

Abstract
A short segment of the disordered p53 transactivation domain (p53TAD) forms an amphipathic helix when bound to the E3 ubiquitin ligase, MDM2. In the unbound p53TAD, this short segment has transient helical secondary structure. Using a method that combines broad sampling of conformational space with re-weighting, it is shown that it is possible to generate multiple, independent structural ensembles that have highly similar secondary structure distributions for both p53TAD and a P27A mutant. Fractional amounts of transient helical secondary structure were found at the MDM2 binding site that are very similar to estimates based directly on experimental observations. Structures were identified in these ensembles containing segments that are highly similar to short p53 peptides bound to MDM2, even though the ensembles were re-weighted using unbound experimental data. Ensembles were generated using chemical shift data (alpha carbon only, or in combination with other chemical shifts) and cross-validated by predicting residual dipolar couplings. We think this ensemble generator could be used to predict the bound state structure of protein interaction sites in IDPs if there are detectable amounts of matching transient secondary structure in the unbound state.
Keywords: chemical shift, ensemble generation, Intrinsically disordered proteins, nuclear magnetic resonance, p53, re-weighting
Abbreviations
- p53TAD
transactivation domain of human p53
- IDP
intrinsically disordered protein
- NMR
nuclear magnetic resonance
- BEGR
broad ensemble generation with re-weighting
- CAΔδ
alpha carbon secondary chemical shifts
- TraDES
trajectory directed ensemble-sampling
- RMSD
root mean square deviation
- PPII
polyproline II.
Introduction
Intrinsically disordered proteins (IDPs) perform essential functions in organisms from all phyla.1-6 IDPs are highly dynamic, do not form tertiary structures, and contain variable amounts of transient secondary structure.1,2,7-9 Generating realistic structural ensembles of even a small IDP represents a major challenge in structural biology.7,8,10-13
Several groups have made substantial progress developing methods to generate structural ensembles of IDPs that are consistent with the available experimental data.7,10,14-31 Most of these methods use a strategy that is similar to the one that we are using 32,33; that is, generate pools of physically realistic structures, use these structures to simulate experimental data, and determine a weight for each structure in the pool based on how well the weighted average, simulated data fits the experimental data. The final ensemble of structures with non-zero weights should have properties that are consistent with the type(s) of experimental data used during the fitting process and may contain features that can be used to rationalize function.
There are some important differences between the methods that are currently available to generate structural ensembles of IDPs. Forman-Kay and collaborators developed a software package, referred to as ENSEMBLE, that determines structure weights using Monte Carlo and attempts to generate the smallest possible ensemble that is simultaneously consistent with multiple forms of experimental data.7,14,22,23,31 Selecting the smallest ensemble that fits the experimental data limits problems associated with overfitting. Another approach developed by Hummer and collaborators reduces overfitting by using simulated annealing to implement a maximum-entropy method.20,34 Blackledge and collaborators have done extensive development and testing of two software packages, Flexible-Meccano and ASTEROIDS, that can generate and re-weight pools of structures.8,19,21,24,26-29,35 The Flexible-Meccano software samples a database of phi and psi angles taken from the loop and coil regions of ordered proteins to provide a representation of the conformational variability of IDPs. After a suitable pool of structures is generated, the ASTEROIDS software uses a genetic algorithm with an iterative procedure to enhance the selection (and subsequent generation) of structures consistent with different forms of nuclear magnetic resonance (NMR) and small-angle x-ray scattering data. There have also been efforts to approximate the accuracy of the ensemble. In particular, Stultz and collaborators developed a Bayesian weighting approach that determines the error associated with all the possible combinations of weights that can be assigned to the structures in the pool.10,15,16 Finally, Vendruscolo and collaborators developed an approach that uses the experimental data as a form of restraint during a molecular simulation.11,36,37
In this study, we use the transactivation domain of the tumor suppressor protein p53 as a model IDP because it has well characterized structural and functional properties. The p53 protein is a transcription factor and cell cycle regulator that determines cell fate in response to DNA damage. Depending on the type and extent of the damage, p53 will activate target genes that will repair the damage, or induce cell-cycle arrest or apoptosis.38,39 The p53 protein is referred to as the “guardian of the genome” and it is mutated and/or dysregulated in most human cancers.38,39 Residues 1-73 of human p53 include a transactivation domain (p53TAD) that is responsible for regulating the transcriptional activity and cellular stability of p53.38,40,41 To perform these functions p53TAD interacts with multiple binding partners including the E3 ubiquitin ligase, MDM2. When bound to MDM2 short segments of p53TAD undergo a disorder to order transition to form amphipathic helices.42,43 Several studies have concluded that p53TAD is an IDP containing some transient helical secondary structure that is localized to the MDM2 binding site.41,44,45
Using our broad ensemble generation with re-weighting (BEGR) method 32,33 we show it is possible to generate multiple, independent structural ensembles of p53TAD that have highly similar secondary structure distributions. The key difference between BEGR and other methods is that we can simultaneously re-weight pools containing over one million structures, which was necessary to obtain converged ensemble properties. Using either alpha carbon (CA) chemical shifts, or CA chemical shifts combined with beta carbon (CB) and carbonyl carbon (CO) chemical shifts from unbound p53TAD, ensembles were generated that contained structures that resemble short peptides of p53TAD bound to MDM2. The BEGR ensembles of p53TAD generated using chemical shifts were cross-validated by predicting residual dipolar coupling data from Blackledge and Fersht.24
Results
Ensemble generation and cross-validation
Primary CA, CB, and CO chemical shifts were measured for human p53TAD and a mutant that changes a highly conserved proline on the C-terminal edge of the MDM2 binding site to alanine (P27A). The chemical shifts were measured in the absence of any binding partners and therefore represent the ensemble average secondary structure of the unbound proteins. These experimental data were then used with our BEGR approach (see Materials and Methods) to generate five independent structural ensembles for both p53TAD and P27A.32 Briefly, the trajectory directed ensemble-sampling (TraDES) program was first used to generate one million member pools of structures for p53TAD and P27A.46 The SPARTA+ program was then used to predict the CA, CB, and CO chemical shifts for these structures.47 Finally, a non-negative least squares fitting procedure was used (accounting for simulation and experimental uncertainty) to simultaneously assign weights to each of the structures in the million member pools to obtain the best fit between the weighted average of the predicted chemical shifts and the experimental chemical shifts (see equation (1)). The weights represent the relative importance of each structure in fitting the experimental data and are used to calculate physical properties of the ensembles (see equation (2)). After the fitting process is complete, all structures with non-zero weights are collected for further analysis and referred to as the BEGR ensemble.
Figure 1 shows that the predicted chemical shifts from individual BEGR ensembles of p53TAD fit the experimental secondary chemical shifts for CA (ΔδCA), CB (ΔδCB), and CO (ΔδCO) with high accuracy. Black bars show the experimental data. Positive CAΔδ and COΔδ values are observed for residues in a helical conformation and negative values are observed for residues in a beta or extended conformation.48,49 The opposite trend is observed for CBΔδ values in regions with beta structure and CBΔδ values are near random coil for helical regions. Figure 1A shows that the MDM2 binding site has some transient helical secondary structure (positive CAΔδ values for residues 17-28) with an estimated population (≈ 28%) that is consistent with previous reports, and Figure 1B shows that this transient helicity increases for P27A ((≈ 64%).24, 41, 45 The red and blue bars in Figures 1A, B show the predicted CAΔδ values for p53TAD and P27A respectively, from a BEGR ensemble that was re-weighted using CA chemical shifts (see Materials and Methods). Figures 1C, D, and E respectively show the secondary shift predictions for CA, CB and CO nuclei when re-weighting is performed using all three carbon chemical shifts. The gray bars show the secondary chemical shift values predicted for a randomly drawn ensemble with no re-weighting. The mean squared errors are shown as a measure of the goodness of fit between the BEGR ensemble results and experiment, and between the random ensembles and experiment.
Figure 1.

BEGR ensembles for p53TAD re-weighted against primary CA chemical shifts. For all panels the experimental values are shown with black bars, values for randomly selected structures are shown with gray bars, and the BEGR ensemble predictions are shown with red bars for panels a, c-e and blue bars for panel b. The mean squared errors (MSE) are shown to quantify the agreement between BEGR and experiment, and between random and experiment. (A) Secondary CA chemical shifts (CAΔδ) for BEGR ensembles of p53TAD re-weighted with using CA chemical shift data, (B) Secondary CA chemical shifts (CAΔδ) for BEGR ensembles of the P27A mutant re-weighted with using CA chemical shift data, (C) Secondary CA chemical shifts (CAΔδ) for BEGR ensembles of p53TAD re-weighted with using CA, CB and CO chemical shift data, (D) Secondary CB chemical shifts (CBΔδ) for BEGR ensembles of p53TAD re-weighted with using CA, CB and CO chemical shift data, (E) Secondary CO chemical shifts (COΔδ) for BEGR ensembles of p53TAD re-weighted with using CA, CB and CO chemical shift data.
Figure 2 shows cross-validation of the BEGR ensembles using experimental residual dipolar couplings (RDCs) obtained from Blackledge and Firsht.24 The five independent BEGR ensembles that were generated for p53TAD, using either CA chemical shifts (Fig. 2A) or CA, CB, and CO chemical shifts (Fig. 2B), were used to predict residual dipolar couplings (RDCs). The predicted RDCs are shown as averages (red lines) with standard deviations (red shading) and the experimental RDCs are show as black lines. The mean squared errors between the BEGR ensemble predictions and the experimental data are also shown. The CA re-weighted results (Fig. 2A) fit the experimental data very well for most of the residues with the largest deviations occurring for residues 58-70, possibly because the experimental data was collected for a larger p53 fragment (residues 1-93). Using CA, CB and CO chemical shifts (Fig. 2B) resulted in poorer fits to the peaks of the RDC (e.g., residues 22-24, 48-49) compared to CA re-weighting, and improved fits near the N-terminal.
Figure 2.
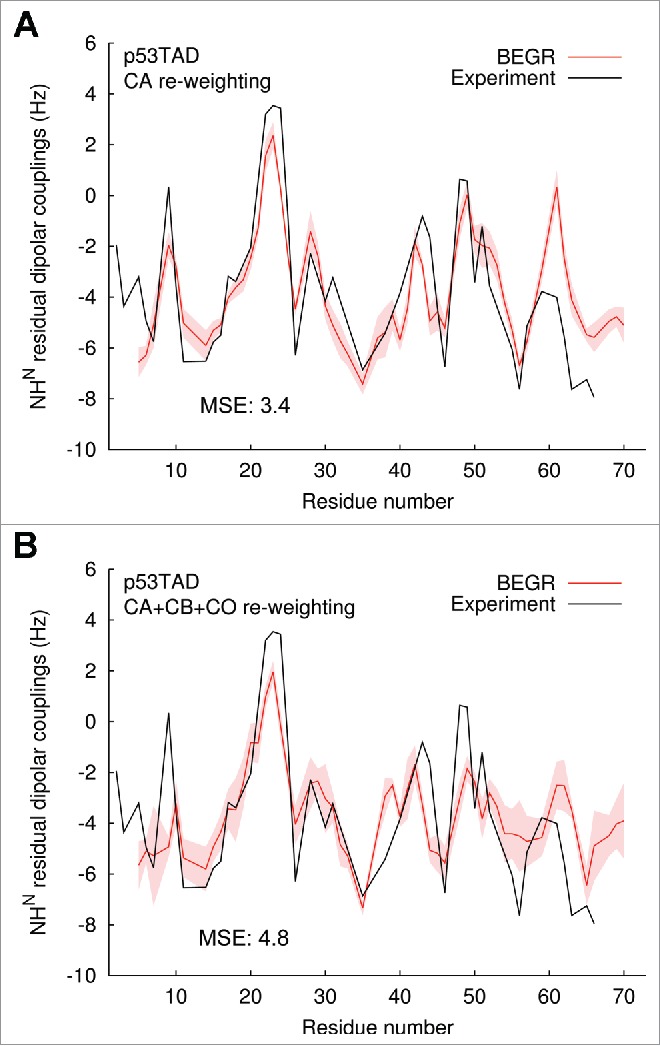
Cross-validation of BEGR ensemble structures. Results are predictions of the residual dipolar coupling data for five independent ensembles were generated using: (A) CA chemical shift data, (B) CA, CB and CO chemical shift data. Averages are shown as red lines with standard deviations as the red shading and should be compared to the experimental data (from reference 24) shown as black lines. The mean squared errors (MSE) are shown to quantify the agreement between the predicted and experimental results.
Using BEGR ensembles to estimate secondary structure populations
Figure 3 shows ensemble averaged population estimates of α helical structure predicted by the five independent BEGR ensembles. These ensembles were generated using CA chemical shift data for unbound p53TAD and P27A. Averages (red line) and standard deviations (light red shading) are given for p53TAD and for P27A (blue line with light blue shading). The black dashed line with gray shading shows averages and standard deviations for a randomly drawn ensemble with no re-weighting. According to the secondary structure predictions from the BEGR ensembles, the P27A mutant has a population of transient helical secondary structure in the MDM2 binding site that is more than double the amount observed for wild type p53TAD. This is consistent with the population estimates predicted directly from the chemical shift data using δ2D (Fig. 4).50
Figure 3.
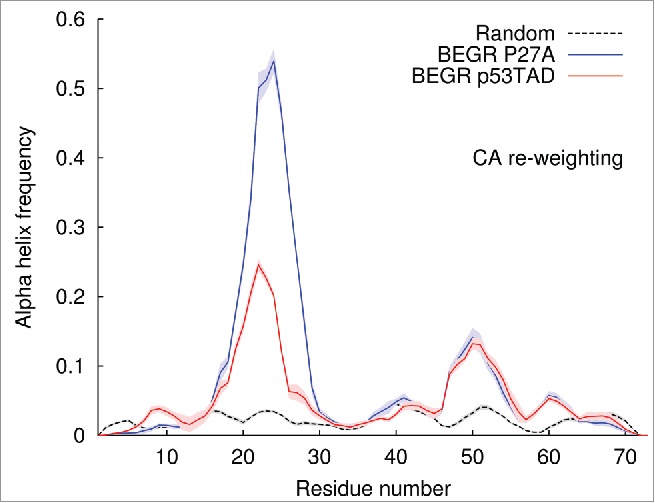
Helicity of five BEGR ensembles for human p53TAD (red) and P27A (blue). The solid lines are the weighted average helicities and the shaded regions are the standard deviations for ensembles re-weighted using CA chemical shifts.
Figure 4.
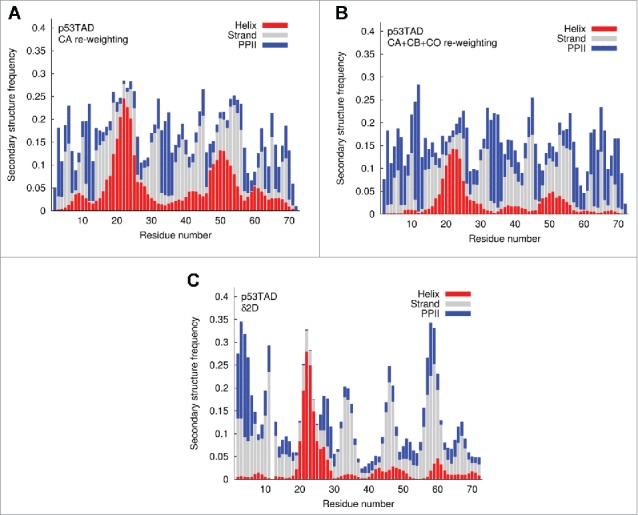
Frequency of alpha helix (red), beta strand (gray) and polyproline II (PPII) helix (blue) predicted by BEGR ensembles and δ2D. (A) Averages of five BEGR ensembles using CA chemical shift data, (B) averages of five BEGR ensembles using CA, CB and CO chemical shift data, (C) δ2D predictions based on using all available chemical shifts.
Figure 4 shows the predicted secondary structure distributions estimated using either the BEGR ensembles of p53TAD (4a,b), or δ2D (4c).50 The average secondary structure properties for alpha helix (red), beta strand (gray) and polyproline II (PPII) helix (blue) are shown for BEGR ensembles re-weighted using CA chemical shifts (Fig. 4A), re-weighted using CA, CB and CO chemical shifts (Fig. 4B), and estimated using δ2D with all available chemical shifts (CA, CB, CO, N, HN; Fig. 4C). Using CA, CB and CO chemical shift data lowers the predicted helical frequency compared to using CA data alone, but does not significantly change the predicted strand or PPII frequencies.
BEGR ensembles contain structures that resemble p53 peptides bound to MDM2
The results in Figure 5A show that p53TAD ensembles, re-weighted using only CA chemical shifts for the unbound protein, contain short helical segments resembling the backbone structure of p53 peptides bound to MDM2. Both panels show histograms of the root mean square deviations (RMSD) measured between the CA atoms of residues 19-24 from all of the structures in the five independent BEGR ensembles of either p53TAD (red line) or P27A (blue line) and a reference structure from the protein data bank of a short p53 peptide bound to MDM2 (1YCR).42,43 The solid lines with shading show the averages and standard deviations of the RMSD, respectively, for the five ensembles of p53TAD and P27A. The black dashed line with gray shading shows the averages and standard deviations for a randomly drawn ensemble with no re-weighting. Figure 5B shows results for re-weighting using CA chemical shifts, and Figure 5B shows results for re-weighting using CA, CB and CO shifts. Figure 5A also contains example structures from the three peaks seen in the p53TAD histogram with the BEGR ensemble structure shown in blue and the 1YCR peptide shown in green. Chou and collaborators have also showed that NMR data for unbound p21 could be used to identify structures in the free ensemble that resemble the bound state.51
Figure 5.
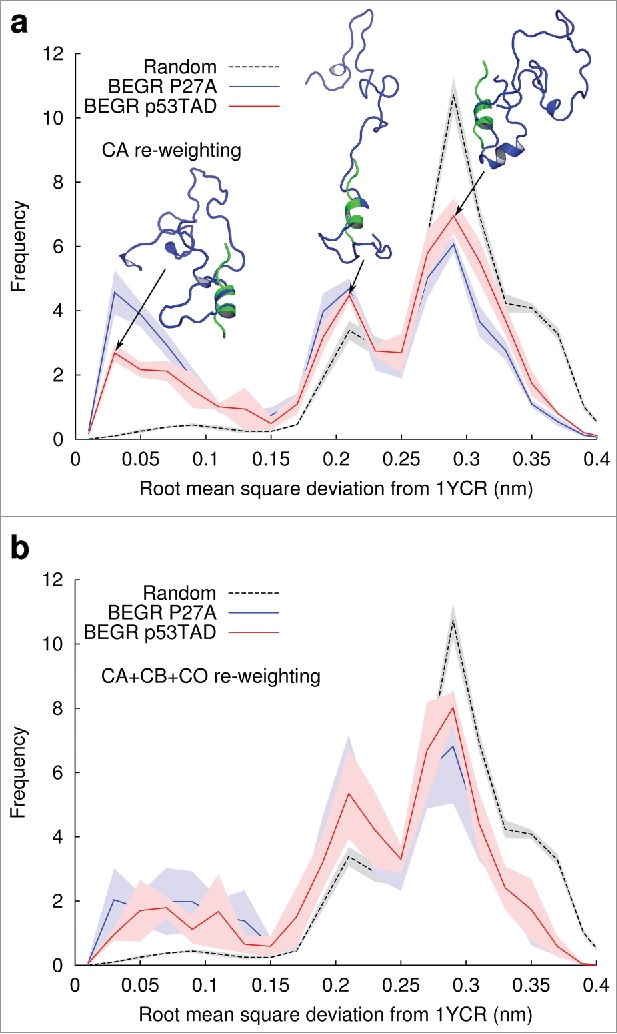
BEGR ensembles contain structures that resemble p53TAD bound to MDM2. Histograms of CA root mean square deviations (RMSD) for five BEGR ensembles are shown. The solid red and blue lines show the average frequency of structures from the five BEGR ensembles of p53TAD and P27A respectively, and the light red and blue shading shows the standard deviations. Dashed black lines with gray shading show the averages and standard deviation, respectively, for randomly selected structures. (A) BEGR ensembles generated using CA chemical shift data, (B) BEGR ensembles generated using CA, CB and CO chemical shift data.
Large pools are necessary to generate ensembles with converged properties
Figure 6 shows how pool size and accounting for uncertainty (using noise) in the re-weighting process changes the convergence of the RMSD histograms for independent ensembles. All the panels in Figure 6 show the averages (red line) with standard deviation (light red shading) for five independent ensembles generated using one million member pools with noise. Comparison is made using the same pool size, shown as averages (black lines) with standard deviation (gray shading), using no noise (Fig. 6A), and using noise with ten thousand (Fig. 6B) and one hundred thousand member pools. Convergence of the ensembles improves as pool size is increased and noise is used.
Figure 6.
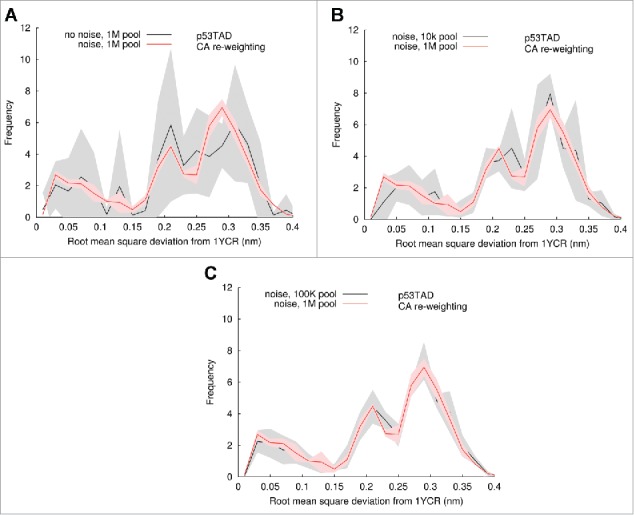
Influence of pool size and noise on BEGR ensemble properties. The red lines with red shading in all panels shows the averages and standard deviations, respectively, of the RMSD between BEGR ensemble structures and the p53 peptide bound to MDM2 (same as red curve in Fig. 5A). The black lines with gray shading show results for BEGR ensembles generated using: (A) no noise with pools containing 106 structures, (B) noise with pools containing 104 structures, (C) noise with pools containing 105 structures.
Discussion
Previous groups have observed that good fits to the experimental data (Fig. 1) can be obtained using ensembles of structures that appear very different.10 In the current study, we relied exclusively on carbon chemical shift data, which has a strong dependence on secondary structure, even the transient secondary structure that is sometimes observed in IDPs.49,52,53 We demonstrate that BEGR ensembles have very similar secondary structure populations as those estimated directly from the primary chemical shifts. The small standard deviation values in Figures 2, 3, and 5 (red or blue shading) show that the local structural properties of the five independent ensembles for p53TAD and P27A are highly similar. To obtain a high level of similarity between independent ensembles it was necessary to use large, one million member pools of structures, and to account for the uncertainty of the simulated and experimental results (see Fig. 6 and Materials and Methods).
In our previous study of the global structural properties of p53TAD using small-angle X-ray scattering data we determined that the weighted average radius of gyration of the BEGR ensembles was ≈ 2.9 nm.32 In the current study we did not include any data that would constrain or relax the tertiary structures of the re-weighted ensembles, and we do not expect any correspondence between the tertiary structures of the BEGR ensembles that were re-weighted using chemical shifts and the real ensembles. This is indeed what we observe. The BEGR ensembles that were re-weighted using CA chemical shifts have an average radius of gyration that is ≈ 2.0 nm. We do not consider this a limitation of the current study since only chemical shifts were used. We did not include other forms of experimental data because we are not convinced that a successful strategy for simultaneously re-weighting data types that report on secondary and tertiary structures has been determined.
Because our method of generating ensembles is analogous to estimating the equilibrium distribution of structures the results do not have any bearing on whether the binding mechanism is occurring through conformational selection or induced folding. Regardless of the binding mechanism an IDP uses, it is expected that the unbound protein will occasionally (or frequently) sample conformations that are similar to the bound structure(s) and that some minimal indication of these structures will be present in the unbound chemical shift data. The method we present for generating structural ensembles can be used to predict the bound state structures of novel IDPs for which this condition is met, at least if they fold into a helix upon binding.
The transient secondary structure (Fig. 4) and RMSD histograms (Fig. 5) for ensembles that were re-weighted using only CA chemical shifts are very similar to those re-weighted using CA, CB, and CO chemical shifts. These results suggest that using only CA chemical shifts may be sufficient to reproduce some of the important structural properties of IDPs. We believe that using CA chemical shifts to generate structural ensembles is desirable since these data have a straightforward relationship with the backbone dihedral angles phi and psi ,48 because they are easier to measure than residual dipolar couplings or paramagnetic relaxation enhancements, and because using only CA chemical shifts could open the door for a minimalist approach to generating structural ensembles of IDPs. The reason that using CA, CB and CO chemical shifts results in poorer fits (larger MSE values) to the experimental chemical shifts (Fig. 1) and the predicted RDCs (Fig. 2), compared using CA only, may be due to our strategy for using multiple forms of data. Future work will include determining the best way to use multiple forms of data.
To determine whether large pools are needed to generate similar structural properties of the BEGR ensembles we calculated the number of structures in BEGR ensembles, generated using different pool sizes, that are similar to (RMSD < 0.1 nm) p53 peptides bound to MDM2. For p53TAD using CA re-weighting, going from pool sizes of 104 to 105 to 106 structures increased the number of bound state structures from an average of 41 to 299 to 754 (factor of 18.4). These RMSD values are based on a comparison to a short five residue helix. To understand how these values would change for a longer helical segment we also calculated the RMSD between the BEGR ensemble structures and a reference structure from the protein data bank of a p53 peptide, corresponding to p53 residues 46-54, bound to RPA70 (2B3G).42 For this longer helical segment, going from 104 to 105 to 106 structures in the pool increased the number of bound state structures from an average of 1.6 to 24 to 60 (factor of 37.5). Using larger pools significantly increases the number of bound state structures in the BEGR ensembles, and this effect is more pronounced for longer helical segments (probably because longer helices are generated at a lower frequency in the pool). From this result we conclude that larger pools are more important for identifying longer helical segments.
In this report we used our BEGR method to generate independent ensembles for an intrinsically disordered protein that have highly similar secondary structure properties, and these properties did not have a strong dependence on the combination of chemical shifts used during the re-weighting. These ensembles also contained structures with segments that were very similar to p53 peptides bound to MDM2 but, as expected, did not have accurate tertiary structure since the experimental data used for BEGR reports only on local structure. Because we were able to consistently reproduce ensembles that had differences in the fractional helicity of the MDM2 binding site expected for p53TAD and P27A, we can also conclude that the BEGR method is sensitive to single amino acid changes that modify the average properties of the structural ensemble. Based on previous work we know that mutating the proline at position 27 increases the binding affinity to MDM2.54,55 This may be related to a reduction in the entropic penalty for binding that is expected for P27A. The sensitivity of BEGR to single mutations is important because the future of drug discovery for IDPs could depend on being able to determine whether and how disease-causing mutations change the structural ensemble.
Materials and Methods
Experimental methods
Samples of human p53TAD (residues 1-73) that were uniformly labeled with either 15N or 15N and 13C were prepared as previously described.45 Samples of the P27A mutant were prepared using the same protocol. NMR experiments on p53TAD and P27A were carried out at 25 °C on a Varian VNMRS 600 MHz spectrometer equipped with a triple resonance, pulsed field, Z-axis gradient cold probe. To make the backbone resonance assignments, sensitivity enhanced 1H-15N HSQC and three dimensional HNCACB and HNCO experiments were performed on the labeled p53TAD (0.40 mM) and P27A (0.35 mM) samples in a 90%H2O/10% D2O, PBS buffer, at a pH of 6.8 (Kay et al., 1992; Kay et al., 1994; Wittekind and Mueller, 1993); see Table 1 for details. All NMR spectra were processed with nmrPipe and analyzed using nmrView.56 To calculate the secondary chemical shifts (Δδ), neighbor corrected random coil values 57 were subtracted from the measured chemical shifts (δ) except for the case where a glycine preceded a proline (G59) and for the two tryptophan residues (W23,W53). Random coil values for these residues were taken from Wishart.58
Table 1.
NMR experiments and number of resonance assignments.
| Experiment/Sample | Nuclei Detected | Sweep Width (Hz) t3 x t2 x t1 | Complex Points t3 x t2 x t1 | 1H-15N (amides) | 13CA | 13CB | 13CO |
|---|---|---|---|---|---|---|---|
| HSQC/p53TAD | 1H-15N | n/a x 7225.4 x 1500.0 | n/a x 512 x 128 | ||||
| HNCACB/p53TAD | 1H-15N, 13CA, 13CB | 7225.4 x 12063.4 x 1500.0 | 512 x 128 x 32 | 60 | 72 | 72 | |
| HNCO/p53TAD | 1H-15N, 13CO | 7225.4 x 3612.7 x 1500.0 | 512 x 128 x 32 | 59 | |||
| HSQC/P27A | 1H-15N | n/a x 7225.4 x 1500.0 | n/a x 512 x 128 | ||||
| HNCACB/P27A | 1H-15N, 13CA, 13CB | 7225.4 x 12063.4 x 1500.0 | 512 x 128 x 32 | 61 | 72 | 72 | |
| HNCO/P27A | 1H-15N 13CO | 7225.4 x 3612.7 x 1500.0 | 512 x 128 x 32 | 57 |
Simulation Methods
Outline of the BEGR method
The BEGR method follows the same basic steps as other methods such as ASTEROIDS 29 and ENSEMBLE.31 The key difference between these methods and the BEGR method that will be explained below is that a non-negative least squares approach is used for re-weighting, rather than Monte Carlo or a genetic algorithm. This allows for simultaneous re-weighting of a very large number (over a million) of structures.
Step 1: Collect experimental data. For the current study CA, CB, and CO chemical shifts were used.
Step 2: Generate a large pool of structures using computer simulation. Pools containing one million p53TAD or P27A structures were generated using the trajectory directed ensemble-sampling (TraDES) software.46 TraDES implements a build-up method to generate structures and was used in the standard sampling mode with commands “seq2trj -t 2” and “trades -l 0 -k T.”
Step 3: Calculate the corresponding experimental data for each structure in the pool. CA, CB, and CO chemical shift values were calculated using SPARTA+.47
Step 4: Determine the BEGR ensemble by re-weighting structures in the pool. The goal of the re-weighting step is to assign each structure in the pool a weight such that the weighted average calculated data has the best possible fit to experimental data. In order to assign these weights, the fit between the calculated spectra and the experimental spectrum was optimized by minimizing the two-norm of the difference between the experimental spectrum and the weighted average simulated spectrum:
| (1) |
Here w is vector of n × 1 structure weights wi that must all be positive and n is the number of structures to be re-weighted (n = 104, 105, or 106 for the results shown here). Iexp is a k × 1 vector containing the experimental data where k is the number of available experimental data points, which will vary between 72 and 203 depending on the combination of chemical shifts used for re-weighting. Isim is a k × n matrix containing the simulated spectra where each column in the matrix is the spectrum for a single structure from the pool.
Re-weighting using non-negative least squares and Gaussian noise
To perform the re-weighting (step 4 above) the raw simulated and experimental primary chemical shifts were imported into GNU Octave respectively as a matrix Isim and vector Iexp and normalized by dividing each element by the maximum value from Iexp. The following steps were then performed:
Gaussian random deviates with a mean of zero and standard deviation of 0.23 ppm (25% of the CA chemical shift RMSD from SPARTA+) were added to the simulated matrix Isim, generating a new matrix Isim_noise. Gaussian random deviates with a mean of zero and standard deviation of 0.05 ppm (25% of approximate experimental uncertainty for CA chemical shifts) were added to the experimental vector Iexp generating a new vector Iexp_noise. The choice of 25% of the simulation and experimental uncertainty allowed for convergence of the secondary structure properties while providing good fits to experimental data, and is consistent with the choice of Blackledge and collaborators.19 Other groups have incorporated uncertainty into their re-weighting procedure using different strategies.15,20,29 We used CB and/or CO shifts with the same importance as CA; the process above was carried out for each shift type and then all shifts used for re-weighting were concatenated to form Isim_noise and Iexp_noise.
A set of weights, w, were determined by running the lsqnonneg command in Octave using Isim_noise and Iexp_noise for the case of using noise, and Isim and Iexp for the case of not using noise (see Eq. (1)). This set of weights was then saved into memory. If an element of Iexp was zero (experimental chemical shift value could not be determined) then that element of Iexp or Iexp_noise and the corresponding row of Isim or Isim_noise were ignored in the re-weighting.
For the case of using noise, the previous 2 steps were repeated until the number of structures in the BEGR ensemble converged. Convergence was determined by counting the number of structures in the BEGR ensemble with non-zero weights every 10 re-weighting steps. If the number of structures changed by less than 5% then the re-weighting process was stopped. This required around 200 re-weighting steps for the results shown in this report for 106 member pools.
The final set of weights was then calculated by averaging the weights obtained during all of the individual re-weighting steps.
To demonstrate how the use of Gaussian noise in BEGR helps prevent overfitting we calculated the square two-norm (goodness of fit) between the chemical shifts generated using 100% of the experimental data for re-weighting and shifts using 90% of the data. For CA re-weighting the square two-norm was 0.60 and 0.31 for no noise and noise respectively, demonstrating that BEGR does a better job predicting the 100% data results when noise is added during re-weighting. This was also true for CA+CB+CO re-weighting (averaged over CA, CB and CO) where the square two-norm was 1.11 and 0.61 for no noise and noise respectively. Use of noise also leads to large ensembles (around 8,000 structures for CA re-weighting and 2,000 for CA+CB+CO re-weighting) which we believe could be an advantage given that a complete description of the ensemble nature of an IDP may require a large number of structures.
The non-negative least squares approach implemented in BEGR is highly efficient. Using one core of a Xeon E5520 processor, a single re-weighting step takes approximately 0.5 s, 6 s, and 41 s for 104, 105 and 106 structure pools respectively. In our tests, both Monte Carlo minimization and genetic algorithms were much slower and became impractical for pools larger than 20,000 (data not shown).
Correcting for pre-proline residues and random coil shifts
In disordered proteins there is a 2.0 ppm upfield chemical shift for the CA of amino acids that precede proline and a 2.8 ppm upfield chemical shift for CO.58 The CAΔδ values calculated by SPARTA+ 59 for p53TAD do not completely correct for this effect (data not shown). To develop a correction for this residual offset we generated a 102 residue sequence containing four residues of each amino acid type followed by a proline, and capped at the C-terminus with two alanines (AAAAPRRRRPNNNNP…YYYYPVVVVPAA). A million structures with this sequence were generated with TraDES and the average chemical shifts for CA, CB, and CO were calculated using SPARTA+. The correction was calculated as the difference between chemical shift values for the i-1 and i-2 residues that precede a proline. In addition, we wanted to use the neighbor corrected random coil library developed by Mulder and colleagues 57 instead of the built in SPARTA+ random coil values. The final primary shifts used for re-weighting with BEGR are given by (SPARTA+ primary shifts) - (SPARTA+ random coil shifts) + (Mulder neighbor corrected random coil shifts) + (pre-proline corrections).
Calculation of secondary structure and RMSD histograms
The BEGR ensemble consists of N structures with corresponding weights wi. Ensemble averaged quantities were calculated using a weighted average formula
| (2) |
where <X> is the desired average and Xi is the value for the i th structure.
Transient secondary structure properties for alpha helix, beta strand and PPII helix (Figs. 3, 4) were calculated using SEGNO.60 The transient secondary structure type for a single residue was calculated using Eq. (2) with for cases when this residue in structure i is consistent with the secondary structure type and otherwise.
The RMSDs between the CA atoms of the p53 peptide from 1YCR and the BEGR ensembles were calculated using the confrms program from GROMACS 4.5.5.61 RMSDs were measured for residues 19-24. Histograms were calculated by multiplying the RMSD values for each BEGR ensemble structures by the corresponding weight of that structure.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
Computational resources were provided in part by the Institute for Bioinformatics and Evolutionary Studies (IBEST) at University of Idaho. NMR facilities were provided by the Florida Center of Excellence for Drug Discovery and Innovation.
Funding
The research was supported by funding to GWD from the American Cancer Society (RSG-07-289-01-GMC) and the National Science Foundation (MCB-0939014). The research was also supported by funding to FMY and GWD from the National Institutes of Health (5R21GM083827), and by funding to FMY from Idaho INBRE.
References
- 1. Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 2005; 6:197-208; PMID:15738986; http://dx.doi.org/ 10.1038/nrm1589 [DOI] [PubMed] [Google Scholar]
- 2. Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 1999; 293:321-31; PMID:10550212; http://dx.doi.org/ 10.1006/jmbi.1999.3110 [DOI] [PubMed] [Google Scholar]
- 3. Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. biological processes and functions of proteins with long disordered regions. J Proteome Res 2007; 6:1882-98; PMID:17391014; http://dx.doi.org/ 10.1021/pr060392u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry 2002; 41:6573-82; PMID:12022860; http://dx.doi.org/ 10.1021/bi012159+ [DOI] [PubMed] [Google Scholar]
- 5. Tompa P. Structure and Function of Intrinsically Disordered Proteins. Boca Raton: Taylor and Francis Group, 2010 [Google Scholar]
- 6. Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci 2002; 11:739-56; PMID:11910019; http://dx.doi.org/ 10.1110/ps.4210102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mittag T, Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr Opin Struct Biol 2007; 17:3-14; PMID:17250999; http://dx.doi.org/ 10.1016/j.sbi.2007.01.009 [DOI] [PubMed] [Google Scholar]
- 8. Schneider R, Huang JR, Yao M, Communie G, Ozenne V, Mollica L, Salmon L, Jensen MR, Blackledge M. Towards a robust description of intrinsic protein disorder using nuclear magnetic resonance spectroscopy. Mol Biosyst 2012; 8:58-68; PMID:21874206; http://dx.doi.org/ 10.1039/c1mb05291h [DOI] [PubMed] [Google Scholar]
- 9. Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr Opin Struct Biol 2009; 19:23-30; PMID:19162471; http://dx.doi.org/ 10.1016/j.sbi.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fisher CK, Stultz CM. Constructing ensembles for intrinsically disordered proteins. Curr Opin Struct Biol 2011; 21:426-31; PMID:21530234; http://dx.doi.org/ 10.1016/j.sbi.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vendruscolo M. Determination of conformationally heterogeneous states of proteins. Curr Opin Struct Biol 2007; 17:15-20; PMID:17239581; http://dx.doi.org/ 10.1016/j.sbi.2007.01.002 [DOI] [PubMed] [Google Scholar]
- 12. Daughdrill GW. Determining structural ensembles for intrinsically disordered proteins. In: VN Uversky, S Longhi, eds. Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation. Hoboken: John Wiley and Sons, Inc., 2010 [Google Scholar]
- 13. Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci 2012; 37:509-16; PMID:22989858; http://dx.doi.org/ 10.1016/j.tibs.2012.08.004 [DOI] [PubMed] [Google Scholar]
- 14. Choy WY, Forman-Kay JD. Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J Mol Biol 2001; 308:1011-32; PMID:11352588; http://dx.doi.org/ 10.1006/jmbi.2001.4750 [DOI] [PubMed] [Google Scholar]
- 15. Fisher CK, Huang A, Stultz CM. Modeling intrinsically disordered proteins with bayesian statistics. J Am Chem Soc 2010; 132:14919-27; PMID:20925316; http://dx.doi.org/ 10.1021/ja105832g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang A, Stultz CM. The effect of a DeltaK280 mutation on the unfolded state of a microtubule-binding repeat in Tau. PLoS Comput Biol 2008; 4:e1000155; PMID:18725924; http://dx.doi.org/ 10.1371/journal.pcbi.1000155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindorff-Larsen K, Kristjansdottir S, Teilum K, Fieber W, Dobson CM, Poulsen FM, Vendruscolo M. Determination of an ensemble of structures representing the denatured state of the bovine acyl-coenzyme a binding protein. J Am Chem Soc 2004; 126:3291-9; PMID:15012160; http://dx.doi.org/ 10.1021/ja039250g [DOI] [PubMed] [Google Scholar]
- 18. Nodet G, Salmon L, Ozenne V, Meier S, Jensen MR, Blackledge M. Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J Am Chem Soc 2009; 131:17908-18; PMID:19908838; http://dx.doi.org/ 10.1021/ja9069024 [DOI] [PubMed] [Google Scholar]
- 19. Ozenne V, Schneider R, Yao M, Huang JR, Salmon L, Zweckstetter M, Jensen MR, Blackledge M. Mapping the potential energy landscape of intrinsically disordered proteins at amino Acid resolution. J Am Chem Soc 2012; 134:15138-48; PMID:22901047; http://dx.doi.org/ 10.1021/ja306905s [DOI] [PubMed] [Google Scholar]
- 20. Rozycki B, Kim YC, Hummer G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure 2011; 19:109-16; PMID:21220121; http://dx.doi.org/ 10.1016/j.str.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RW, Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc Natl Acad Sci U S A 2005; 102:17002-7; PMID:16284250; http://dx.doi.org/ 10.1073/pnas.0506202102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsh JA, Forman-Kay JD. Structure and disorder in an unfolded state under nondenaturing conditions from ensemble models consistent with a large number of experimental restraints. J Mol Biol 2009; 391:359-74; PMID:19501099; http://dx.doi.org/ 10.1016/j.jmb.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 23. Marsh JA, Neale C, Jack FE, Choy WY, Lee AY, Crowhurst KA, Forman-Kay JD. Improved structural characterizations of the drkN SH3 domain unfolded state suggest a compact ensemble with native-like and non-native structure. J Mol Biol 2007; 367:1494-510; PMID:17320108; http://dx.doi.org/ 10.1016/j.jmb.2007.01.038 [DOI] [PubMed] [Google Scholar]
- 24. Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci U S A 2008; 105:5762-7; PMID:18391200; http://dx.doi.org/ 10.1073/pnas.0801353105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc 2005; 127:476-7; PMID:15643843; http://dx.doi.org/ 10.1021/ja044834j [DOI] [PubMed] [Google Scholar]
- 26. Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 2007; 129:5656-64; PMID:17411046; http://dx.doi.org/ 10.1021/ja069124n [DOI] [PubMed] [Google Scholar]
- 27. Jensen MR, Markwick PR, Meier S, Griesinger C, Zweckstetter M, Grzesiek S, Bernado P, Blackledge M. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure 2009; 17:1169-85; PMID:19748338; http://dx.doi.org/ 10.1016/j.str.2009.08.001 [DOI] [PubMed] [Google Scholar]
- 28. Jensen MR, Salmon L, Nodet G, Blackledge M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J Am Chem Soc 2010; 132:1270-2; PMID:20063887; http://dx.doi.org/ 10.1021/ja909973n [DOI] [PubMed] [Google Scholar]
- 29. Ozenne V, Bauer F, Salmon L, Huang JR, Jensen MR, Segard S, Bernado P, Charavay C, Blackledge M. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics 2012; 28:1463-70; PMID:22613562; http://dx.doi.org/ 10.1093/bioinformatics/bts172 [DOI] [PubMed] [Google Scholar]
- 30. Lowry DF, Stancik A, Shrestha RM, Daughdrill GW. Modeling the accessible conformations of the intrinsically unstructured transactivation domain of p53. Proteins 2007; 71:587-98; PMID:17972286; http://dx.doi.org/ 10.1002/prot.21721 [DOI] [PubMed] [Google Scholar]
- 31. Krzeminski M, Marsh JA, Neale C, Choy WY, Forman-Kay JD. Characterization of disordered proteins with ENSEMBLE. Bioinformatics 2013; 29:398-9; PMID:23233655; http://dx.doi.org/ 10.1093/bioinfor-matics/bts701 [DOI] [PubMed] [Google Scholar]
- 32. Daughdrill GW, Kashtanov S, Stancik A, Hill SE, Helms G, Muschol M, Receveur-Brechot V, Ytreberg FM. Understanding the structural ensembles of a highly extended disordered protein. Mol Biosyst 2012; 8:308-19; PMID:21979461; http://dx.doi.org/ 10.1039/c1mb05243h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kashtanov S, Borcherds W, Wu H, Daughdrill GW, Ytreberg FM. Using chemical shifts to assess transient secondary structure and generate ensemble structures of intrinsically disordered proteins. Methods Mol Biol 2012; 895:139-52; PMID:22760318; http://dx.doi.org/ 10.1007/978-1-61779-927-3_11 [DOI] [PubMed] [Google Scholar]
- 34. Boura E, Rozycki B, Herrick DZ, Chung HS, Vecer J, Eaton WA, Cafiso DS, Hummer G, Hurley JH. Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc Natl Acad Sci U S A 2011; 108:9437-42; PMID:21596998; http://dx.doi.org/ 10.1073/pnas.1101763108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwalbe M, Ozenne V, Bibow S, Jaremko M, Jaremko L, Gajda M, Jensen Malene R, Biernat J, Becker S, Mandelkow E, et al. Predictive atomic resolution descriptions of intrinsically disordered hTau40 and α-synuclein in solution from NMR and small angle scattering. Structure 2014; 22:238-49; PMID:24361273; http://dx.doi.org/ 10.1016/j.str.2013.10.020 [DOI] [PubMed] [Google Scholar]
- 36. Cavalli A, Camilloni C, Vendruscolo M. Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J Chem Phys 2013; 138:094112; PMID:23485282; http://dx.doi.org/ 10.1063/1.4793625 [DOI] [PubMed] [Google Scholar]
- 37. Camilloni C, Vendruscolo M. Statistical mechanics of the denatured state of a protein using replica-averaged metadynamics. J Am Chem Soc 2014; 136:8982-91; PMID:24884637; http://dx.doi.org/ 10.1021/ja5027584 [DOI] [PubMed] [Google Scholar]
- 38. Bargonetti J, Manfredi JJ. Multiple roles of the tumor suppressor p53. Curr Opin Oncol 2002; 14:86-91; PMID:11790986; http://dx.doi.org/ 10.1097/00001622-200201000-00015 [DOI] [PubMed] [Google Scholar]
- 39. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; http://dx.doi.org/ 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- 40. Woods DB, Vousden KH. Regulation of p53 function. Exp Cell Res 2001; 264:56-66; PMID:11237523; http://dx.doi.org/ 10.1006/excr.2000.5141 [DOI] [PubMed] [Google Scholar]
- 41. Lee H, Mok KH, Muhandiram R, Park KH, Suk JE, Kim DH, Chang J, Sung YC, Choi KY, Han KH. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem 2000; 275:29426-32; PMID:10884388; http://dx.doi.org/ 10.1074/jbc.M003107200 [DOI] [PubMed] [Google Scholar]
- 42. Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc Natl Acad Sci U S A 2005; 102:15412-7; PMID:16234232; http://dx.doi.org/ 10.1073/pnas.0504614102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996; 274:948-53; PMID:8875929; http://dx.doi.org/ 10.1126/science.274.5289.948 [DOI] [PubMed] [Google Scholar]
- 44. Dawson R, Muller L, Dehner A, Klein C, Kessler H, Buchner J. The N-terminal domain of p53 is natively unfolded. J Mol Biol 2003; 332:1131-41; PMID:14499615; http://dx.doi.org/ 10.1016/j.jmb.2003.08.008 [DOI] [PubMed] [Google Scholar]
- 45. Vise PD, Baral B, Latos AJ, Daughdrill GW. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res 2005; 33:2061-77; PMID:15824059; http://dx.doi.org/ 10.1093/nar/gki336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feldman HJ, Hogue CW. Probabilistic sampling of protein conformations: new hope for brute force? Proteins 2002; 46:8-23; PMID:11746699; http://dx.doi.org/ 10.1002/prot.1163 [DOI] [PubMed] [Google Scholar]
- 47. Shen Y, Bax A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR 2010; 48:13-22; PMID:20628786; http://dx.doi.org/ 10.1007/s10858-010-9433-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Methods Enzymol 2001; 338:3-34; PMID:11460554; http://dx.doi.org/ 10.1016/S0076-6879(02)38214-4 [DOI] [PubMed] [Google Scholar]
- 49. Dyson HJ, Wright PE. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem 2002; 62:311-40; PMID:12418108; http://dx.doi.org/ 10.1016/S0065-3233(02)62012-1 [DOI] [PubMed] [Google Scholar]
- 50. Camilloni C, De Simone A, Vranken WF, Vendruscolo M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 2012; 51:2224-31; PMID:22360139; http://dx.doi.org/ 10.1021/bi3001825 [DOI] [PubMed] [Google Scholar]
- 51. Yoon MK, Venkatachalam V, Huang A, Choi BS, Stultz CM, Chou JJ. Residual structure within the disordered C-terminal segment of p21(Waf1/Cip1/Sdi1) and its implications for molecular recognition. Protein Sci 2009; 18:337-47; PMID:19165719; http://dx.doi.org/ 10.1002/pro.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR 1994; 4:171-80; PMID:8019132; http://dx.doi.org/ 10.1007/BF00175245 [DOI] [PubMed] [Google Scholar]
- 53. Wishart DS, Sykes BD. Chemical shifts as a tool for structure determination. Methods Enzymol 1994; 239:363-92; PMID:7830591; http://dx.doi.org/ 10.1016/S0076-6879(94)39014-2 [DOI] [PubMed] [Google Scholar]
- 54. Li C, Pazgier M, Li C, Yuan W, Liu M, Wei G, Lu WY, Lu W. Systematic mutational analysis of peptide inhibition of the p53-MDM2/MDMX interactions. J Mol Biol 2010; 398:200-13; PMID:20226197; http://dx.doi.org/ 10.1016/j.jmb.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zondlo SC, Lee AE, Zondlo NJ. Determinants of specificity of MDM2 for the activation domains of p53 and p65: proline27 disrupts the MDM2-binding motif of p53. Biochemistry 2006; 45:11945-57; PMID:17002294; http://dx.doi.org/ 10.1021/bi060309g [DOI] [PubMed] [Google Scholar]
- 56. Johnson BAR, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR 1994; 4:603-14; PMID:22911360; http://dx.doi.org/ 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 57. Tamiola K, Acar B, Mulder FA. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J Am Chem Soc 2010; 132:18000-3; PMID:21128621; http://dx.doi.org/ 10.1021/ja105656t [DOI] [PubMed] [Google Scholar]
- 58. Wishart DS. Interpreting protein chemical shift data. Prog Nucl Magn Reson Spectrosc 2011; 58:62-87; PMID:21241884; http://dx.doi.org/ 10.1016/j.pnmrs.2010.07.004 [DOI] [PubMed] [Google Scholar]
- 59. Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR 2007; 38:289-302; PMID:17610132; http://dx.doi.org/ 10.1007/s10858-007-9166-6 [DOI] [PubMed] [Google Scholar]
- 60. Cubellis M, Cailliez F, Lovell S. Secondary structure assignment that accurately reflects physical and evolutionary characteristics. BMC Bioinformatics 2005; 6:S8; PMID:16351757; http://dx.doi.org/ 10.1186/1471-2105-6-S4-S8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem 2005; 26:1701-18; PMID:16211538; http://dx.doi.org/ 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]