

This article opens a series of short comments on paradoxes and wonders of the protein intrinsic disorder phenomenon. Here, the “prevalence of exceptionality” paradox is introduced in a form of a brief historical overview that shows a progression in understanding of the natural abundance of intrinsically disordered proteins from the early days, when these biologically active proteins without unique structures were taken as rare exceptions, to the current days, when the prevalence of intrinsically disordered proteins (IDPs) in various proteomes and biological processes is a well-recognized reality.
In one of the first systematic works on IDPs published in 1997,1 Dunker et al. searched PDB for proteins containing at least one intrinsically disordered region (IDR) longer than seven 7 residues. There, IDRs were defined as regions of missing electron density in the corresponding crystal structures (since disorder leads to incoherent X-ray scattering and subsequent absence of electron density in the solved structure), and, depending on their length, were partitioned into short, medium and long data sets, denoted as SIDR (7–21 amino acids), MIDR (22–44 amino acids), and LIDR (45 or more amino acids), respectively.1 The SIDR dataset contained 38 disordered segments from 34 proteins with 411 disordered amino acids and 11,050 total amino acids; MIDR set contained 22 disordered segments from 20 proteins with 464 disordered amino acids and 4,764 total amino acids; and LIDR set contained 7 regions from 7 proteins with 465 disordered amino acids and 2,069 total amino acids.1 In the subsequent study, the set of seven 7 LIDR proteins with the X-rayX-ray-characterized regions of disorder was extended to include seven 7 proteins shown to be disordered by NMR, with the total number of disordered residues in that set being 677 amino acids.2 Uversky et al. compiled a list of 91 IDPs characterized by NMR, circular dichroism or other biophysical techniques.3 Proteins in that study were completely disordered, belonging to the sub-class of natively unfolded proteins that do not have any (or almost any) residual structure. Those IDPs ranged in length from 49 to 1,827 residues and the total number of disordered residues in that set was 17,318 amino acids.4 In 2001, it has been reported that the list of experimentally validated IDPs characterized by NMR or X-ray, or circular dichroism can be extended to 150 entries that contained 17,417 disordered residues5 and a year later, this set was further extended to total 157 proteins with 18,833 disordered residues6 A subsequent search of X-rayX-ray crystal structures and the literature have further expanded this list to more than 200 proteins that contain disordered regions of 30 consecutive residues or longer as characterized by X-ray crystallography, proteolytic digestion or other physical analyses such as NMR or circular dichroism.7-9 Recently, the exhaustive literature analysis revealed that the current list of experimentally validated IDPs includes ∼1,150 non-redundant proteins (DeForte S., Uversky V.N., manuscript in preparation)
Careful analysis and comparison of the non-redundant sets of ordered and disordered proteins (where IDPs/IDRs were characterized by different experimental techniques, such X-rayX-ray crystallography, NMR and CD) revealed that IDRs share at least some common sequence features over many proteins and that amino acid sequences of IDPs/IDRs are different from those of ordered protein and domains.2,10 Finding numerous proteins with the experimentally characterized regions of disorder and recognizing that amino acid sequences of IDPs/IDRs and ordered protein and domains are significantly different opened a possibility for the development of rather accurate predictors of intrinsic disorder. One of the extremely useful features of the computation tools is their applicability for the large scale analyses of various datasetdata sets and proteomes. In attempt to have an educated guess on the natural abundance of intrinsic disorder, Romero et al. developed neural network predictors of protein disorder using primary sequence information and applied these tools to the Swiss Protein Database11 With more than 15,000 proteins being predicted to contain disordered regions of at least 40 consecutive amino acids, and with more than 1,000 proteins having especially high disorder scores11 that analysis resulted in shocking and completely unexpected conclusion – disordered proteins are not as rare as it was originally expected, suggesting that the presence of intrinsic disorder in a protein is not an exception, but a rule!
Several subsequent studies, where various computational tools were applied to the different large datasets, databases, and genomes, provided very strong support to these first observations. In one of the first study of that kind, the commonness of IDPs/IDPRs was estimated by predicting disorder for whole genomes containing both known and putative protein sequences.12 The analyzed in that study proteins belonged to 3131 g genomes from 3 kingdoms of life, and the percentage of sequences in each genome with segments predicted to have ≥40 consecutive disordered residues by one of the early PONDR® predictors was used to gain an overview of proteomic disorder.12 This analysis revealed that the eukaryotes exhibited more disorder than either the prokaryotes or the archaea, with C. elegans, A. thaliana, S. cerevisiae, and D. melanogaster being predicted to have 52–67% their proteins with long IDRs, and with bacteria and archaea being predicted to have 16–45% and 26–51% proteins with such long IDRs, respectively.4,12 Later, using a conservative disorder classifier, DISOPRED2, Ward et al. revealed that putative, long (>3030 residue) IDR can be found in 2.0% of archaean, 4.2% of eubacterial and 33.0% of eukaryotic proteins.13 The difference in numbers of IDR-containing proteins generated by these two 2 studies was explained by the difference in the false positive rates, which were at the level of 16% for disordered segments longer than 40 residues in the PONDR®-based study and with false positive rates estimated to be lower than 0.5% on long disordered segments in the DISOPRED2-based study.13 Alternative analysis of several whole proteomes utilizing binary disorder predictors; i.e., predictors that indicate if a query protein is expected to be ordered or disordered as a whole and are based on the net charge-hydropathy distribution and disorder prediction score distribution, and by a corresponding consensus-based method revealed that approximately 4.5% of Yersinia pestis, 5% of Escherichia coli K12, 6% of Archaeoglobus fulgidus, 8% of Methanobacterium thermoautotrophicum, 23% of Arabidopsis thaliana, and 28% of Mus musculus proteins were expected to be mostly disordered.4
There were several subsequent studies dedicated to the large-scale analyses of the abundance of intrinsic disorder. For example, the abundance of IDPs and IDRs in 53 archaea species14 or in 332 prokaryotic proteomes15 was evaluated. Later, the completed proteomes of 3,484 species from three 3 domains of life (archaea, bacteria and eukaryotes) and from viruses were evaluated by PONDR® VSL2 for the presence of IDPs and IDRs.16 Results of this analysis are shown in Figure 1 that represents the correlation between the intrinsic disorder content and proteome size for 3,484 species from viruses, archaea, bacteria and eukaryotes and clearly indicates that (a) viruses are characterized by the widest spread of the proteome disorder content (the percentage of disordered residues ranges from 7.3% in human coronavirus NL63 to 77.3% in Avian carcinoma virus); (b) eukaryotic proteomes are typically more disordered than proteomes of archaea and bacteria; and (c) there is a well-defined gap between the prokaryotes and eukaryotes in the plot of fraction of disordered residues on proteome size.16 The presence of this gap, where almost all eukaryotes have 32% or more disordered residues, whereas the large majority of the prokaryotic species have 27% or fewer disordered residues, suggested in transition from the morphologically less-complex prokaryotes to the morphologically more-complex eukaryotes the gain in the complexity of cellular morphology was compensated by a leap in intrinsic disorder content16 Very recently, a broad and detailed computational analysis of 6,438,736 proteins from 965 complete proteomes (59 archaea, 471 bacterial, 110 eukaryotic, and 325 viral proteomes) was performed using arguably more accurate consensus-based disorder predictions.17 In this work too, high natural abundance of disorder was observed, with higher prevalence of IDPs/IDPRs being found in eukaryotic proteomes.17 Walsh et al. analyzed 25,833 UniProt proteins with disorder annotations from the X-ray crystallographic data using a total of 11 fast disorder predictors with different disorder flavors, and showed that these crystallizable proteins are expected to contain 350,858 disordered residues combined in 23,566 short and 3,439 long (>2020 residues) IDRs.18
Figure 1.
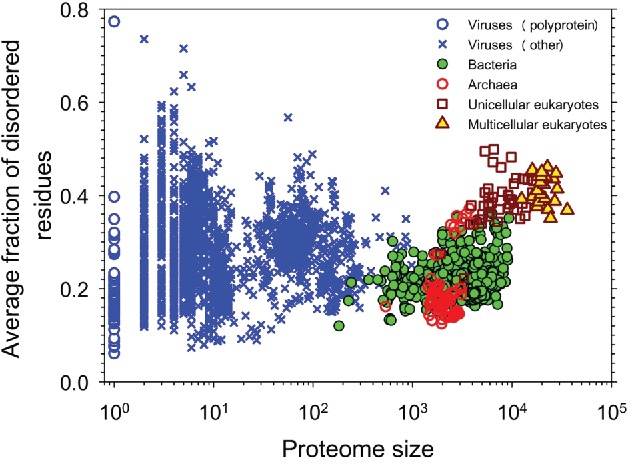
Correlation between the intrinsic disorder content and proteome size for 3,484 species from viruses, archaea, bacteria, and eukaryotes. Each symbol indicates a species. There are totally six 6g groups of species: viruses expressing one polyprotein precursor (open blue circles), other viruses (blue crosses), bacteria (green circles), archaea (red open circles), unicellular eukaryotes (brown open squares), and multicellar eukaryotes (yellow triangles). Each viral polyprotein was analyzed as a single polypeptide chain, without parsing it into the individual proteins before predictions. The proteome size is the number of proteins in the proteome of that species shown in the log base. The average fraction of disordered residues is calculated by averaging the fraction of disordered residues of each sequence over the all sequences of that species. Disorder prediction is evaluated by PONDR® VSL2B. Based this plot is based on data published in ref.16
To conclude this journey from the “rare exceptions” to the “exceptionally abundant exceptions” and the “prevalence of exceptionality,” a D2P2 database19 and MobiDB platform20,21 have to be mentioned. D2P2 is a Database of Disordered Protein Prediction that is available at http://d2p2.pro and represents predicted disorder information for 10,429,761 proteins from 1,765 complete proteomes.19 Disorder propensities of all these proteins are pre-computed by several disorder predictors and their variants (PONDR® VLXT, PONDR® VSL2b, PrDOS, PV2, Espritz, and IUPred). The output is further enhanced by presenting positions of posttranslational modifications, disorder-based binding sites, and locations of conserved functional domains.19 MobiDB (http://mobidb.bio.unipd.it/) is a database of intrinsically disordered and mobile proteins that provides the most complete picture on different flavors of disorder in protein structures covering all UniProt sequences (currently over 80 million).20,21 This platform represents outputs of 10 disorder predictors (three 3 ESpritz flavors, two 2 IUPred flavors, two 2 DisEMBL flavors, GlobPlot, PONDR® VSL2b and JRONN) and also generates a consensus annotation and classification for long disordered regions.20,21 In addition to showing disorder predispositions of UniProt proteins, MobiDB includes annotations of their posttranslational modifications, linear motifs, and Pfam domains. Furthermore, it shows experimental protein-protein interactions from STRING (http://string-db.org/), with binding partners being also annotated with their disorder contents.20,21
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Dunker AK. Identifying disordered regions in proteins from amino acid sequences. IEEE International Conference on Neural Networks Houston, Texas, USA: IEEE Service Center, 1997:90-5 [Google Scholar]
- 2.Garner E, Cannon P, Romero P, Obradovic Z, Dunker AK. Predicting Disordered Regions from Amino Acid Sequence: Common Themes Despite Differing Structural Characterization. Genome Inform Ser Workshop Genome Inform 1998; 9:201-13; PMID:11072336 [PubMed] [Google Scholar]
- 3.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000; 41:415-27; PMID:11025552; http://dx.doi.org/ 10.1002/1097-0134(20001115)41:3%3c415::AID-PROT130%3e3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 4.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005; 44:1989-2000; PMID:15697224; http://dx.doi.org/ 10.1021/bi047993o [DOI] [PubMed] [Google Scholar]
- 5.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins 2001; 42:38-48; PMID:11093259; http://dx.doi.org/ 10.1002/1097-0134(20010101)42:1%3c38::AID-PROT50%3e3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- 6.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins 2003; 52:573-84; PMID:12910457; http://dx.doi.org/ 10.1002/prot.10437 [DOI] [PubMed] [Google Scholar]
- 7.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, et al.. Intrinsically disordered protein. J Mol Graph Model 2001; 19:26-59; PMID:11381529; http://dx.doi.org/ 10.1016/S1093-3263(00)00138-8 [DOI] [PubMed] [Google Scholar]
- 8.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry 2002; 41:6573-82; PMID:12022860; http://dx.doi.org/ 10.1021/bi012159+ [DOI] [PubMed] [Google Scholar]
- 9.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem 2002; 62:25-49; PMID:12418100; http://dx.doi.org/ 10.1016/S0065-3233(02)62004-2 [DOI] [PubMed] [Google Scholar]
- 10.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput 1998:473-84; PMID:9697205 [PubMed] [Google Scholar]
- 11.Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Garner E, Guilliot S, Dunker AK. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput 1998:437-48; PMID:9697202 [PubMed] [Google Scholar]
- 12.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform 2000; 11:161-71; PMID:11700597 [PubMed] [Google Scholar]
- 13.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004; 337:635-45; PMID:15019783; http://dx.doi.org/ 10.1016/j.jmb.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 14.Xue B, Williams RW, Oldfield CJ, Dunker AK, Uversky VN. Archaic chaos: intrinsically disordered proteins in Archaea. BMC Syst Biol 2010; 4 Suppl 1:S1; PMID:20522251; http://dx.doi.org/ 10.1186/1752-0509-4-S1-S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burra PV, Kalmar L, Tompa P. Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PLoS One 2010; 5:e12069; PMID:20711457; http://dx.doi.org/ 10.1371/journal.pone.0012069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn 2012; 30:137-49; PMID:22702725; http://dx.doi.org/ 10.1080/07391102.2012.675145 [DOI] [PubMed] [Google Scholar]
- 17.Peng Z, Yan J, Fan X, Mizianty MJ, Xue B, Wang K, Hu G, Uversky VN, Kurgan L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci 2015; 72:137-51; PMID:24939692; http://dx.doi.org/ 10.1007/s00018-014-1661-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh I, Giollo M, Di Domenico T, Ferrari C, Zimmermann O, Tosatto SC. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics 2015; 31:201-8; PMID:25246432; http://dx.doi.org/ 10.1093/bioinformatics/btu625 [DOI] [PubMed] [Google Scholar]
- 19.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L, et al.. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res 2013; 41:D508-16; PMID:23203878; http://dx.doi.org/ 10.1093/nar/gks1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Domenico T, Walsh I, Martin AJ, Tosatto SC. MobiDB: a comprehensive database of intrinsic protein disorder annotations. Bioinformatics 2012; 28:2080-1; PMID:22661649; http://dx.doi.org/ 10.1093/bioinformatics/bts327 [DOI] [PubMed] [Google Scholar]
- 21.Potenza E, Di Domenico T, Walsh I, Tosatto SC. MobiDB 2.0: an improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res 2015; 43:D315-20; PMID:25361972; http://dx.doi.org/ 10.1093/nar/gku982 [DOI] [PMC free article] [PubMed] [Google Scholar]