

Abstract
Many human proteins are predicted to contain intrinsically disordered regions (IDRs), yet their occurrence in enzymes is notably rare. Human pancreatic glucokinase (GCK) is one of a small, but growing number of enzymes shown to possess an IDR. In this commentary, we summarize the results of recent biophysical studies that provide evidence for a functionally significant disorder-order transition within the IDR of GCK during the enzyme's catalytic cycle. High-resolution NMR studies indicate that kinetic cooperativity in GCK results from glucose-mediated millisecond conformational dynamics within the structurally heterogeneous and partially disordered small domain of this monomeric enzyme, whereby the precise timescale of these motions is critical for the manifestation of the kinetic cooperativity effect. GCK provides an excellent case study for understanding how structural and dynamic alterations within an IDR enable novel regulatory mechanisms. These studies also establish GCK as a model system for investigating the functional consequences of disorder and conformational heterogeneity in enzymatic systems in general.
Keywords: allosteric regulation, enzymes, glucokinase, intrinsically disordered regions, intrinsically disordered proteins, kinetic cooperativity, NMR
The textbook view of proteins describes them as well-folded, stable 3-dimensional structures that may possess limited flexibility at the termini, loops and interfacial regions. The advent of solution-based biophysical methods including nuclear magnetic resonance and single-molecule spectroscopy has led to a growing appreciation that many proteins are more dynamic than is implied by the traditional view. It is now accepted that proteins are best described by an ensemble of interconverting structural states. At one extreme of this ensemble view of protein “structure” are intrinsically disorder proteins (IDPs). IDPs and their associated intrinsically disordered regions (IDRs) are generally capable of sampling large portions of conformational space. IDRs typically lack a unique, stable tertiary structure and they are often found to be involved in the regulation of various cellular processes.1-9 The functional benefits of IDRs for transcription factors and other proteins with multiple binding partners have become quite clear. Moreover, it is becoming more apparent how the absence of structure can lead to the formation of amyloid fibrils that are associated with certain degenerative diseases, such as Alzheimer, Down syndrome, Parkinson, and prion disease.6-9
Enzymes are often viewed through the traditionalist lens of protein structure, namely X-ray crystallography, which has provided a tremendous amount of invaluable structural information about proteins in their crystalline state. As a consequence, enzymes are often used as prototypical examples of the structure-function paradigm, in which the 3D structure of a protein determines its function. Surprisingly few examples of partially or wholly disordered enzymes have been described in the literature. They include the urease accessory protein UreG, Ribonuclease T1, an acylphosphatase from Sulfolobus solfataricus, and a circularly permutated dihydrofolate reductase from Escherichia coli.10,11 A particularly noteworthy example of an intrinsically disordered enzyme is the designed monomer of chorismate mutase (mMjCM), which appears to be the first example of an enzyme that retains high catalytic efficiency in the absence of a well-ordered 3-dimensional conformation.12
The ability of proteins to sample multiple conformational sub-states has long been known to play a role in the regulation of enzyme function. For example, ligand-mediated conformational transitions lie at the center of classical allosteric models. Nevertheless, these traditional models tend to describe cooperativity on the basis of equilibrium shifts between static endpoint protein structures that emerged from early crystallographic studies of allosteric systems.13,14 Later, the possibility that long-range communication can be mediated though dynamic mechanism(s), which rely on conformational entropic contributions without changes in the structure was put forth.15 More recently, the concept of dynamic allostery has been emphasized, in which allosteric regulation is manifested as changes in the width of conformational distributions experienced by proteins. As more allosteric systems are identified, the dynamic view of allostery is becoming more prevalent.16,17 Functional characteristics of IDRs such as structural plasticity, enrichment of post-translational modification sites, and low intrinsic ligand binding affinity are beneficial for the requirements of allostery. In the new dynamic view, allosteric inhibitors or activators can achieve precise regulation via modulation, or population shift, of the ensemble distribution.16,17
Human pancreatic glucokinase (GCK) represents an excellent model system to study the role of disorder and conformational heterogeneity to regulation. GCK is a glucose-phosphorylating enzyme that functions as the glucose sensor of the functions as the body's glucose sensor. GCK function is tightly regulated by the amount of glucose present in the bloodstream via a cooperative kinetic response to glucose.18 The rate of glucose phosphorylation catalyzed by GCK follows a sigmoidal dependence upon glucose concentrations, rather than the classical hyperbolic dependence displayed by Michaelis-Menten enzymes. The sigmoidicity in the kinetics of GCK represents the hallmark of allosteric regulation. However, this type of regulation is different from the thermodynamic allostery observed for ligand binding to multimeric proteins such as hemoglobin. Since GCK is monomeric with one active site, early mechanisms postulated that kinetic cooperativity couldn't be achieved unless at least 2 structurally different conformations of the enzyme existed in solution.19,20 These mechanism(s) were formulated with conformational heterogeneity in mind at a time when proteins and especially enzymes were regarded as static structures. Conformational heterogeneity and slow conformational changes have been the common postulate in both the Mnemonic and Ligand Induced Slow Transition (LIST) models that describe kinetic cooperativity. In the Mnemonic model conformational heterogeneity is less emphasized compared to the LIST model. Ainslie et al,20 were the first to observe slow conformational changes accompanying glucose binding using rapid kinetics measurements and fluorescence spectroscopy and postulated that multiple conformations of the enzyme are capable of participating in the catalytic step. Although other kinetic models have been proposed to account for the complex kinetic curves of glucose binding, elucidation of the 3D structure of GCK remained elusive until 2004.18,21
The interest in understanding the molecular mechanism of GCK's regulation spiked with the realization that this enzyme could serve as a therapeutic target for patients suffering from Maturity-onset Diabetes of the Young 2 (MODY 2) and Permanent Neonatal Diabetes Mellitus (PNDM). Pharmaceutical companies around the globe have invested in the discovery of small molecules that can act as GCK activators. This work also resulted in the determination of the GCK crystal structure in the unliganded state and in the activator- and glucose-bound forms, which provided a static description of the catalytic process. These structures also revealed the degree of conformational changes experienced by the small domain between the unliganded and glucose-bound states. In the unliganded structure, amino acid region 151–180 lacks electron density while in the glucose-bound structure, this IDR folds into a β-hairpin.21 Even with the availability of at least 29 crystal structures of GCK, some of which have depicted a different opening angle between the large and the small domain, the detailed molecular mechanism by which glucose regulates GCK function remained still unanswered. A few questions that remained unanswered by the static structural studies included: is conformational heterogeneity in GCK a result of a simple rigid body hinge motion, which would explain the different opening angles, or are there more complex conformational dynamics processes at play to produce the regulatory effect? What is the physiological role of 151–180 IDR in GCK function or regulation?
Our recent work21 provides new insights about how conformational heterogeneity, coupled with conformer interconversion on the proper timescale, is exploited by GCK to generate precise enzyme regulation. GCK cooperativity is portrayed in terms of large-scale, glucose-mediated conformational transitions based upon the NMR spectroscopic properties of 1713 C-isotopically labeled isoleucine methyl groups and 3 tryptophan side chains. It was found that GCK displays both conformational heterogeneity and disorder. Amino acid region 151–180 contains 2 reporters, Ile-159 and Ile-163, both of which display chemical shifts in the disordered region of the 2D1H-13C HMQC NMR spectrum in the absence of glucose. Glucose addition leads to folding of this IDR into a β-hairpin, consistent with the X-ray crystallographic studies. The authors postulate that the 151–180 IDR, together with the α-13 helix, form a path by which the allosteric site communicates with the active site. In the crystal structure of glucose-bound GCK, 151–180 IDR is folded into a β-hairpin and forms specific H-bonding interactions with glucose in the active site via Lys-169 and hydrophobic interactions with the α-13 helix and allosteric activator via Ile-159 and Val-452. Binding of the activator molecule might stabilize the interaction between the α-13 helix and IDR residues, which is then communicated to the active site through Lys-169. In support of this hypothesis, structural stabilization of the α-13 helix via mutations leads to chemical shifts of residues Ile-159 and Ile-163 away from the disorder chemical shift region of the 2D1H-13C HMQC spectrum.
In addition, the study by Larion et al.22 finds that the small domain of unliganded GCK samples a broad conformational ensemble on the timescale predicted to give rise to conformational non-equilibration and kinetic cooperativity. A model in which GCK generates its cooperative kinetic response at low glucose concentrations by using a millisecond conformational exchange cycle of the small domain as a “time-delay loop” was proposed. At high glucose concentrations, this loop is bypassed, providing a unique mechanism to regulate the activity of human GCK under physiological conditions (Fig. 1). Additional evidence to support the existence of extensive dynamics in GCK comes from fast H/D exchange NMR studies. 80% of the signal is lost over the first 2 hours of acquisition, suggesting flexibility and solvent exposure of amide backbone moieties.23 Transient-state kinetic studies using fluorescence spectroscopy, together with the NMR work, identified multiple transitions upon glucose binding and directly detected heterogeneity in enzyme conformation.23,24 Quantification of the timescales associated with conformational transitions in GCK is currently under way. Preliminary results suggest that the timescales observed for these transitions coincide with the predicted timescale that would lead to the kinetic cooperativity effect. Conformational heterogeneity could also explain the ability of GCK to bind multiple proteins such as the glucokinase regulatory protein (GKRP) and/or BAD.
Figure 1.
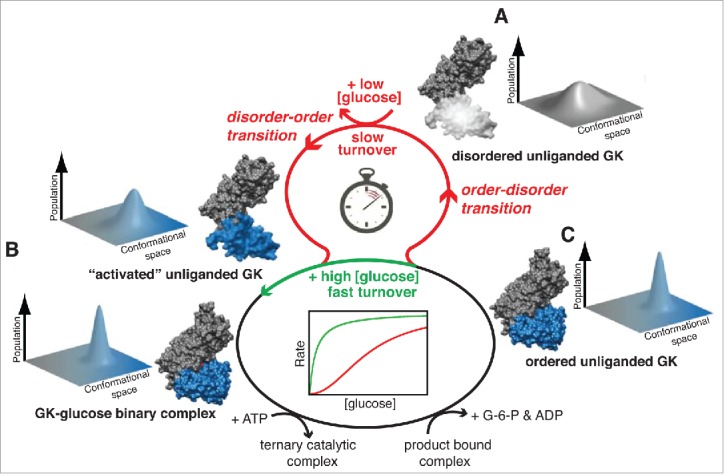
Role of intrinsic disorder for kinetic cooperativity of GCK. (A) Disorder-order transitions involving a conformational ensemble experienced by the small domain of GCK on a timescale comparable to the turnover rate constant. (B) GCK activation via binding of small molecule allosteric activators or via mutations leads to narrowing in the conformer distribution and folding of 151–180 IDR into a β-hairpin. (C) Glucose concentration controls the flow into the time-delay loop leading to a sigmoidal kinetic profile. Figure taken from Ref. Twenty-two.
In the future it will be of interest to quantify the degree of conformational heterogeneity in the unliganded state of the enzyme, but such quantification will not be straightforward especially using ensemble-averaged methods. Single molecule studies could shed some light on this issue. The structural description of the unliganded conformers that are physiologically relevant will be most informative. In addition, it will be worthwhile to investigate if the amino acid region 151–180 is the only disordered region in GCK or if other regions display partial disorder in solution. The extent to which the unliganded crystal structure represents a catalytically relevant conformational state and not a stabilized conformation inside the crystal is also of interest, since only one crystal structure of unliganded GCK has been reported.
Perspective
Since the first reviews were published bringing attention to the potential roles of IDPs for protein function, many IDPs have been reported and the link between a lack of structure in these proteins and their function has been rationalized by adopting a dynamic view.1-3,6,25-30 For enzymes, the number of known IDRs is still small, which in part could be also due to the primary use of crystallography for the determination of the structure of these large proteins. In the future, with the further development of existing and novel complementary techniques, such as in-cell NMR or single molecule based methods that allow the direct identification and structural characterization of IDRs in their physiologically relevant environment, it would not come as a surprise if more intrinsically disordered enzymes (IDEs) will be identified.31 Although the existence of molten globule enzymes with high catalytic efficiency already challenges the traditional view, in which efficient catalysis is correlated with increased structural pre-organization, the role of disorder for enzyme function or regulation in those systems is still to be established. Here, we bring into focus the first example in which conformational heterogeneity and the associated lifetimes play an active role in generating kinetic cooperativity via transitions on the millisecond timescale. Although a quantitative description of this heterogeneity in terms of the conformational substates involved is still to be determined, it is clear that it is predominantly localized to the small domain and generates a time delay that leads to the kinetic cooperativity effect. In the future, it will be interesting to see how common such slow transitions between multiple conformers are in other enzymes with regulatory roles.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the American Heart Association (http://www.heart.org/ HEARTORG/; grant 12POST12040344 to ML), the NSF (http://www.nsf.gov/; grant MCB-1330150 to RB), and the NIH (http://www.nih.gov/; grant 1R01DK081358 to BGM).
References
- 1. Protasova NYu, Kireeva ML, Murzina NV, Murzin AG, Uversky VN, Gryaznova OI, Gudkov AT. Circularly permuted dihydrofolate reductase of E. coli has functional activity and a destabilized tertiary structure. Protein Eng 1994; 11:1373-7; PMID:7700869; http://dx.doi.org/ 10.1093/protein/7.11.1373 [DOI] [PubMed] [Google Scholar]
- 2. Leontiev VV, Uversky VN, Gryaznova OI, Gudkov AT. Effect of single amino-acid replacement on the T4 phage lysozyme stability. Two. point mutations Asp10His, Asn101Asp, Arg148Ser lead to the molten globule state. Biofizika 1993; 38:606-10; PMID: 8364063 [PubMed] [Google Scholar]
- 3. Uversky VN, Ptitsyn OB. “Partly folded” state, a new equilibrium state of protein molecules: four-state guanidinium chloride-induced unfolding of betalactamase at low temperature. Biochem 1994; 33: 2782-91; PMID: 8130190; http://dx.doi.org/ 10.1021/bi00176a006 [DOI] [PubMed] [Google Scholar]
- 4. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT: Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004; 337:635-45; PMID:15019783; http://dx.doi.org/ 10.1016/j.jmb.2004.02.002 [DOI] [PubMed] [Google Scholar]
- 5. Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L., et al. Nucleic Acids Res 2013; 41:D508; PMID:23203878; http://dx.doi.org/ 10.1093/nar/gks1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 1999; 293:321-31; PMID:10550212; http://dx.doi.org/ 10.1006/jmbi.1999.3110 [DOI] [PubMed] [Google Scholar]
- 7. Dunker AK, Obradovic Z, The protein trinity—linking function and disorder. Nat Biotechnol 2001; 19:805-6; PMID:11533628; http://dx.doi.org/ 10.1038/nbt0901-805 [DOI] [PubMed] [Google Scholar]
- 8. Uversky VN, Dunker AK. Understanding protein non-folding. Biochimica Biophysica Acta 2010; 1804:1231-64; PMID:20117254; http://dx.doi.org/ 10.1016/j.bbapap.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci 2013; 22:693-724; PMID:23553817; http://dx.doi.org/ 10.1002/pro.2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Musiani F, Ippoliti E, Micheletti C, Carloni P, Ciurli S. Conformational fluctuations of UreG, an intrinsically disordered enzyme. Biochem 2013; 52:2949-54; PMID: 23560717; http://dx.doi.org/ 10.1021/bi4001744 [DOI] [PubMed] [Google Scholar]
- 11. Schulenburg C, Hilvert D. Protein conformational isorder and enzyme catalysis. Top Curr Chem 2013; 337:41-68; PMID:23536241; http://dx.doi.org/ 10.1007/128_2012_411 [DOI] [PubMed] [Google Scholar]
- 12. Roca M, Messer B, Hilvert D, Warshel A. On the relationship between folding and chemical landscapes in enzyme catalysis. PNAS 2008; 105:13877-82; PMID:18779576; http://dx.doi.org/ 10.1073/pnas.0803405105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: a plausible model. J Mol Biol 1965; 12:88-118; PMID:14343300; http://dx.doi.org/ 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 14. Koshland DE, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochem 1996; 5: 365-85; PMID:5938952; http://dx.doi.org/ 10.1021/bi00865a047 [DOI] [PubMed] [Google Scholar]
- 15. Cooper A, Dryden D. Allostery without conformational change. a plausible model. Eur Biophys J 1984; 11:103-9; PMID:6544679; http://dx.doi.org/ 10.1007/BF00276625 [DOI] [PubMed] [Google Scholar]
- 16. Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature 2014; 508 (7496):331-9; PMID:24740064; http://dx.doi.org/ 10.1038/nature13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferreon ACM, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature 2013; 498 (7454):390-4; PMID:23783631; http://dx.doi.org/ 10.1038/nature12294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larion M, Miller BG. Homotropic allosteric regulation in monomeric mammalian glucokinase. Arch Biochem Biophysics 2012; 519:103-11; PMID:22107947; http://dx.doi.org/ 10.1016/j.abb.2011.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richard J, Meunier J-C, Buc J. Regulatory behavior of monomeric enzymes: the mnemonical enzyme concept. Eur J Biochem 1974; 49:195-208; PMID:4459141; http://dx.doi.org/ 10.1111/j.1432-1033.1974.tb03825.x [DOI] [PubMed] [Google Scholar]
- 20. Ainslie GR, Jr, Shill JP, Neet KE. (1972) Transients and cooperativity. A slow transition model for relating transients and cooperative kinetics of enzymes. J Biol Chem 1974; 247:7088-96; PMID:4343169 [PubMed] [Google Scholar]
- 21. Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 2004; 12: 429-38; PMID:15016359; http://dx.doi.org/ 10.1016/j.str.2004.02.005 [DOI] [PubMed] [Google Scholar]
- 22. Larion M, Salinas RK, Bruschweiler-Li L, Miller BG, Brüschweiler R. Order-disorder transitions govern kinetic cooperativity and allostery in monomeric human glucokinase. PLoS Biol 2012; 10(12):1-9; PMID:23271955; http://dx.doi.org/ 10.1371/journal.pbio.1001452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larion M, Salinas RK, Bruschweiler-Li L, Brüschweiler R, Miller BG. Direct evidence of conformational heterogeneity in human pancreatic glucokinase from high-resolution nuclear magnetic resonance. Biochem 2010; 49 (37):7969-71; PMID:20735087; http://dx.doi.org/ 10.1021/bi101098f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Larion M, Miller BG. Global fit analysis of glucose binding curves reveals a minimal model for kinetic cooperativity in human glucokinase. Biochem 2010; 49 (41):8902-11; PMID:20828143; http://dx.doi.org/ 10.1021/bi1008672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough JG, Sponer J, Jones DT, et al. Classification of intrinsically disordered region and proteins. Chem Rev 2013; 114(13):6589-631; PMID:24773235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci U S A 1996; 93:11504-9; PMID:8876165; http://dx.doi.org/ 10.1073/pnas.93.21.11504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins 2000; 41: 415-27; PMID:11025552; http://dx.doi.org/ 10.1002/1097-0134(20001115)41:3%3c415::AID-PROT130%3e3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 28. Uversky VN. Natively unfolded proteins: a point where biology waits for physics, Protein Sci 2002; 11:739-56; PMID:11910019; http://dx.doi.org/ 10.1110/ps.4210102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem 2002; 62: 25-49; PMID:12418100; http://dx.doi.org/ 10.1016/S0065-3233(02)62004-2 [DOI] [PubMed] [Google Scholar]
- 30. Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett 2005; 579: 3346-54; PMID:15943980; http://dx.doi.org/ 10.1016/j.febslet.2005.03.072 [DOI] [PubMed] [Google Scholar]
- 31. Instrumental analysis of intrinsically disordered proteins. Assessing Structure and Conformation. 2010; (Eds Vladimir N. Uversky and Sonia Longhi Wiley and Sons, Inc, New Jersey. [Google Scholar]