


Abstract
Differential expression analysis of RNA sequencing (RNA-seq) data typically relies on reconstructing transcripts or counting reads that overlap known gene structures. We previously introduced an intermediate statistical approach called differentially expressed region (DER) finder that seeks to identify contiguous regions of the genome showing differential expression signal at single base resolution without relying on existing annotation or potentially inaccurate transcript assembly.
We present the derfinder software that improves our annotation-agnostic approach to RNA-seq analysis by: (i) implementing a computationally efficient bump-hunting approach to identify DERs that permits genome-scale analyses in a large number of samples, (ii) introducing a flexible statistical modeling framework, including multi-group and time-course analyses and (iii) introducing a new set of data visualizations for expressed region analysis. We apply this approach to public RNA-seq data from the Genotype-Tissue Expression (GTEx) project and BrainSpan project to show that derfinder permits the analysis of hundreds of samples at base resolution in R, identifies expression outside of known gene boundaries and can be used to visualize expressed regions at base-resolution. In simulations, our base resolution approaches enable discovery in the presence of incomplete annotation and is nearly as powerful as feature-level methods when the annotation is complete.
derfinder analysis using expressed region-level and single base-level approaches provides a compromise between full transcript reconstruction and feature-level analysis. The package is available from Bioconductor at www.bioconductor.org/packages/derfinder.
INTRODUCTION
The increased flexibility of RNA sequencing (RNA-seq) has made it possible to characterize the transcriptomes of a diverse range of experimental systems, including human tissues (1–3), cell lines (4,5) and model organisms (6,7). The goal of many experiments involves identifying differential expression with respect to disease, development or treatment. In experiments using RNA-seq, RNA is sequenced to generate short ‘reads’ (36–200+ base pairs). These reads are aligned to a reference genome, and this alignment information is used to quantify the transcriptional activity of both annotated (present in databases like Ensembl) and novel transcripts and genes.
The ability to quantitatively measure expression levels in regions not previously annotated in gene databases, particularly in tissues or cell types that are difficult to ascertain, is one key advantage of RNA-seq over hybridization-based assays like microarray technologies. As complicated transcript structures are difficult to completely characterize using short read sequencing technologies (8), the most mature statistical methods used for RNA-seq analysis rely on existing annotation for defining regions of interest—such as genes or exons—and counting reads that overlap those regions (9). These counts are then used as measures of gene expression abundance for downstream differential expression analysis (10–18). Unfortunately, the gene annotation may be incorrect or incomplete, which can affect downstream modeling of the number of reads that cross these defined features.
We previously proposed an alternative statistical model for finding differentially expressed regions (DERs) that first identifies regions that show differential expression signal and then annotates these regions using previously annotated genomic features (19). This analysis framework first proposed using coverage tracks (i.e. the number of reads aligned to each base in the genome) to identify differential expression signal at each individual base and merges adjacent bases with similar signal into candidate regions. However, the software for our first version was limited to small sample sizes, the ability to interrogate targeted genomic loci and comparisons between only two groups.
Here, we expand the DER finder framework to permit the analysis of larger sample sizes with more flexible statistical models across the genome. This paper introduces a comprehensive software package called derfinder built upon base-resolution analysis, which performs coverage calculation, preprocessing, statistical modeling, region annotation and data visualization. This software permits differential expression analysis at both the single base level, resulting in direct calculation of DERs (20), and a feature summarization we introduce here call ‘expressed region’ (ER)-level analysis. We show that ER analysis allows us to perform base resolution analysis on larger scale RNA-seq data sets using the BrainSpan project (21) and Genotype-Tissue Expression (GTEx) project data (3) to demonstrate that derfinder can identify differential expression signal in regions outside of known annotation without assembly. We use these DERs to illustrate the post-discovery annotation capabilities of derfinder and label each DER as exonic, intronic, intergenic or some combination of those labels. We show that some of these DERs we identify are outside of annotated protein coding regions and would not have been identified using gene or exon counting approaches.
In the GTEx data, we identify DERs that differentiate heart (left ventricle), testis and liver tissues for eight subjects. There are many potential reasons for this observed intronic expression including intron retention, background levels of mis-transcription or incomplete protein-coding annotation. A subset of these strictly intronic ERs are associated with tissue differences, even conditional on the expression of the nearest annotated protein-coding region. However, we point out that intronic expression may be artifactual and our package permits visualization and discovery of potential expression artifacts not possible with other packages.
Finally, using simulated differentially expressed transcripts, we demonstrate that when transcript annotation is correct, derfinder is nearly as powerful as exon-count based approaches with statistical tests performed by limma (16) (or DESeq2 (14), edgeR-robust (13)) and ballgown (22) after summarizing the information using Rsubread (13) and StringTie (23), respectively. Finally, we also demonstrate that when annotation is incomplete, derfinder can be substantially more powerful than methods that rely on a complete annotation.
MATERIALS AND METHODS
Overview of R implementation
We chose to implement derfinder entirely in the R statistical environment www.R-project.org/. Our software includes upstream pre-processing of BAM and/or BigWig files into base-resolution coverage. At this stage the user can choose to summarize the base resolution coverage into feature-level counts and apply popular feature-level RNA-seq differential expression analysis tools like DESeq2 (14), edgeR-robust (13), limma (15,16) and voom (17).
derfinder can be used to identify regions of differential expression agnostic to existing annotation (Figure 1). This can be done with either the expressed regions (ER)-level or single base-level approaches, described in detail in the following subsection and Supplementary Section 2.1. The resulting regions can then be visualized to identify novel regions and filter out potential artifacts.
Figure 1.

An overview of the derfinder suite. The derfinder software package includes functions for processing and normalizing coverage per sample, performing statistical tests to identify differentially expressed regions (DERs), labeling those regions with known annotation and visualizing the results across groups.
After differential expression analysis, derfinder can plot DERs using base-resolution coverage data by accessing the raw reads within DERs for posthoc analysis like clustering and sensitivity analyses. We have also created a lightweight annotation function for quickly annotating DERs based on existing transcriptome annotation, including the UCSC knownGene hg19, Ensembl p12 and Gencode v19 databases as well as newer versions.
Vignettes with detailed instructions and examples are available through the Bioconductor pages for derfinder and derfinderPlot. The main functions for the expressed region and single base-level approaches are further described in Supplementary Section 1.1.
Expressed region level analysis
In the expressed region approach, we compute the mean coverage for all base pairs from all the samples and filter out those below a user specified cutoff. Contiguous bases passing this filtering step are then considered a candidate region (Figure 2A). Then for each sample, we sum the base-level coverage for each such region in order to create an expression matrix with one row per region and one column per sample. This matrix can then be used with feature-level RNA-seq differential expression analysis tools. The statistical model used for the differential expression is a general F-statistic model as shown in Figure 1, Step 4 and Equation (1). In Equation (1), yij is the scaled log2 mean base-level coverage for the expressed region i and sample j. The model is completed by an intercept term αi, n group effects βi, m adjustment variable effects γi and measurement error ε. The F-statistic is derived from comparing this alternative model against a null model without the βi terms as in Supplementary Methods 2.1. The model can allow time-course analyses, two group comparisons or multi-group comparisons, thus making derfinder flexible. Examples of the latter case are shown in Methods Sections 2.4.1 and 2.4.2.
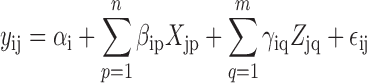 |
(1) |
Figure 2.

Finding regions via expressed region-level approach on chromosome 5 with BrainSpan data set. (A) Mean coverage with segments passing the mean cutoff (0.25) marked as regions. (B) Raw coverage curves superimposed with the candidate regions. Coverage curves are colored by brain region and developmental stage (NCX: Neocortex: Non-NCX: Non-neocortex, CBC: cerebellum, F: fetal, P: postnatal). (C) Known exons (dark blue) and introns (light blue) by strand for genes and subsequent transcripts in the locus. The DERs best support the GABRA6 transcript with a red star, indicating the presence of a differentially expressed transcript.
Annotation and ‘genomic state’ objects
We have implemented a ‘genomic state’ framework to efficiently annotate and summarize resulting regions, which assigns each base in the genome to exactly one state: exonic, intronic or intergenic, based on any existing or user-defined annotation (e.g. UCSC, Ensembl, Gencode). At each base, we prioritize exon > intron > unannotated across all annotated transcripts.
Overlapping exons of different lengths belonging to different transcripts are reduced into a single ‘exonic’ region, while retaining merged transcript annotations. We have a second implementation that further defines promoters and divides exonic regions into coding and untranslated regions (UTRs) that may be useful for the user to more specifically annotate regions – this implementation prioritizes coding exon > UTR > promoter > intron > unannotated.
Data processing for results in main manuscript
BrainSpan data
BigWig files for all 487 samples across 16 brain regions were downloaded from the BrainSpan website (21). The samples for HSB169.A1C, HSB168.V1C and HSB168.DFC were dropped due to quality issues. Based on exploratory analyses the coverage was assumed to be reads-per-million mapped reads in this data set. We set the coverage filter to 0.25 for both the single base-level and ER-level derfinder approaches. Since the coverage is already adjusted to reads per million mapped reads, we did not include a library size adjustment term in the single base-level derfinder analysis (see Supplementary Section 2.1 for details on this adjustment term). The details for the single base-level derfinder analysis are described further in Supplementary Section 2.2. For the ER-level approach we only considered regions longer than 5 base-pairs.
We sought to identify differences in expression across brain region (neocortical regions: DFC, VFC, MFC, OFC, M1C, S1C, IPC, A1C, STC, ITC, V1C and non-neocortical regions: HIP, AMY, STR, MD and CBC) and developmental stage (fetal versus postnatal). We therefore fit the following region-by-stage interaction alternative model, which included main effects for fetal versus postnatal (binary) and categorical brain region variable (15 region indicators, relative to A1C), and interaction terms for each brain region and developmental stage. This resulted in a total of 32 terms in the model (intercept; 16 main effects, 15 interaction terms). In Equation (2), yij is the scaled log2 mean base-level coverage for the expressed region i and sample j as in Equation (1); that is yij = log2(meancoverageij + 1). The model is completed by an intercept term αi, a indicator variable for fetal status βi, m indicators variables γ for the brain region and m interaction variables ζ between fetal status and brain region. The term εij represents residual error.
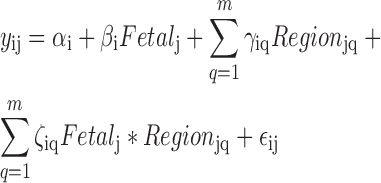 |
(2) |
We compared the above model to an intercept-only model where using the lmFit function from limma (15,16). The P-values for the ER-level DERs were adjusted via the Bonferroni method and those with adjusted P-values less than 0.05 were determined to be significant. We then calculated the mean coverage for each significant expressed region DERs in each sample, resulting in a mean coverage matrix (DERs by samples) and we performed principal component analysis (PCA) on this log2-transformed matrix (after adding an offset of 1).
Once the DERs were identified, we identified which of them overlap ENCODE blacklisted regions of the genome (4) using the file at http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDacMapabilityConsensusExcludable.bed.gz. For identifying which DERs overlap lincRNAs, we used EnsDb.Hsapiens.v75 (24), which can also be used for a variety of transcript types. We then performed the gene ontology (GO) analysis for the DERs using GOstats (25) using as background all genes that are within 5 kb of an ER.
GTEx data
We selected samples from individuals that had data from heart (left ventricle), liver and testis tissues with RNA Integrity Number (RIN) values greater than 7. Eight subjects matched this criteria and we selected only 1 sample if their tissue was analyzed more than once, leaving us with 24 samples. The data were aligned using Rail-RNA (26) version 0.2.1 with the code as described at www.github.com/nellore/runs. We created a normalized mean BigWig file for these 4 samples adjusted for library sizes of 40 million reads. We then identified the ERs using a cutoff of 5 using the function railMatrix from derfinder version 1.5.19.
For each expressed region greater than 9 bp, we assigned its annotation status by using a genomic state object created with the Ensembl GRCh38.p5 database. We then performed PCA on the log2-transformed matrix (after adding an offset of 1) separately for strictly exonic and strictly intronic ERs. Using limma (15,16) functions lmFit, ebayes we fit an intercept-only null model and an alternative model with coefficients for tissue differences. For each ER we calculated a F-statistic and determined whether it was differentially expressed by tissue using a Bonferroni adjusted P-value cutoff of 0.05.
For the conditional expression analysis, we found the nearest exonic ER for each intronic ER using the distanceToNearest function from GenomicRanges (27). For each intronic ER we fitted two linear regression models for the log2-transformed coverage matrix (after adding an offset of 1). For the alternative model we used as covariates two tissue indicator variables (Heart as the reference) and the coverage from the nearest strictly exonic ER as shown in Equation (3) for ER i and sample j. For the null model we only used the coverage from the nearest exonic ER. We calculated an F-statistic using the anova function that tests whether β1i or β2i are equal to 0 and used a Bonferroni adjusted P-value cutoff of 0.05 to identify which intronic ERs had differential expression adjusting for the coverage at the nearest exonic ER.
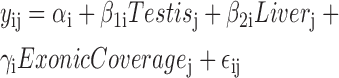 |
(3) |
Simulated data
We simulated 100 bp paired-end reads (250 bp fragments, sd = 25) with polyester (28) for two groups with five samples per group from human chromosome 17 with uniform error rate of 0.005 and replicated this process three times. One-sixth of the transcripts were set to have higher expression (2x) in group 2, a sixth to have lower expression in group 2 (1/2x) and the remaining two-thirds to be equally expressed in both groups. Given a RNA-seq experiment with 40 million paired-end reads, assuming that all transcripts are equally expressed we would expect 1 989 247 of them to be from chromosome 17 based on the length of all exons using the known transcripts UCSC knownGene hg19 annotation. We used this information and the transcript length to assign the number of reads per transcript in chromosome 17 and generated the number of reads with the NB function from polyester with mean μ and size (see the rnbinom function from the stats package) equal to 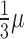 . This resulted in an average of 2 073 682 paired-end reads per sample. For each simulation replicate, paired-end reads were aligned to the hg19 reference genome using HISAT version 0.1.6-beta (29) and Rail-RNA version 0.2.2b (26). We created a GTF file using all known transcripts from chromosome 17 as well as one with 20% of the transcripts missing (8.28% of exons missing). Using these two GTF files we performed transcript quantification with StringTie version 1.2.1 (23) as well as exon counting allowing multiple overlaps with the featureCounts function from Rsubread version 1.21.4 (13). ERs were determined with derfinder version 1.5.19 functions regionMatrix and railMatrix, respectively, from the HISAT BAM and Rail-RNA BigWig output using a mean cutoff of 5 for libraries adjusted to 80 million single-end reads. Count matrices resulting from featureCounts and derfinder were analyzed with limma (16), DESeq2 (14) and edgeR-robust (18) controlling the false discovery rate (FDR) at 5% and testing for differences between the two groups of samples. We used ballgown version 2.2.0 (22) to perform differential expression tests using coverage at the transcript and exon levels, controlling the FDR at 5%.
. This resulted in an average of 2 073 682 paired-end reads per sample. For each simulation replicate, paired-end reads were aligned to the hg19 reference genome using HISAT version 0.1.6-beta (29) and Rail-RNA version 0.2.2b (26). We created a GTF file using all known transcripts from chromosome 17 as well as one with 20% of the transcripts missing (8.28% of exons missing). Using these two GTF files we performed transcript quantification with StringTie version 1.2.1 (23) as well as exon counting allowing multiple overlaps with the featureCounts function from Rsubread version 1.21.4 (13). ERs were determined with derfinder version 1.5.19 functions regionMatrix and railMatrix, respectively, from the HISAT BAM and Rail-RNA BigWig output using a mean cutoff of 5 for libraries adjusted to 80 million single-end reads. Count matrices resulting from featureCounts and derfinder were analyzed with limma (16), DESeq2 (14) and edgeR-robust (18) controlling the false discovery rate (FDR) at 5% and testing for differences between the two groups of samples. We used ballgown version 2.2.0 (22) to perform differential expression tests using coverage at the transcript and exon levels, controlling the FDR at 5%.
The 3 900 transcripts from chromosome 17 are composed in total by 39 338 exons (15 033 unique). To avoid ambiguous truth assignments, we used only the 3 868 that overlap only 1 transcript and assigned the truth status based on whether that transcript was set to have a high or low expression on group 2 for the replication replicate under evaluation. We assessed the different pipelines by checking if these 3 868 exons overlapped at least one differentially expressed unit: exons (featureCounts and ballgown), transcripts (ballgown) and ERs (derfinder), respectively. We then calculated the empirical power, false discovery rate and false positive rate.
RESULTS
Overview of the derfinder package
The derfinder package includes functions for several stages in the analysis of data from an RNA-sequencing experiment (Figure 1).
First, derfinder includes functions for pre-processing coverage data from BAM files or bigWig coverage files. The base-level coverage data for multiple samples can be loaded and filtered since most bases will show zero or very low coverage across most samples. Then, the software allows for definition of contiguous regions that show average coverage levels above a certain threshold. These ERs are non-overlapping subsets of the genome that can then be counted to arrive at a matrix with an expression value for each region in each sample. Alternatively, the software provides options for counting exons or genes for use in more standard analysis pipelines.
Next, derfinder can be used to perform statistical tests on the region level expression matrix. These tests can be carried out using any standard package for differential expression of RNA-seq data including edgeR (10,12), DESeq (11), DESeq2 (14) or limma-voom (17).
derfinder can then be used to annotate the DERs. We have developed functions that label each region according to whether it falls entirely in a previously annotated protein coding exon (exonic), entirely inside a previously annotated intronic region (intronic) or outside of any previously annotated gene (intragenic). The software also will report any region that overlaps any combination of those types of regions.
Finally, data from an expressed region analysis can be visualized using different visualization approaches. While region-level summaries can be plotted versus known phenotypes, derfinder also provides functions to plot base resolution coverage tracks for multiple samples, labeled with color according to phenotype.
We now provide more detail on each of these steps.
Finding ERs
The first step in a derfinder analysis is to identify ERs. Reads should be aligned using any splicing aware alignment tool such as TopHat2 (30), HISAT (29) or Rail-RNA (26).
Base resolution coverage information can be read directly from the BAM files that are produced by most alignment software (26,29,30). This process can be parallelized across multiple cores to reduce computational time. An alternative is to read bigWig (31) coverage files. Recent alignment software such as Rail-RNA (26) produces these files directly, or they can be created using samtools (32) or produced using the derfinder package. Reading BigWig files can produce significant computational and memory advantages over reading from BAM files.
The coverage information represents the number of reads that covers each genomic base in each sample. derfinder first filters out bases that show low levels of expression across all samples. Since most genomic bases are not expressed, this filtering step can reduce the number of bases that must be analyzed by up to 90%, reducing both CPU and memory usage. We originally proposed performing a statistical test for every base in the genome (19) and this approach is still supported by the derfinder package for backwards compatibility (Supplementary Section 1.3).
Here, we focus on a new approach based on the bump-hunting methodology for region level genomic analysis (33) (Figure 2). This approach first calculates ERs across the set of observed samples. For each base, the average, potentially library size-adjusted, coverage is calculated across all samples in the data set. This generates a vector of (normalized) mean level expression measurements across the genome. Then an average-coverage cutoff is applied to this mean coverage vector to identify bases that show minimum levels of expression. An expressed region is any contiguous set of bases that has expression above the mean expression cutoff.
The next step is to count the number of reads (including fractions of reads) that overlap each expressed region. As we have pointed out previously (19) that counting expression in genes and exons is complicated by overlapping annotation. ERs are non-overlapping, so this means that each read can be unambiguously assigned to the appropriate region.
ER level statistical tests
The result of the ER step is a coverage matrix with each row corresponding to one ER and each column corresponding to one sample. This count matrix can then be analyzed using statistical models that have been developed for gene or exon counts such as limma (15,16), voom (17), edgeR-robust (18) and DESeq2 (14). We emphasize that unlike other feature-level counting approaches, our approach is annotation-agnostic: ERs are defined empirically using the observed sample data and coverage threshold. So if there is sufficient expression in a region outside of previously annotated genes, it will be quantified and analyzed with our approach.
Visualizing DERs
After statistical modeling, derfinder produces a set of DERs with summary statistics per region. They are stored as a GRanges object (27) and can be visualized with a range of packages from the Bioconductor suite. We have also developed several visualization tools specific to the derfinder approach.
These plots can be made at different levels of summarization. First, the derfinder and derfinderPlot packages provide a range of visualizations of coverage tracks at single base resolution. These plots can be used to identify coverage patterns that may diverge from annotated protein-coding regions. For example, using the GTEx example we can visualize genes that have consistently high intronic expression as shown in Figure 3. We show several examples of genes known to be functionally important in heart—LBD3 and MYOZ2 (Figure 3A and B) (34,35) and liver—HGD and UPB1 (Figure 3C and D) (36,37). The coverage profiles can provide additional insight into transcription, and well as potential technical artifacts, beyond the level of annotated genes, exons and transcripts, which we include in our base-resolution plots.
Figure 3.

Coverage plots for the average coverage levels for the GTEx example. Average coverage profile for heart (blue), liver (red) and testis (green) from the GTEx example near genes: (A) LDB3, (B) MYOZ2, (C) HGD and (D) UPB1.
DERs can be grouped into larger regions by distance, which can be useful to identify potentially systematic artifacts such as coverage dips (Figure 4), perhaps due to sequence composition. Visualizing the base-level coverage for a set of nearby candidate DERs can reveal patterns that explain why one DER is sometimes fragmented into two or more shorter DERs. Coverage dips (Figure 4), spikes and data quality in general can affect the borders of the candidate DERs. Some artifacts can be discarded, like candidate DERs inside repetitive regions. Base-pairs inside repetitive regions available in repeat masker tracks can be flagged and filtered out from the analysis. Other known potentially problematic regions of the genome, like those with extreme GC content or mappability issues can also be filtered out, either before identifying candidate DERs or post-hoc.
Figure 4.

Example of a coverage dip. Mean coverage per group for the BrainSpan data set for a region that results in two DERs for a single exon due to a coverage dip. The genome segment shown corresponds to the DERs cluster ranked 15th in terms of overall signal by the single base-level approach applied to the BrainSpan data set.
Annotating DERs
The DERs can be annotated to their nearest gene or known feature using bumphunter (33). The basic approach is to overlap DERs genomic coordinates with the genomic coordinates of known genomic features. By default, derfinder labels each identified region as exonic, intronic, intragenic or some combination of those three labels.
A region may overlap multiple genomic features (say an exon and the adjacent intron). Using this information, candidate DERs can further be compared to known gene annotation tables (Methods Section 2.3) to identify potentially novel transcription events. Using this information, visualizations of specific loci for overlap with annotation can be made with derfinderPlot. The regions can be exported to CSV files or other file formats for follow-up and downstream analyses. We have also developed a complementary R package for creating reproducible reports incorporating the annotation and visualization steps of the derfinder pipeline called regionReport (38).
Application: large-scale expression analysis at base resolution
We used derfinder to detect regions that were differentially expressed across the lifespan in the human brain. We applied derfinder to the BrainSpan RNA-seq coverage data (Methods Section 2.4.1), a publicly available data set consisting of 484 postmortem samples across 16 brain regions from 40 unique individuals that collectively span the full course of human brain development (21). We used the expressed region approach described above for this analysis. For comparison we applied the single-based resolution approach previously utilized on independent dorsolateral prefrontal cortex RNA-seq data (20) (Supplementary Section 1.4).
We identified 174 610 ERs across the 484 samples with mean across-sample normalized coverage > 0.25, which constituted 34.57 megabases of expressed sequence. The majority (81.7%) of these ERs were labeled as strictly exonic while only a small subset (5.4%) were strictly non-exonic by Ensembl annotation. These ERs largely distinguished the fetal and postnatal samples using PCA – the first principal component explained 40.6% of the variance of the mean coverage levels and separated these developmental stages across all brain regions. This separation was consistent regardless of the annotation status of the DERs including in the strictly intronic regions (Figure 5 and Supplementary Figure S1). The separation between brain regions in intronic regions may be due to noisy or incorrect splicing (39) or may be due to missing annotation (19) or mistaken sequencing of pre-mRNA. The base resolution visualizations available as part of derfinder and derfinderPlot make it possible to explore to determine if it is biology or artifacts driving these expression differences.
Figure 5.

Principal components analysis reveals clusters of samples in the BrainSpan data set. (Left) First two principal components (PCs) with samples colored by sample type (F: Fetal or P: Postnatal) and shape given by brain region using only the strictly intronic ERs. Analysis of other subsets of ERs produce similar results (Supplementary Figure S1). (Right) Boxplots for PCs 1 and 2 by brain region (NCX: neocortex, HIP: hippocampus, AMY: amygdala, STR: striatum, MD: thalamus, CBC: cerebellum) and sample type with non-neocortex brain decomposed into its specific regions. Using the single base-level approach (Supplementary Figure S2) produces similar results as shown in Supplementary Figure S3.
The PCA plots also appear to show patterns consistent with potential artifacts such as batch effects (40) (Figure 5). Regardless, the new ER approach we present here provides options for analysts who wish to discover patterns of expression outside of known annotation on hundreds of samples – an analysis of this scope and scale was unfeasible with earlier versions of our single base resolution software (19).
Using statistical models where expression levels were associated with developmental stage (fetal versus postnatal) and/or brain region (Methods Section 2.4.1), we found that 129 278 ERs (74%) were differentially expressed by brain region and/or developmental stage at the ER-level controlling the family-wise error rate (FWER) at < 5% via Bonferroni correction. We controlled the FWER instead of the FDR due to the expected large effects between the developmental stages and/or brain regions. The 129 278 ER-level DERs overlapped a total of 17 525 Ensembl genes (13 016 with gene symbols), representing a large portion of the known transcriptome. Of the significant ER-level DERs, 93 355 (72.2%) overlapped at least 1 significant single base-level DER (Supplementary Section 1.4). Lack of overlap results from almost half (45.2%) of single base-level DERs having an average coverage lower than the expression cutoff determining ERs (0.25). For example, there was high expression only in the samples from a few brain regions, or only one development period.
Decreasing the cutoff that defines the ERs from 0.25 to 0.1 results in a larger number of regions (217 085) that have a higher proportion of non-exonic sequence (12.1%), suggesting that the choice of this expression cutoff requires some initial exploratory data analysis as shown in Supplementary Section 1.5. Increasing the cutoff reduces the number of ERs (Supplementary Figure S4A) and their lengths (Supplementary Figure S4B). With increasing cutoffs, the fraction of known exons present in the ERs is reduced (Supplementary Figure S4C) while increasing the percent of ERs that overlap known exons (Supplementary Figure S4D). We recommend using a cutoff that balances these factors (Supplementary Figure S4), such as 0.25 in this particular data set.
We highlight the utility of the ER-level analysis (using the original 0.25 cutoff) to identify regions differentially expressed within subsets of the data by analyzing brain regions within a single developmental period. We identified 1170 ERs that were differentially expressed comparing striatum versus hippocampus samples in the fetal developmental stage. These DERs mapped to 293 unique genes. Genes more highly expressed in the striatum include ARPP-21, previously shown to localize in the basal ganglia (41), and dopamine receptor genes DRD1 and DRD2 (42). Genes more highly expressed in the hippocampus in fetal life were strongly enriched for neurodevelopmental genes including FZD7 (43), ZBTB18 (44) and NEUROD1 (45). The ER-level analysis therefore permits subgroup analysis without the need to rerun the full derfinder single base-level pipeline – another improvement over previous versions of single base resolution analysis software (19).
DERs are non-standard in the sense that they don't necessarily match with known exons. Depending on the application, you might be interested in filtering out DERs that overlap problematic regions of the genome. This can be done prior to defining the ERs or once the candidate DERs have been identified. In the BrainSpan application, only 0.086% of the 129 278 DERs overlap ENCODE blacklisted regions (4) and 1.58% overlap lincRNAs. Similarly one can check if the DERs overlap other known features of interest. The genes overlapped by the DERs are enriched for GO terms such as neuron differentiation (GO:0030182, P-value 4.13e-15), neurogenesis (GO:0022008, P-value 4.62e-14) and neuron projection development (GO:0031175, P-value 1.4e-12) among other terms associated to neuronal development.
Identification of ERs that differentiate tissues using a subset of the GTEx data
We selected a subset of subjects from the GTEx project (3) that had RNA-seq data from heart (left ventricle), liver and testis, specifically the eight subjects with samples that had RINs greater 7, given RIN's impact on transcript quantification (46). Using only one sequencing library from each subject aligned with Rail-RNA (26), we applied the ER-level derfinder approach with a cutoff of 5 normalized reads (after normalizing coverage to libraries of 40 million reads). We found a total of 163 674 ERs with lengths greater than 9 base-pairs. Figure 6A shows that 118 795 (72.6%) of the ERs only overlapped known exonic regions of the genome using the Ensembl GRCh38.p5 database (47).
Figure 6.

GTEx ERs analysis using 24 samples from the heart (left ventricle), liver and testis for 8 subjects. (A) ERs (longer than 9 bp) overlapping known annotation based on GRGh38.p5 (hg38). 72.6% of the ERs only overlap known exons (strictly exonic) while 10.4% only overlap known introns (strictly intronic). (B) First two PCs with samples colored by sample type (red: liver, blue: heart, green: testis) using only the strictly exonic ERs. (C) First two PCs with samples colored by sample type using only the strictly intronic ERs. The sign change of the second principal component is simply a rotation and the results are consistent between the strictly exonic and strictly intronic ERs.
We performed PCA on the log2 adjusted coverage matrix using just the 118 795 strictly exonic ERs (Figure 6B). Here, the first two PCs explain 56.8% and 21.6% of the variance, respectively, and show three distinct clusters of samples that correspond to the tissue of the sample. We found that the 16 985 (10.4%) ERs (Figure 6A) that only overlap annotated introns can also differentiate tissues using PCA, as shown in Figure 6C. The total percent of variance explained by the first two principal components is slightly lower (44.4 + 26.6% = 71% versus 56.8 + 21.6% = 78.4%) when using only the strictly intronic ERs versus the strictly exonic ERs. This may represent a different biological signal and/or potentially noisy splicing (as in Figure 3B), but we use this example to illustrate the potential to use derfinder to explore regions outside of known annotation.
Using limma (15,16) to test for differential expression between tissues (Supplementary Methods Section 2.4.2) we found that 42 880 (36.1%) of the strictly exonic ERs and 4 401 (25.9%) of the strictly intronic ERs were differentially expressed (FWER of 5% via Bonferroni correction). Overall 59 776 (36.5%) of the ERs were differentially expressed between tissues. Given the similar global patterns of expression between annotated and unannotated ERs, we considered the scenario that the strictly intronic ERs were differentially expressed between tissues in the same pattern as the nearest exonic ERs due to possible run-off transcription events. To assess this scenario we fitted a conditional regression for each strictly intronic ER adjusting for the coverage of the nearest strictly exonic ER. A total of 749 (4.4%) of the strictly intronic ERs differentiate tissues while adjusting for the coverage at the nearest exonic ER at a FWER of 5%. Figure 7A and B shows an example where the expression is similar between tissues in the nearest exonic ER but there is a clear tissue difference in the intronic ER with testis having higher expression than the other two tissues. Figure 7C and D shows different patterns between the intronic and exonic ERs where in the exonic ER the expression is lowest in the heart, higher in liver and slightly higher at the testis. However, in the intronic ER, liver is the tissue that has the lowest expression. These results suggest that expression at unannotated sequence could have biological relevance beyond local annotated exonic sequence.
Figure 7.

Differential expression on strictly intronic ERs adjusting for expression on the nearest strictly exonic ER. Boxplots (A and C) and region coverage plots (B and D) for two strictly intronic ERs showing differential expression signal adjusting for the nearest exonic ER. Boxplots show the log2 adjusted coverage for the strictly intronic ERs by tissue with the corresponding boxplot for the nearest strictly exonic ERs. The P-value shown is for the differential expression between tissues on the intronic ERs conditional on the expression values for the nearest exonic ERs. The distance to the nearest strictly exonic ER and the gene symbol are shown below. The region coverage plots are centered at the strictly intronic ER with the neighboring 2 kb and 5 kb for (C) and (D), respectively. (A and B) Expression on the exonic ER is fairly similar between the groups but different on the intronic ER. (C and D) Expression on the exonic ER has an increasing pattern from heart to liver to testis but has a different pattern on the intronic ER.
Simulation results
We lastly performed a simulation study to evaluate the statistical properties of derfinder with and without complete annotation. To compare derfinder against feature-level alternatives, we simulated reads for 2 groups, 10 samples in total (5 per group) with 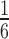 of the transcripts having higher and
of the transcripts having higher and 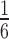 lower expression in group 2 versus group 1 at fold changes of 2x and
lower expression in group 2 versus group 1 at fold changes of 2x and 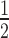 x, respectively. Reads were simulated from chromosome 17 using polyester (28) with the total number of reads matching the expected number given paired-end library with 40 million reads (Supplementary Methods Section 2.4.3). We used HISAT (29) to align the simulated reads and summarized them using either featureCounts from the Rsubread package (13) or StringTie (23) and performed the statistical tests on the resulting coverage matrices using limma and ballgown, (22) respectively. We performed the ballgown statistical test at the exon-level as well as the transcript-level. We performed the feature-level analyses using the complete annotation and with an annotation set missing 20% randomly selected transcripts (8.28% unique exons missing). We then used derfinder to find the ERs from the same HISAT alignments as well as from Rail-RNA (26) output and performed the statistical test with limma. For all statistical tests, we controlled the FDR at 5% and repeated the simulation three times.
x, respectively. Reads were simulated from chromosome 17 using polyester (28) with the total number of reads matching the expected number given paired-end library with 40 million reads (Supplementary Methods Section 2.4.3). We used HISAT (29) to align the simulated reads and summarized them using either featureCounts from the Rsubread package (13) or StringTie (23) and performed the statistical tests on the resulting coverage matrices using limma and ballgown, (22) respectively. We performed the ballgown statistical test at the exon-level as well as the transcript-level. We performed the feature-level analyses using the complete annotation and with an annotation set missing 20% randomly selected transcripts (8.28% unique exons missing). We then used derfinder to find the ERs from the same HISAT alignments as well as from Rail-RNA (26) output and performed the statistical test with limma. For all statistical tests, we controlled the FDR at 5% and repeated the simulation three times.
Table 1 shows the range of the empirical power, false positive rate (FPR) and FDR for all these methods based on the three simulation replicates. derfinder’s expressed region approach resulted in overlapping empirical power ranges to the exon-level methods that are supplied the complete annotation. The exon-level methods had a 18 to 27% loss in power when using the incomplete annotation set compared to the complete set even though only 8.28% of the unique exons were missing. derfinder, being annotation-agnostic, does not rely on having the complete annotation but did show increased FPR and FDR compared to the exon-level methods. We recommend performing sensitivity analyses of the cutoff parameter used for defining ERs or the FDR control in the statistical method used to determine which ERs are differentially expressed (i.e. DERs). Transcript-level analyses had the lowest FPR and FDR but also the lowest power. Note that we only performed transcript expression quantification with StringTie and did not use the data to determine new transcripts. Doing so resulted in a much larger transcript set than originally present in the data: 3 900 in the original set versus 15 920 (average for the three replicates using the complete annotation).
Table 1. Minimum and maximum empirical power, FPR and FDR observed from the three simulation replicates for each analysis pipeline. Ballgown analyses were done at either the exon or transcript levels. Pipelines that rely on annotation were run with the full annotation or with 20% of the transcripts missing (8.28% exons missing). Count matrices were analyzed with limma, DESeq2 and edgeR-robust (Supplementary Table S2). FDR of 5% was targeted.
| Power | FPR | FDR | Annotation complete | Aligner | Summary method | Statistical method |
|---|---|---|---|---|---|---|
| (93.6–94.2) | (6.4–9.3) | (12.8–16.5) | HISAT | derfinder | limma | |
| (93.7–94.2) | (6.5–9.1) | (12.5–16.1) | Rail-RNA | derfinder | limma | |
| (69–77.6) | (2.5–3.3) | (6–7.7) | No | HISAT | featureCounts | limma |
| (94.4–95.1) | (3.1–4.5) | (6.5–7.5) | Yes | HISAT | featureCounts | limma |
| (68.4–77) | (2.8–3) | (5.5–8.3) | No | HISAT | StringTie | ballgown-exon |
| (93.7–94.6) | (3.6–4) | (5.9–7.8) | Yes | HISAT | StringTie | ballgown-exon |
| (53.2–60) | (0.6–2.2) | (1.4–8.1) | No | HISAT | StringTie | ballgown-trans |
| (67.2–71.9) | (0.6–1.1) | (1.4–3.2) | Yes | HISAT | StringTie | ballgown-trans |
Supplementary Section 1.6.1 shows the results when using DEseq2 or edgeR-robust for performing the statistical tests. Figure 8 shows the mean empirical power against the observed FDR for the different combinations of methods when controlling the FDR at 1%, 5%, 10%, 15% and 20%. Results with derfinder are among the set with the highest empirical power, at the cost of a higher observed FDR than what was controlled for.
Figure 8.

Mean empirical power versus observed FDR across the three simulation replicates for a combination of statistical and summary methods. For FDR cutoffs of 1, 5, 10, 15 and 20% the mean empirical power and FDR across the 3 simulation replicates is displayed for the combination of statistical method (ballgown at exon or transcript level, limma, DESeq2, edgeR-robust) the summary method (derfinder, featureCounts (fC), StringTie (sT)) and whether the annotation used was complete or not (complete, incomplete).
Identifying ERs uses computational resources and runs in similar time to summarization steps required for the exon-level pipelines used in this simulation (Supplementary Section 1.6.2) and is the fastest when using BigWig files such as those produced by Rail-RNA. These results suggest that the derfinder approach performs well when differentially expressed features overlap known annotation and appear in unannotated regions of the genome. If you are only interested in studying known regions, other methods have better FDR control than derfinder as shown in Figure 8.
DISCUSSION
Here, we introduced the derfinder statistical software for performing genome-scale annotation-agnostic RNA-seq differential expression analysis. This approach utilizes coverage-level information to identify DERs at the expressed region or single base-levels, and then generates useful summary statistics, visualizations and reports to further inspect and validate candidate regions. derfinder's statistical model is flexible to allow answering any biological question related to differential expression analysis, such as multi-group comparisons and time-course analyses.
The reduced dependence on the transcriptome annotation permits the discovery of novel regulated transcriptional activity, such as the expression of intronic or intergenic sequences, which we highlight in publicly available RNA-seq data and our previous derfinder application (20). As shown with a subset of GTEx, strictly intronic ERs can differentiate tissues when adjusting for the expression from the nearest exonic expressed region, suggesting that some intronic DERs may represent signal beyond run-off transcription. Furthermore, the structure of DERs across a given gene can permit the direct identification of differentially expressed transcripts (e.g. Figure 2C), providing useful information for biologists running validation experiments. Lastly, this software and statistical approach may be useful for RNA-seq studies on less well-studies species, where transcript annotation is especially likely to be incomplete.
We hypothesize that many ERs, particularly in polyA+ data sets, likely belong to novel transcript isoforms, antisense expression, retained introns or extended UTRs that can relate to novel insights into particular biological questions. For example, identifying extensive differentially expressed intronic ERs might point to deficits in splicing or a potential novel transcript isoform with extended exonic boundaries related to the outcome of interest. Or, differentially expressed intergenic ERs might point to previously uncharacterized regulatory RNAs like polyadenylated lincRNAs that might be related to the outcome of interest. As described in the introduction, we have previously demonstrated the utility of the DER finder approach in the human brain, which suggested that previously unannotated expressed sequence was developmentally regulated, expressed in other brain regions and cell types and associated with clinical risk for schizophrenia (20).
The software pipeline, starting with BAM or BigWig files, and ending with lists of DERs, reports and visualizations, runs at comparable speeds to existing RNA-seq analysis software. Given the appropriate computing resources, derfinder can scale to analyze studies with several hundred samples. For such large studies, it will be important to correct for batch effects and potentially expand derfinder's statistical model for base-level covariates. This approach provides a powerful intermediate analysis approach that combines the benefits of feature counting and transcript assembly to identify differential expression without relying on existing gene annotation.
AVAILABILITY
The derfinder vignettes detail how to use the software and its infrastructure. The latest versions are available at www.bioconductor.org/packages/derfinder. The code and log files detailing the versions of the software used for all the analyses described in this paper is available at the Supplementary Website: leekgroup.github.io/derSupplement.
Acknowledgments
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health. Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI/SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171) and Science Care, Inc. (X10S172). The Laboratory, Data Analysis and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to The Broad Institute, Inc. Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The raw data (sequencing reads and phenotype data) used for the analyses described in this manuscript were obtained from SRA accession number phs000424.v6.p1 on 10/07/2015.
Author's contributions: A.E.J., J.T.L., R.A.I. conceived the software. L.C.T. wrote the software under the supervision of J.T.L. and A.E.J. L.C.T analyzed the data with the supervision of J.T.L. and A.E.J. A.N., C.W. and B.L. helped with the GTEx data analysis. All authors contributed to writing the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [1R01GM105705 to J.T.L.]; Consejo Nacional de Ciencia y Tecnología México [351535 to L.C.T.]; A.E.J. was supported by 1R21MH109956.
Conflict of interest statement. None declared.
REFERENCES
- 1.Farrell C.M., O'Leary N.A., Harte R.A., Loveland J.E., Wilming L.G., Wallin C., Diekhans M., Barrell D., Searle S. M.J., Aken B., et al. Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res. 2014;42:D865–D872. doi: 10.1093/nar/gkt1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ENCODE Project Consortium. Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dillman A.A., Hauser D.N., Gibbs J.R., Nalls M.A., McCoy M.K., Rudenko I.N., Galter D., Cookson M.R. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci. 2013;16:499–506. doi: 10.1038/nn.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daines B., Wang H., Wang L., Li Y., Han Y., Emmert D., Gelbart W., Wang X., Li W., Gibbs R., et al. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res. 2011;21:315–324. doi: 10.1101/gr.107854.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steijger T., Abril J.F., Engström P.G., Kokocinski F., Abril J.F., Akerman M., Alioto T., Ambrosini G., Antonarakis S.E., RGASP Consortium et al. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao Y., Smyth G.K., Shi W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 14.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 17.Law C.W., Chen Y., Shi W., Smyth G.K. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X., Lindsay H., Robinson M.D. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 2014;42:e91. doi: 10.1093/nar/gku310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frazee A.C., Sabunciyan S., Hansen K.D., Irizarry R.A., Leek J.T. Differential expression analysis of RNA-seq data at single-base resolution. Biostatistics. 2014;15:413–426. doi: 10.1093/biostatistics/kxt053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaffe A.E., Shin J., Collado-Torres L., Leek J.T., Tao R., Li C., Gao Y., Jia Y., Maher B.J., Hyde T.M., et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat. Neurosci. 2015;18:154–161. doi: 10.1038/nn.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.BrainSpan Atlas of the Developing Human Brain. 2011. http://www.brainspan.org/ Funded by ARRA Awards 1RC2MH089921-01, 1RC2MH090047-01, and 1RC2MH089929-01.
- 22.Frazee A.C., Pertea G., Jaffe A.E., Langmead B., Salzberg S.L., Leek J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015;33:243–246. doi: 10.1038/nbt.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rainer J. EnsDb.Hsapiens.v75: Ensembl based annotation package. 2015. http://www.bioconductor.org/packages/EnsDb.Hsapiens.v75 R package version 0.99.12. [Google Scholar]
- 25.Falcon S., Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 26.Nellore A., Collado-Torres L., Jaffe A. E., Alquicira-Hernández J., Wilks C., Pritt J., Morton J., Leek J.T., Langmead B. Rail-RNA: Scalable analysis of RNA-seq splicing and coverage. Bioinformatics. 2016:btw575. doi: 10.1093/bioinformatics/btw575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M., Carey V. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frazee A.C., Jaffe A.E., Langmead B., Leek J.T. Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics. 2015;31:2778–2784. doi: 10.1093/bioinformatics/btv272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim D., Langmead B., Salzberg S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D. BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics. 2010;26:2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaffe A.E., Murakami P., Lee H., Leek J.T., Fallin M.D., Feinberg A.P., Irizarry R.A. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 2012;41:200–209. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selcen D., Engel A.G. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann. Neurol. 2005;57:269–276. doi: 10.1002/ana.20376. [DOI] [PubMed] [Google Scholar]
- 35.Osio A., Tan L., Chen S.N., Lombardi R., Nagueh S.F., Shete S., Roberts R., Willerson J.T., Marian A.J. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2007;100:766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duncan A.W., Hanlon Newell A.E., Bi W., Finegold M.J., Olson S.B., Beaudet A.L., Grompe M. Aneuploidy as a mechanism for stress-induced liver adaptation. J. Clin. Investig. 2012;122:3307–3315. doi: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakamoto T., Sakata S.F., Matsuda K., Horikawa Y., Tamaki N. Expression and properties of human liver beta-ureidopropionase. J. Nutr. Sci. Vitaminol (Tokyo) 2001;47:132–138. doi: 10.3177/jnsv.47.132. [DOI] [PubMed] [Google Scholar]
- 38.Collado-Torres L., Jaffe A.E., Leek J.T. regionReport: Interactive reports for region-level and feature-level genomic analyses [version2; referees: 2 approved, 1 approved with reservations] F1000Research. 2016;4:1–10. doi: 10.12688/f1000research.6379.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pickrell J.K., Pai A.A., Gilad Y., Pritchard J.K. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leek J.T., Scharpf R.B., Bravo H.C., Simcha D., Langmead B., Johnson W.E., Geman D., Baggerly K., Irizarry R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouimet C.C., Hemmings H.C., Greengard P. ARPP-21, a cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions. II. Immunocytochemical localization in rat brain. J. Neurosci. 1989;9:865–875. doi: 10.1523/JNEUROSCI.09-03-00865.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cachope R., Cheer J.F. Local control of striatal dopamine release. Front. Behav. Neurosci. 2014;8:1–7. doi: 10.3389/fnbeh.2014.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melchior K., Weiss J., Zaehres H., Kim Y.-m., Lutzko C., Roosta N., Hescheler J., Müschen M. The WNT receptor FZD7 contributes to self-renewal signaling of human embryonic stem cells. Biol. Chem. 2008;389:897–903. doi: 10.1515/BC.2008.108. [DOI] [PubMed] [Google Scholar]
- 44.Tatard V.M., Xiang C., Biegel J.A., Dahmane N. ZNF238 is expressed in postmitotic brain cells and inhibits brain tumor growth. Cancer Res. 2010;70:1236–1246. doi: 10.1158/0008-5472.CAN-09-2249. [DOI] [PubMed] [Google Scholar]
- 45.Poulin G., Turgeon B., Drouin J. NeuroD1/beta2 contributes to cell-specific transcription of the proopiomelanocortin gene. Mol. Cell. Biol. 1997;17:6673–6682. doi: 10.1128/mcb.17.11.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gallego Romero I., Pai A.A., Tung J., Gilad Y. RNA-seq: impact of RNA degradation on transcript quantification. BMC Biol. 2014;12:1–13. doi: 10.1186/1741-7007-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]