1 with functional impact (Figure 8D and E) estimated by using MutationAssessor (66). Pathway analyses showed that SPTA1-METLL1 could influence three hallmark pathways including genomic instability, sustained angiogenesis and inflammation & evading immune (P = 0.0105, P = 0.0020 and P = 0.0094, one-sided Wilcoxon rank sum test, Figure 8G). We thus speculated that the combination of defects of SPTA1 and METTL1 was likely to be a high-risk clinical factor and they might act (Figure 8F) as cooperative driver genes contributing to the malignant progression of GBM. Further functional experiments are required to validate our findings.
Figure 8.

METTL1 and SPTA1 are potential cooperative drivers in GBM. (A) Kaplan-Meier curves for sample groups based on METTL1 and SPTA1 co-alteration in GBM. (B) Kaplan–Meier curves for sample groups based on either METTL1 copy number amplification or SPTA1 mutation in GBM. (C) The correlation of METTL1 copy number level and its mRNA expression. The red line denotes fitted curve based on correlation. (D) SPTA1 mutation distribution in gene body. The red, blue and green rectangle denotes SH3-1 pfama, Ca2+ insensitive EF hand, and spectrin pfama on SPTA1 protein, respectively. (E) The functional effect of mutations for SPTA1. And mutations with functional impact includes nonsense mutations, frame shift indels and missense mutations that are categorized as high or median effects by MutationAssessor. (F) The overlap between samples carrying METTL1 copy number amplification and SPTA1 mutation. P value is calculated by Chi-square test. (G) Activity difference for three hallmark pathways affected by METTL1-SPTA1 cooperation in different sample groups.
Case study: cooperative events in breast cancer
PIK3CA-EIF3H is a unique cooperative event in BRCA
Of note, only one cooperative event PIK3CA-EIF3H was identified in BRCA. The cooperative event was linked to several clinically relevant genes (such as ERBB2, TP53 and GATA3) and seven pathways involving six cancer hallmarks (Supplementary Figure S12A–C), and was associated with a poor survival in BRCA patients (P < 0.0001, log-rank test, Supplementary Figure S12d). The role of PIK3CA in BRCA has been well studied, and as for EIF3H, it was previously demonstrated as a driver gene within 8q23 amplicon and had effects on cell growth, survival and transformation in breast cancer (67). We next asked whether PIK3CA-EIF3H was a potential prognostic factor in the two main molecular subtypes (i.e. Basal and Luminal) of BRCA (68). We divided all BRCA patients into two subtypes (Basal and Luminal) based on ER and PR status and found that the cooperative event could be associated with overall survival in both subtypes (P = 0.0442 and P = 0.0009, log-rank test, Supplementary Figure S12E).
DISCUSSION
We developed an integrated method, HCOC, for identifying cooperative genomic alteration events in human cancer. Applying HCOC to 3753 patients from 12 cancer types yield a total of 1199 cooperative events with high heterogeneity across human cancers. We constructed the landscape of cooperative networks within and across cancers utilizing the cooperative events identified. We found that these hallmark associated cooperative events were always coupled with genomic alterations of several famous cancer drivers. Notably, patient classification based on cooperative events could provide additional prognostic information complementary with clinical and molecular features. As a case study, the diverse genomic patterns of cooperative events in GBM revealed multiple molecular mechanisms of gene cooperation underlying glioblastoma. The cooperative events in GBM allowed us to reveal a high-risk subgroup of GBM patients that were significantly enriched for SPTA1-METTL1 cooperation representing a novel high-risk prognostic factor. These two genes were seriously underestimated when considered in isolation. This study represented one of the largest analysis of somatic cooperative aberrations and demonstrated that the integration of omics data can be used to characterize cooperative alterations during tumorigenesis.
One advantage of HCOC is the consideration of genetic context of gene cooperation. Each cooperative event is linked to a specific genomic pattern of some important genes with recurrent alterations. This specific genomic context depicted certain genetic dependence between mutated genes and the cooperative event. As in previous study, in melanomas with BRAF V600E mutations, concurrent mutational inactivation of the PTEN and RB1 exhibited diminished dependence upon BRAF signaling and were mutually exclusive with loss of CDKN2A (27). The cooperative event and the specific genomic context form a unique combination of genomic aberrations, like the orchestration of genomic alterations, which can help to precisely dissect the mechanisms contributing to cancer initiation and progression. For example, despite that EGFR and PDGFRA were reported as members of a mutually exclusive module in GBM, in our result PDGFRA displayed an elevated alteration frequency when the co-alteration of EGFR-MDM2 occurred (P = 0.0017, one-sided binomial test, Supplementary Figure S13). The fact that both EGFR and PDGFRA were upstream regulators of MDM2 may explain this conditional co-occurrence.
Moreover, these cooperative events were highly associated with some cancer hallmarks. Interestingly, the cooperative events could lead to the changes of hallmark activities to a larger extent than single alterations (Figure 3D), consistent with a phenomenon of epistasis, i.e., a mutation is selectively advantageous only in the context of other mutations (19). In fact, certain oncogenic mutations can mandate that specific cellular pathways be targeted by subsequent mutations (20). Epistatic interaction has been demonstrated to play a crucial role in cancer genome evolution. Previous protein-engineering studies demonstrated that epistatic interactions could limit the potential mutational trajectories and enforce ratchet-like constraints by inhibiting the reversibility of the tumor evolutionary process (20,69). During the development of cancer, distinct tumor subclones evolve in parallel and gradually acquire various hallmark capabilities by accumulating different genetic alterations and, in the end, gain a significant fitness advantage and further expand to dominant lineages (70). Therefore, the cooperative network constituted of all cooperative events in human cancers may approximately depict the tumor heterogeneity and reflect diverse evolution branches during tumorigenesis. In our results, TP53-IDH1 and TP53-RB1 were both identified in GBM, but their affected patients, genetic contexts and associated cancer hallmarks were quite different (Figure 6C–E). Since TP53 mutation was an early clonal event in many cancers including GBM (71,72), subsequent IDH1 and RB1 mutation may indicate different evolution paths.
Importantly, these cooperative events could be as potential new biomarkers for patient stratification. Specially, SPTA1-METTL1 cooperation in GBM characterized a subgroup with the worst survival, while single alterations can not. METTL1 has been reported to influence the sensitivity of an anti-carcinogen (5-fluorouracil) in cancer cells (73), and mutations of SPTA1 were observed in different stages of lung cancer, such as stages before and after chemotherapy (74). We speculated that METTL1-SPAT1 cooperation might exert an adverse effect on anti-cancer therapy. Classification based on all cooperative events could also provide implications for clinical outcomes in most cancer types (Figure 4), however, it was not the case when based on single alterations (Supplementary Figure S10). Tumor subtypes based on single drivers may be confounded by epistasis (75) or gene pleiotropy (76), which can be alleviated by gene cooperation. Besides, due to the links of cooperative events to evolutional branches, it's understandable that cooperative events allowed us to reveal subtypes with distinct evolution paths. Such subtypes have a significant clinical benefit. Our results suggest that gene cooperation is able to depict the underlying mechanism underlying tumorigenesis, and may be superior when translated into clinical practice. Note that the identified cooperative events in our study were associated with a specific pattern of background marker genes, suggesting a possibility that the effects of the cooperative events on cancer hallmarks and clinical outcomes may be mainly caused by the background marker genes. To test the possibility, we evaluated the effects of the marker genes on the hallmarks and clinical outcomes in the samples without co-alteration of cooperative events (for details, see Supplementary Methods). The results showed that in most cases the cooperative events, rather than the marker genes, have major effects on the cancer hallmarks and overall survival.
A previous study characterized the pre-disease state based on time-series gene expression data, allowing to diagnose whether a new individual approaches to the pre-disease state or not (77). With increasing numbers of time-series genomic data available in cancer, we can use the concept of dynamical network biomarkers to detect early-warning cooperative signals in cancer, which will be crucial to further understand the progression of cancer and facilitate early clinical diagnosis.
In our study, we integrated copy number alteration and point mutations, which can comprehensively capture the DNA damage upon certain genes. Nonetheless, point mutations and copy number alterations cannot always be considered similar. Moreover, all point mutations are not equal. Some mutations lead to gain-of-function, while some others lead to loss-of-function. We thus classified mutations into gain-of-function and loss-of-function, and determined the dominant type of mutations for each gene (see Supplementary Methods for details). By reapplying HCOC, we observed that 91.4% cooperative pairs across cancers were shared with previous results. Especially, the results were exactly the same in CR, OV, BRCA, LAML and BLCA (Supplementary Table S10).
In summary, our study presents a comprehensive characterization of genomic cooperative events in 12 cancer types, which extends our knowledge of interdependence among cancer genes and enables to discovery novel oncogenic genes. Our analyses shed new insights on molecular mechanisms underlying tumorigenesis, and offer implications for clinical diagnosis, prognosis and therapeutic strategies. Ultimately, we expect that our approach will be applicable to other cancer genomic data and our results will be helpful for achieving the goal of precise medicine.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National High Technology Research and Development Program of China (863 Program) [2014AA021102] (in part); National Program on Key Basic Research Project (973 Program) [2014CB910504]; National Natural Science Foundation of China [91439117, 61473106, 61573122]; Wu lien-teh youth science fund project of Harbin medical university [WLD-QN1407]; Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education, and the Funds for the Graduate Innovation Fund of Heilongjiang Province [YJSCX2015-8HYD]. Funding for open access charge: National High Technology Research and Development Program of China (863 Program) [2014AA021102] (in part); National Program on Key Basic Research Project (973 Program) [2014CB910504]; National Natural Science Foundation of China [91439117, 61473106, 61573122]; Wu lien-teh youth science fund project of Harbin medical university [WLD-QN1407]; Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education, and the Funds for the Graduate Innovation Fund of Heilongjiang Province [YJSCX2015-8HYD].
Conflict of interest statement. None declared.
REFERENCES
- 1.Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M., et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bozic I., Antal T., Ohtsuki H., Carter H., Kim D., Chen S., Karchin R., Kinzler K.W., Vogelstein B., Nowak M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aktipis C.A., Boddy A.M., Gatenby R.A., Brown J.S., Maley C.C. Life history trade-offs in cancer evolution. Nat. Rev. Cancer. 2013;13:883–892. doi: 10.1038/nrc3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gatenby R.A., Cunningham J.J., Brown J.S. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nat. Commun. 2014;5:5499. doi: 10.1038/ncomms6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeang C.H., McCormick F., Levine A. Combinatorial patterns of somatic gene mutations in cancer. FASEB J. 2008;22:2605–2622. doi: 10.1096/fj.08-108985. [DOI] [PubMed] [Google Scholar]
- 6.Bredel M., Scholtens D.M., Harsh G.R., Bredel C., Chandler J.P., Renfrow J.J., Yadav A.K., Vogel H., Scheck A.C., Tibshirani R., et al. A network model of a cooperative genetic landscape in brain tumors. JAMA. 2009;302:261–275. doi: 10.1001/jama.2009.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elsum I.A., Yates L.L., Pearson H.B., Phesse T.J., Long F., O'Donoghue R., Ernst M., Cullinane C., Humbert P.O. Scrib heterozygosity predisposes to lung cancer and cooperates with KRas hyperactivation to accelerate lung cancer progression in vivo. Oncogene. 2014;33:5523–5533. doi: 10.1038/onc.2013.498. [DOI] [PubMed] [Google Scholar]
- 8.Eckel-Passow J.E., Lachance D.H., Molinaro A.M., Walsh K.M., Decker P.A., Sicotte H., Pekmezci M., Rice T., Kosel M.L., Smirnov I.V., et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 2015;372:2499–2508. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins R.B., Blair H., Ballman K.V., Giannini C., Arusell R.M., Law M., Flynn H., Passe S., Felten S., Brown P.D., et al. A t(1;19) (q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- 10.Klijn C., Bot J., Adams D.J., Reinders M., Wessels L., Jonkers J. Identification of networks of co-occurring, tumor-related DNA copy number changes using a genome-wide scoring approach. PLoS Comput. Biol. 2010;6:e1000631. doi: 10.1371/journal.pcbi.1000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Y., Wang H., Qin Y., Zhang Y., Zhao W., Qi L., Zhang Y., Wang C., Guo Z. Network analysis of genomic alteration profiles reveals co-altered functional modules and driver genes for glioblastoma. Mol. bioSyst. 2013;9:467–477. doi: 10.1039/c2mb25528f. [DOI] [PubMed] [Google Scholar]
- 12.Liu J., Zhao D., Fan R. Shared and unique mutational gene co-occurrences in cancers. Biochem. Biophys. Res. Commun. 2015;465:777–783. doi: 10.1016/j.bbrc.2015.08.086. [DOI] [PubMed] [Google Scholar]
- 13.Cui Q. A network of cancer genes with co-occurring and anti-co-occurring mutations. PLoS One. 2010;5:e13180. doi: 10.1371/journal.pone.0013180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J., Zhang Y., Shen X., Zhu J., Zhang L., Zou J., Guo Z. Finding co-mutated genes and candidate cancer genes in cancer genomes by stratified false discovery rate control. Mol. bioSyst. 2011;7:1158–1166. doi: 10.1039/c0mb00211a. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y., Yang D., Zou J., Ma W., Wu R., Zhao W., Zhang Y., Xiao H., Gong X., Zhang M., et al. Systematic interpretation of comutated genes in large-scale cancer mutation profiles. Mol. Cancer Ther. 2010;9:2186–2195. doi: 10.1158/1535-7163.MCT-10-0022. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J., Wu L.Y., Zhang X.S., Zhang S. Discovery of co-occurring driver pathways in cancer. BMC Bioinformatics. 2014;15:271. doi: 10.1186/1471-2105-15-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciriello G., Cerami E., Sander C., Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Meur N., Gentleman R. Modeling synthetic lethality. Genome Biol. 2008;9:R135. doi: 10.1186/gb-2008-9-9-r135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore J.H. A global view of epistasis. Nat. Genet. 2005;37:13–14. doi: 10.1038/ng0105-13. [DOI] [PubMed] [Google Scholar]
- 20.Yates L.R., Campbell P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Podsypanina K., Politi K., Beverly L.J., Varmus H.E. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5242–5247. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill R.M., Kuijper S., Lindsey J.C., Petrie K., Schwalbe E.C., Barker K., Boult J.K., Williamson D., Ahmad Z., Hallsworth A., et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. 2015;27:72–84. doi: 10.1016/j.ccell.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wasserman W.W., Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 25.Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing F., Persaud Y., Pratilas C.A., Taylor B.S., Janakiraman M., She Q.B., Gallardo H., Liu C., Merghoub T., Hefter B., et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene. 2012;31:446–457. doi: 10.1038/onc.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez-Garcia F., Villagrasa P., Matsui J., Kotliar D., Castro V., Akavia U.D., Chen B.J., Saucedo-Cuevas L., Rodriguez Barrueco R., Llobet-Navas D., et al. Integration of genomic data enables selective discovery of breast cancer drivers. Cell. 2014;159:1461–1475. doi: 10.1016/j.cell.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamosh A., Scott A.F., Amberger J., Valle D., McKusick V.A. Online Mendelian Inheritance in Man (OMIM) Hum. Mutat. 2000;15:57–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 30.Becker K.G., Barnes K.C., Bright T.J., Wang S.A. The genetic association database. Nat. Genet. 2004;36:431–432. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 31.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine D.M., Haynor D.R., Castle J.C., Stepaniants S.B., Pellegrini M., Mao M., Johnson J.M. Pathway and gene-set activation measurement from mRNA expression data: the tissue distribution of human pathways. Genome Biol. 2006;7:R93. doi: 10.1186/gb-2006-7-10-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leiserson M.D., Blokh D., Sharan R., Raphael B.J. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput. Biol. 2013;9:e1003054. doi: 10.1371/journal.pcbi.1003054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stingl J., Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat. Rev. Cancer. 2007;7:791–799. doi: 10.1038/nrc2212. [DOI] [PubMed] [Google Scholar]
- 37.Nowell P.C. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 38.Kumar N., Rehrauer H., Cai H., Baudis M. CDCOCA: a statistical method to define complexity dependence of co-occuring chromosomal aberrations. BMC Med. Genomics. 2011;4:21. doi: 10.1186/1755-8794-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ulz P., Heitzer E., Speicher M.R. Co-occurrence of MYC amplification and TP53 mutations in human cancer. Nat. Genet. 2016;48:104–106. doi: 10.1038/ng.3468. [DOI] [PubMed] [Google Scholar]
- 40.Huang Z.Y., Baldwin R.L., Hedrick N.M., Gutmann D.H. Astrocyte-specific expression of CDK4 is not sufficient for tumor formation, but cooperates with p53 heterozygosity to provide a growth advantage for astrocytes in vivo. Oncogene. 2002;21:1325–1334. doi: 10.1038/sj.onc.1205206. [DOI] [PubMed] [Google Scholar]
- 41.Chow L.M., Endersby R., Zhu X., Rankin S., Qu C., Zhang J., Broniscer A., Ellison D.W., Baker S.J. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19:305–316. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loghavi S., Zuo Z., Ravandi F., Kantarjian H.M., Bueso-Ramos C., Zhang L., Singh R.R., Patel K.P., Medeiros L.J., Stingo F., et al. Clinical features of de novo acute myeloid leukemia with concurrent DNMT3A, FLT3 and NPM1 mutations. J. Hematol. Oncol. 2014;7:74. doi: 10.1186/s13045-014-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn J.S., Kim H.J., Kim Y.K., Lee S.S., Jung S.H., Yang D.H., Lee J.J., Kim N.Y., Choi S.H., Jung C.W., et al. DNMT3A R882 mutation with FLT3-ITD positivity is an extremely poor prognostic factor in patients with Normal-Karyotype acute myeloid leukemia after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2015;22:61–70. doi: 10.1016/j.bbmt.2015.07.030. [DOI] [PubMed] [Google Scholar]
- 44.Siristatidis C., Sergentanis T.N., Vogiatzi P., Kanavidis P., Chrelias C., Papantoniou N., Psaltopoulou T. In vitro maturation in women with vs. without polycystic ovarian syndrome: a systematic review and meta-analysis. PLoS One. 2015;10:e0134696. doi: 10.1371/journal.pone.0134696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu P., Cheng H., Santiago S., Raeder M., Zhang F., Isabella A., Yang J., Semaan D.J., Chen C., Fox E.A., et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat. Med. 2011;17:1116–1120. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong T.N., Ramsingh G., Young A.L., Miller C.A., Touma W., Welch J.S., Lamprecht T.L., Shen D., Hundal J., Fulton R.S., et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weaver J.M., Ross-Innes C.S., Shannon N., Lynch A.G., Forshew T., Barbera M., Murtaza M., Ong C.A., Lao-Sirieix P., Dunning M.J., et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat. Genet. 2014;46:837–843. doi: 10.1038/ng.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mullany L.K., Liu Z., King E.R., Wong K.K., Richards J.S. Wild-type tumor repressor protein 53 (Trp53) promotes ovarian cancer cell survival. Endocrinology. 2012;153:1638–1648. doi: 10.1210/en.2011-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hertzler-Schaefer K., Mathew G., Somani A.K., Tholpady S., Kadakia M.P., Chen Y., Spandau D.F., Zhang X. Pten loss induces autocrine FGF signaling to promote skin tumorigenesis. Cell Rep. 2014;6:818–826. doi: 10.1016/j.celrep.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu J., Sammons M.A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B.W., Shilatifard A., et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–211. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lord C.J., Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 52.Futcher A.B. Copy number amplification of the 2 micron circle plasmid of Saccharomyces cerevisiae. J. Theoret. Biol. 1986;119:197–204. doi: 10.1016/s0022-5193(86)80074-1. [DOI] [PubMed] [Google Scholar]
- 53.Girardini J.E., Walerych D., Del Sal G. Cooperation of p53 mutations with other oncogenic alterations in cancer. Sub-cell. Biochem. 2014;85:41–70. doi: 10.1007/978-94-017-9211-0_3. [DOI] [PubMed] [Google Scholar]
- 54.Oda K., Stokoe D., Taketani Y., McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–10673. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 55.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang W., Zeng T., Liu X., Chen L. Diagnosing phenotypes of single-sample individuals by edge biomarkers. J. Mol. Cell Biol. 2015;7:231–241. doi: 10.1093/jmcb/mjv025. [DOI] [PubMed] [Google Scholar]
- 57.Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 58.Ping Y., Zhang H., Deng Y., Wang L., Zhao H., Pang L., Fan H., Xu C., Li F., Zhang Y., et al. IndividualizedPath: identifying genetic alterations contributing to the dysfunctional pathways in glioblastoma individuals. Mol. bioSyst. 2014;10:2031–2042. doi: 10.1039/c4mb00289j. [DOI] [PubMed] [Google Scholar]
- 59.Xiao Y., Ping Y., Fan H., Xu C., Guan J., Zhao H., Li Y., Lv Y., Jin Y., Wang L., et al. Identifying dysfunctional miRNA-mRNA regulatory modules by inverse activation, cofunction, and high interconnection of target genes: a case study of glioblastoma. Neuro-oncology. 2013;15:818–828. doi: 10.1093/neuonc/not018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noushmehr H., Weisenberger D.J., Diefes K., Phillips H.S., Pujara K., Berman B.P., Pan F., Pelloski C.E., Sulman E.P., Bhat K.P., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sturm D., Witt H., Hovestadt V., Khuong-Quang D.A., Jones D.T., Konermann C., Pfaff E., Tonjes M., Sill M., Bender S., et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 62.Ozawa T., Riester M., Cheng Y.K., Huse J.T., Squatrito M., Helmy K., Charles N., Michor F., Holland E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vartanian A., Singh S.K., Agnihotri S., Jalali S., Burrell K., Aldape K.D., Zadeh G. GBM's multifaceted landscape: highlighting regional and microenvironmental heterogeneity. Neuro-oncology. 2014;16:1167–1175. doi: 10.1093/neuonc/nou035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ping Y., Deng Y., Wang L., Zhang H., Zhang Y., Xu C., Zhao H., Fan H., Yu F., Xiao Y., et al. Identifying core gene modules in glioblastoma based on multilayer factor-mediated dysfunctional regulatory networks through integrating multi-dimensional genomic data. Nucleic acids research. 2015;43:1997–2007. doi: 10.1093/nar/gkv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reva B., Antipin Y., Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pittman A.M., Naranjo S., Jalava S.E., Twiss P., Ma Y., Olver B., Lloyd A., Vijayakrishnan J., Qureshi M., Broderick P., et al. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS Genet. 2010;6:e1001126. doi: 10.1371/journal.pgen.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gatza M.L., Silva G.O., Parker J.S., Fan C., Perou C.M. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 2014;46:1051–1059. doi: 10.1038/ng.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bridgham J.T., Ortlund E.A., Thornton J.W. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–519. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greaves M., Maley C.C. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Attolini C.S., Cheng Y.K., Beroukhim R., Getz G., Abdel-Wahab O., Levine R.L., Mellinghoff I.K., Michor F. A mathematical framework to determine the temporal sequence of somatic genetic events in cancer. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17604–17609. doi: 10.1073/pnas.1009117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Misra N., Szczurek E., Vingron M. Inferring the paths of somatic evolution in cancer. Bioinformatics. 2014;30:2456–2463. doi: 10.1093/bioinformatics/btu319. [DOI] [PubMed] [Google Scholar]
- 73.Okamoto M., Fujiwara M., Hori M., Okada K., Yazama F., Konishi H., Xiao Y., Qi G., Shimamoto F., Ota T., et al. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet. 2014;10:e1004639. doi: 10.1371/journal.pgen.1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iwakawa R., Kohno T., Totoki Y., Shibata T., Tsuchihara K., Mimaki S., Tsuta K., Narita Y., Nishikawa R., Noguchi M., et al. Expression and clinical significance of genes frequently mutated in small cell lung cancers defined by whole exome/RNA sequencing. Carcinogenesis. 2015;36:616–621. doi: 10.1093/carcin/bgv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burrell R.A., McGranahan N., Bartek J., Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 76.Sivakumaran S., Agakov F., Theodoratou E., Prendergast J.G., Zgaga L., Manolio T., Rudan I., McKeigue P., Wilson J.F., Campbell H. Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet. 2011;89:607–618. doi: 10.1016/j.ajhg.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen L., Liu R., Liu Z.P., Li M., Aihara K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2012;2:342. doi: 10.1038/srep00342. [DOI] [PMC free article] [PubMed] [Google Scholar]