

Abstract
Injury to the meninges is not uncommon after traumatic brain injury (TBI), yet minimal research has been directed toward understanding the relevant biology. After a concussive event, the meninges are observed to abnormally enhance on post-contrast magnetic resonance imaging (MRI) in some patients, but not all. The aim of this work is to identify genes differentially expressed in patients with meningeal injury. Patients presenting to the emergency room with suspected TBI received a standard research MRI and blood draw within 48 h of injury. Two groups of patients were included: those with and without abnormal enhancement of the meninges on post-contrast MRI, both without other imaging findings. Groups were compared on microarray gene expression in peripheral blood samples using Affymetrix (Santa Clara, CA) and Partek Genomics Suite (Partek, Inc., St. Louis, MO) software (false discovery rate, <0.05). Forty patients were enrolled with a time from injury to MRI/blood draw of 16.8 h (interquartile range, 7.5–24.1). We observed 76 genes to be differentially expressed in patients with meningeal injury compared to those without, such as receptor for Fc fragment of IgA, multiple C2 domains, transmembrane 2, and G-protein-coupled receptor 27, which have been previously associated with initiating inflammatory mediators, phagocytosis, and other regulatory mechanisms. Post-contrast MRI is able to detect meningeal injury and has a unique biological signature observed through gene expression. These findings suggest that an acute inflammatory response occurs in response to injury to the meninges following a concussion.
Keywords: : brain trauma, concussion, gene expression, MRI
Introduction
The meninges, often given minimal consideration in the pathogenesis of traumatic brain injury (TBI), have recently come into the spotlight with the recognition that the central nervous system CNS may contain a functional lymphatic system.1 The meninges house a highly complex vasculature that is critical for the delivery of nutrients to the brain, removal of waste, and response of sentinel inflammatory cells. Injury to the meninges after a TBI leads to bleeding between the layers of the meninges, resulting in a subarachnoid, subdural, or epidural hemorrhage. Abnormal enhancement of the meninges on post-contrast magnetic resonance imaging (MRI) can be observed in the absence of other neuroimaging findings and is suggestive of traumatic meningeal injury (TMI).
Evidence from a mouse model of mild TBI (mTBI) that involved minor injury to the meninges resulted in a rapid inflammatory response, and neuronal death through oxidative stress,2 raising concern that TMI may not be innocuous. Changes in gene expression post-TMI can provide clues as to the specific pathways that interact to promote recovery or a secondary injury.3–5 Gene expression profiles of these cells, including that of microglia, are communicated through interactions by neuroimmune pathways that coordinate immune responses in the periphery.6 In considering the role the meninges play in mediating inflammation and clearance of substances from the cerebral spinal fluid (CSF), TMI may be a discrete phenotype of TBI with potential deleterious downstream consequences. Here, we describe the differential gene expression in peripheral blood collected from a cohort of acute patients, differentiated by the presence or absence of TMI.
Methods
Population
This study was reviewed and approved by the appropriate human patient protection authorities at the National Institutes of Health, Uniform Services University of the Health Sciences, Johns Hopkins Suburban Hospital, and MedStar Washington Hospital Center. All patients or surrogates provided informed consent before any study procedure. Patients were enrolled in the Center for Neuroscience and Regenerative Medicine (CNRM) Traumatic Head Injury Neuroimaging Classification protocol (NCT01132937) if they sustained a head injury and presented within 48 h of the event to the emergency department of MedStar Washington Hospital Center (WHC; Washington, DC) or Johns Hopkins Suburban Hospital (SH; Bethesda, MD), level 1 and level 2 trauma centers, respectively. A computed tomography (CT) scan was obtained as clinically indicated. After consent, patients were imaged with MRI and blood was collected. Clinical history, presentation, symptoms, and disposition were captured prospectively on standardized case-report forms.
For this analysis, patients were selected who 1) were below the age of 70 (group matched), 2) received contrast agent, 3) aside from TMI, were absent of trauma-related findings on both clinical CT and research MRI, and 4) had blood collection performed at time of MRI. mild TBI is defined by the published guidelines of the American Congress of Rehabilitation Medicine, which include:
• Loss of consciousness of approximately 30 min or less
• An initial Glasgow Coma Scale (GCS) of 13–15
• Post-traumatic amnesia not greater than 24 h
Based on these guidelines, all of the patients included in this study qualify as having sustained an mTBI.
Images were reviewed prospectively for trauma-related findings; TMI was determined as abnormal enhancement of the meninges with a positive post-contrast fluid-attenuated inversion recovery (FLAIR) and no other imaging findings, and the TMI– group consisted of a negative MRI and a negative CT.
Magnetic resonance imaging
Research MRI was obtained as soon as possible after presentation and as performed on a 1.5 Tesla (T; GE Medical Systems, Milwaukee, WI) and a 3T (Philips, Cleveland, OH) at SH and WHC, respectively. The imaging protocol was standardized and took approximately 25 min to execute. It consisted of a diffusion tensor imaging sequence with derived isotropic diffusion weighted imaging and apparent diffusion coefficient map, two T2*-weighted sequences, FLAIR, three-dimensional/T1 weighted with 1-mm isotropic voxels, dynamic susceptibility contrast perfusion imaging, post-contrast T1, and post-contrast FLAIR (FLAIR-post). Sequence parameters were adjusted as best possible to produce similar appearing contrasts across field strength.
Patients were administered a single dose of a gadolinium-based contrast agent; 0.1 mmol/kg of gadopentetate dimeglumine (Bayer HealthCare Pharmaceuticals, Whippany, NJ) or gadobenate dimeglumine (Braco Diagnostics, Monroe Township, NJ), depending on site policy. Contrast was administered using a power injector at 5 mL/sec through a 22-18-gauge needle placed in the antecubital vein. The post-contrast FLAIR was obtained approximately 5 min after contrast agent administration and took approximately 2 min to acquire.
Sample collection
Sample was collected as close to the time of research MRI as possible. Approximately 2.5 mL of blood were collected into an RNA PAXgene tube. After collection, the PAXgene tubes were inverted eight times and then placed at room temperature for approximately 1 h, and then at −200C for 2 h before placement into an −800C freezer until assayed.
Messenger RNA acquisition, quantitation, and hybridization
PAXgene blood RNA tubes were processed with PAXgene™ Blood RNA Kits (PreAnalytiX; Qiagen, Hombrechtikon, Switzerland) for RNA extraction. The NanoDrop DN-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE) and the Agilent Bioanalyzer 2100 eukaryotic total RNA Nano assay (Agilent Technologies, Inc., Santa Clara, CA) were used to evaluate the quality and quantity of extracted RNA. The 260/280 ratio ranged from 2.03 to 2.34, and the RNA integrity numbers (RINs) were greater than 7.0 in all samples. According to standards,7 an RIN of 6.0 or greater is indicative of admissible quality, and no samples were excluded based on this criterion. Using the GeneChip (GC) 3' IVT Express kit, each RNA (100 ng) sample was reverse transcribed, converted to biotinylated complementary RNA, and hybridized to Affymetrix HG-U133 Plus 2.0 microarrays (Affymetrix, Santa Clara, CA), which contain 54,675 probe sets representing more than 38,500 coding genes. All assays were undertaken based on standard Affymetrix protocols.
Clinical measures
The GCS was obtained during the clinical assessment, and the Neurobehavioral Symptom Inventory (NSI) was administered to all patients by trained research staff. The GCS is a 15-point clinical evaluation performed by a clinician and is the most commonly used TBI assessment used in emergency room settings.8 This measure consists of three subcomponents, which include ocular, verbal, and motor function. The GCS helps assess for loss of consciousness and other behavioral differences post-injury and has been measured for validity and reliability in previous research.9,10
The NSI is a 22-item self-report questionnaire to assess for post-concussive symptoms. This measure has both a high internal consistency (total alpha = 0.95; subscale alpha = 0.88–0.92) and reliability (r = 0.88–0.93).11 Each question asks for the participant to rate the severity of each symptom, such as nausea, headache, and balance, based on a 5-point scale (none, mild, moderate, severe, and very severe). Severity of symptoms for the patient is assessed by adding up all the responses, providing a range of 0–88 to determine their NSI score.
Statistical analysis
All analytical procedures performed on microarray data were conducted with Partek Genomics Suite software (version 6.6; Partek Inc., St. Louis, MO). Interrogating probes were imported, and corrections for background signal were applied using the robust multi-array average method, with additional corrections applied for the GC content of probes. Probe sets were standardized using quantile normalization, and expression levels of each probe underwent log-2 transformation to yield distributions of data that more closely approximated normality.
Parameters for identifying differentially expressed genes between groups were then identified using analysis of variance of each probe set's expression level as a function of grouping variable: TMI+ compared to TMI– while adjusting for batch effect. A repeated-measures comparison was made in each group. Parameters for significant differential gene expression between the two groups consisted of a p value corrected for multiple comparisons using a false discovery rate <0.05 and a fold change (increase or decrease) of 1.5.
Two-tailed chi-square tests and independent t-tests were used to test group differences that might affect the main analysis using SPSS software (SPSS, Inc., Chicago, IL). The analysis investigated the relationship between TMI+ and TMI– patients' gene expression. Independent t-tests were used to examine between-group differences on outcome variables. Mann-Whitney's U test was used to calculate statistical significance between groups for nonparametric measures, as indicated in Table 1. The confidence level was set at p = 0.05 for all analyses. Data reported are median (interquartile range; IQR).
Table 1.
Demographics and Characteristics of Patients Dichotomized Based on Presence (TMI+) or Absence (TMI–) of Abnormal Enhancement on Post-Contrast FLAIR MRI
| Variable | TMI+ | TMI− | p value |
|---|---|---|---|
| No. | 17 | 23 | |
| Male | 13 (76.5) | 15 (65.2) | 0.443 |
| Age | 39.8 (22.7–57.8) | 38.0 (28.3–46.3) | 0.694 |
| Caucasian | 15(88.2) | 20 (87.0) | 0.395 |
| Latino/Hispanic | 3 (17.6) | 5 (21.7) | 0.749 |
| Post-traumatic amnesia | 13 (76.5) | 12 (52.2) | 0.117 |
| Post-traumatic amnesia duration | 0.503 | ||
| N/A | 4 (23.5) | 11 (47.8) | |
| 1 sec–10 min | 7 (41.2) | 5 (21.7) | |
| 10 min–30 min | 2 (11.8) | 2 (8.7) | |
| 30 min–1 h | 3 (17.6) | 2 (8.7) | |
| 1–12 h | 1 (5.9) | 2 (8.7) | |
| Unknown | 0 (0.0) | 1 (4.3) | |
| Loss of consciousness | 14 (82.4) | 13 (56.5) | 0.126 |
| Loss of consciousness duration | 0.153 | ||
| N/A | 2 (11.8) | 9 (39.1) | |
| <1 min | 1 (5.9) | 3 (13.0) | |
| 1–29 min | 9 (52.9) | 6 (26.1) | |
| Unknown | 5 (29.4) | 5 (21.7) | |
| GCS <15 | 0 (0) | 5 (23) | 0.062 |
| NSI Cumulative Score | 15 (13–17) | 12 (7–16) | 0.146 |
| Injury mechanism | 0.767 | ||
| Acceleration/deceleration | 3 (17.6) | 5 (21.7) | |
| Direct impact (blow to head) | 7 (41.2) | 6 (26.1) | |
| Direct impact (head against object) | 4 (23.5) | 5 (21.7) | |
| Fall (ground floor) | 2 (11.8) | 3 (13.0) | |
| Fall (height >1 m) | 1 (5.9) | 4 (17.4) | |
| Hours from injury to blood draw | 20 (16–26) | 11 (5–21) | 0.021 |
| Hours from injury to MRI scan | 21 (14–26) | 12 (5–21) | 0.023 |
Data are median (Q1–Q3) or n (%).
Bold values signify p < 0.05 statistical significance.
TMI, traumatic meningeal injury; FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging; N/A, not applicable; GCS, Glasgow comma scale; NSI, Neurobehavioral Symptom Inventory.
To identify gene networks and interactions associated with a biological mechanism QIAGEN's Ingenuity® Pathway Analysis (IPA; version 2014-07-10; IPA®; QIAGEN, Redwood City, CA; www.qiagen.com/ingenuity) was performed on differentially expressed genes. IPA runs the complete list of differentially expressed genes between the TMI+ and TMI– groups and identifies focus genes that are known to interact with other genes based on ingenuity data. A Fisher's exact test identifies highly inter-related genes within the pathway. To quantify the significance, the amount of connections used by a focus gene within a pathway is compared to other significant genes within the database. Each network is assigned a score based on the probability of the gene interaction occurring within the pathway. This score is calculated using the following equation:
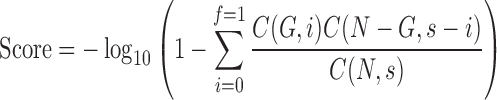 |
In this equation, N is the number of genes in the network, C(n,k) is the binomial coefficient, and G are the focus genes in s, which represents the pathway with f focus genes.12
Last, hierarchical clustering was used to investigate whether expression levels of significant genes could be used to correctly predict whether the patients had sustained a TMI. To calculate this, the top 15 genes were clustered using Partek Genomics Suite. With no classification provided (TMI+ vs. TMI–), patients were clustered based on gene expression. A chi-squared test was used to measure how well the resulting patient clusters fit the original TBI classifications.
Results
Demographic and clinical characteristics
Forty patients were included; 17 patients were positive for TMI and the remaining 23 were negative (Fig. 1). Demographic characteristics are described in Table 1. Overall, the sample was primarily male (70.0%), Caucasian (87.5%), and the median age was 38 (IQR, 26.7–49.9) years in age. GCS scores for the two groups both had median scores of 15 and NSI scores of 15 and 12 for the TMI+ and TMI– groups, respectively. The distribution of GCS were as follows: 15 (77.5%), 14 (10.0%), and 13 (2.5%), with 4 patients not having a GCS collected. Patients were identified based on clinical presentation, and no patients included had a GCS score below 13. Patients in the TMI+ group tended to have been imaged earlier. TMI was most frequently observed in the falx cerebri (n = 15), followed by frontal, temporal, parietal, occipital, tentorium, and regions of the pons, which each occurred in 3 or less patients. Loss of consciousness was observed in 82.4% of TMI+ patients and 56.5% of TMI– patients. Post-traumatic amnesia occurred in 76.5% of TMI+ patients and 52.2% of TMI– patients.
FIG. 1.

TMI+ versus TMI– scans. Example of magnetic resonance imaging obtained from 2 paitents in the study, with and without evidence of TMI. Shown are the GRE which is sensitive to blood, and DWI which is sentitive to ischemia, and pre- and post contrast FLAIR used to detect TMI. Red arrows indicated abnormal enhancment of meningese seen on post-contrast FLAIR in the falx cerebri and along the anterior convexity of the frontal lobe. DWI, diffusion weighted image; FLAIR, fluid-attenuated inversion recovery; GRE, gradient recalled echo; TMI, traumatic meningeal injury.
We report that 76 genes were differentially expressed in the TMI+ group in comparison to the TMI– group, with 44 genes upregulated (Table 2) and 32 downregulated (Table 3). Of these, there were four genes with a change of 1.5-fold or higher (Fig. 2): LOC100134822 (1.62); immunoglobulin A (IgA); Fc receptor (FcαR) (1.58); multiple C2 domains; transmembrane 2 (MCTP2) (1.54); and G-protein-coupled receptor 27 (GPR27) (1.52). IPA analysis revealed three distinguishable networks, all of which received a score above 30. These networks are associated with cellular development, organismal injury and abnormalities, and cellular organization.
Table 2.
Upregulated Gene Expression
| Gene symbol | Gene name | Fold change | p value |
|---|---|---|---|
| LOC100134822 | Uncharacterized LOC100134822 | 1.62426 | 5.05E-05 |
| FCαR | Fc fragment of IgA, receptor for | 1.58147 | 4.81E-08 |
| MCTP2 | Multiple C2 domains, transmembrane 2 | 1.54058 | 1.85E-05 |
| GPR27 | G-protein-coupled receptor 27 | 1.52042 | 5.88E-05 |
| QPCT | Glutaminyl-peptide cyclotransferase | 1.44518 | 4.22E-05 |
| BCL6 | B-cell CLL/lymphoma 6 | 1.4346 | 3.22E-05 |
| KIF1B | Kinesin family member 1B | 1.43429 | 5.07E-05 |
| BST1 | Bone marrow stromal cell antigen 1 | 1.36696 | 1.69E-05 |
| KIF13A | Kinesin family member 13A | 1.34934 | 4.92E-05 |
| UGCG | UDP-glucose ceramide glucosyltransferase | 1.34156 | 1.28E-06 |
| SKAP2 | Src kinase associated phosphoprotein 2 | 1.33539 | 3.21E-07 |
| IL10RB-AS1 | IL10RB antisense RNA 1 (head to head) | 1.33457 | 4.48E-06 |
| ZNF281 | Zinc finger protein 281 | 1.32713 | 5.41E-05 |
| MTF1 | Metal-regulatory transcription factor 1 | 1.27487 | 1.43E-05 |
| JHDM1D | Jumonji C domain containing histone demethylase 1 homolog D (S. cerevisiae) | 1.23287 | 8.42E-06 |
| NPL | N-acetylneuraminate pyruvate lyase (dihydrodipicolinate synthase) | 1.21864 | 9.96E-06 |
| USP3 | Ubiquitin specific peptidase 3 | 1.20802 | 8.32E-07 |
| FLJ36848 | Uncharacterized LOC647115 | 1.20694 | 1.55E-05 |
| CTSZ | Cathepsin Z | 1.19916 | 1.28E-05 |
| GSR | Glutathione reductase | 1.1836 | 2.66E-05 |
| DENND5A | DENN/MADD domain containing 5A | 1.18162 | 5.99E-05 |
| LOC401324 | Uncharacterized LOC401324 | 1.17641 | 2.21E-05 |
| BLOC1S1 | Biogenesis of lysosomal organelles complex-1, subunit 1 | 1.17169 | 8.31E-07 |
| FAM83H | Family with sequence similarity 83, member H | 1.16839 | 2.14E-05 |
| MAPK15 | Mitogen-activated protein kinase 15 | 1.15884 | 3.37E-05 |
| NBLA00301 | Nbla00301 | 1.13391 | 1.25E-05 |
| C8orf17 | Chromosome 8 open reading frame 17 | 1.12907 | 6.23E-06 |
| OFCC1 | Orofacial cleft 1 candidate 1 | 1.12843 | 4.95E-06 |
| FA2H | Fatty acid 2-hydroxylase | 1.12621 | 1.34E-05 |
| AMPD3 /// LOC100130460 | Adenosine monophosphate deaminase 3 /// uncharacterized LOC100130460 | 1.12446 | 4.53E-05 |
| CDC42 | Cell division cycle 42 | 1.11818 | 4.91E-05 |
| PPM1E | Protein phosphatase, Mg2+/Mn2+ dependent, 1E | 1.0928 | 6.40E-05 |
| HES5 | Hairy and enhancer of split 5 (Drosophila) | 1.08581 | 3.57E-05 |
| “LIM” | LIM and senescent cell antigen-like domains 3 /// LIMS3-LOC440895 readthrough /// LIM a | 1.08375 | 6.47E-05 |
LIM, LIMS3 /// LIMS3-LOC440895 /// LIMS3L /// LOC100288570 /// LOC100507334 /// LOC440895.
Table 3.
Downregulated Gene Expression
| Gene symbol | Gene name | Fold change | p value |
|---|---|---|---|
| EIF3C /// EIF3CL | Eukaryotic translation initiation factor 3, subunit C /// eukaryotic translation initia | −1.37763 | 2.85E-06 |
| CD79A | CD79a molecule, immunoglobulin-associated alpha | −1.32909 | 3.00E-05 |
| SPIB | Spi-B transcription factor (Spi-1/PU.1 related) | −1.26792 | 6.40E-05 |
| ZBTB40 | Zinc finger and BTB domain containing 40 | −1.26737 | 2.46E-05 |
| UTP20 | UTP20, small subunit (SSU) processome component, homolog (yeast) | −1.25077 | 7.07E-08 |
| ORMDL3 | ORM1-like 3 (S. cerevisiae) | −1.24365 | 4.49E-05 |
| WDR74 | WD repeat domain 74 | −1.24295 | 8.60E-06 |
| SCFD2 | Sec1 family domain containing 2 | −1.23579 | 4.06E-05 |
| TRAV30 /// TRAV30 | T-cell receptor alpha variable 30 /// NULL | −1.22505 | 5.48E-07 |
| NOC2L | Nucleolar complex associated 2 homolog (S. cerevisiae) | −1.22447 | 3.92E-08 |
| NCOA5 | Nuclear receptor coactivator 5 | −1.21903 | 5.98E-05 |
| C22orf46 | Chromosome 22 open reading frame 46 | −1.19363 | 3.87E-08 |
| MCF2L | MCF.2 cell line derived transforming sequence-like | −1.17744 | 1.83E-09 |
| C1orf123 | Chromosome 1 open reading frame 123 | −1.17403 | 1.80E-05 |
| HN1L | Hematological and neurological expressed 1-like | −1.17389 | 1.78E-06 |
| RABL2A /// RABL2B | RAB, member of RAS oncogene family-like 2A /// RAB, member of RAS oncogene family-like | −1.17347 | 3.91E-06 |
| HLCS | Hyolocarboxylase synthetase (biotin-(proprionyl-CoA-carboxylase (ATP-hydrolysing)) ligase) | −1.16346 | 2.45E-07 |
| GLG1 | Golgi glycoprotein 1 | −1.16291 | 3.80E-05 |
| ARHGAP5 | Rho GTPase activating protein 5 | −1.1609 | 4.50E-06 |
| TMEM99 | Transmembrane protein 99 | −1.16045 | 2.64E-06 |
| IL23R | Interleukin 23 receptor | −1.15507 | 3.03E-07 |
| TMEM231 | Transmembrane protein 231 | −1.14928 | 1.83E-06 |
| TFDP2 | Transcription factor Dp-2 (E2F dimerization partner 2) | −1.14359 | 1.42E-07 |
| ZNF16 | Zinc finger protein 16 | −1.13429 | 1.59E-05 |
| SLC41A3 | Solute carrier family 41, member 3 | −1.13164 | 1.20E-05 |
| YEATS2 | YEATS domain containing 2 | −1.12893 | 9.85E-06 |
| SENP3 | SUMO1/sentrin/SMT3 specific peptidase 3 | −1.10727 | 4.83E-05 |
FIG. 2.
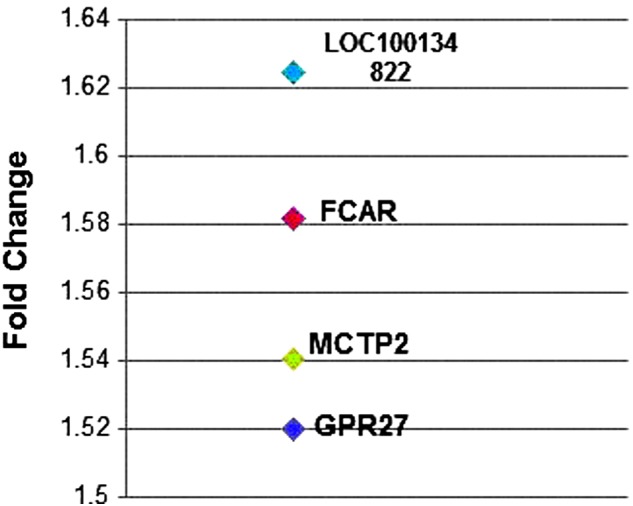
Top four differentially expressed genes. Of the 76 genes found to be differentially expressed in the TMI+ group in comparison to the TMI– group, four were above a 1.5-fold change. These genes have been previously associated with inflammatory mechanisms, phagocytosis, and other regulatory systems. These findings suggest that gene expression profiles may inform a diagnostic biomarker of TMI. TMI, traumatic meningeal injury.
Hierarchical clustering was used to cluster patients based on expression levels of the top 15 differentially expressed genes. A heat map (Fig. 3) illustrates the differential expression of these genes between TMI+ and TMI– groups. Predicted TBI classification based on gene clustering is compared to actual diagnoses, with 70% of patients correctly assigned to their respective groups (p = 0.015).
FIG. 3.

Hierarchical cluster. A hierarchical cluster analysis was able to correctly predict 70% of patients TMI groupings based on their gene expression changes. This suggests that gene expression biomarkers have a utility that are diagnostic of TMI. TMI, traumatic meningeal injury.
Discussion
The gene-expression data presented here suggests that traumatic injury to the meninges, as evidenced by imaging showing TMI, has a distinct biological signature that is inflammatory in nature. We report this finding in two groups of patients presenting to an emergency department in the acute period post-trauma, all of whom are similar in the severity of TBI and type of injury, discriminated solely on the presence or absence of injury to the meninges. This design allows for us to isolate genes specific to TMI, while controlling for genes related to acute mTBI. Specifically, the genes expressed differentially in the TMI group, including FcαR and GPR27, point toward an inflammatory response that may be specific to the meninges. Evidence from pre-clinical work2 has shown that minor traumatic injury to the meninges results in a rapid inflammatory neuronal response that is characterized by free radical release, and ultimately neuronal death, implicating inflammation in the neuronal processes that are associated with TMI. Should this hold true in humans, TMI and the related biological pathways identified through gene expression may offer an opportunity to learn about pathological mechanisms of concussions and TBIs, which will ultimately lead to the development of appropriate diagnostic applications as well as therapeutic interventions.
A variety of genes were differentially expressed in patients with TMI, with many being related to inflammation. The gene expression analysis revealed that 76 coding genes are differentially expressed between patients with and without TMI. Of these genes, four were found to have a fold change greater than 1.5, including LOC100134822 (1.62), FcαR (also known as CD89) (1.58), MCTP2 (1.54), and GPR27 (1.52). These genes have been previously associated with inflammatory mechanisms and other regulatory systems.13–15 In addition, IPA analysis revealed that FcαR, MCTP2, and GPR27 are involved in networks associated with inflammatory pathways that relate to cellular development. Specifically, we report that expression of FcαR is more than 50% greater in TMI patients. FcαR, more commonly known as CD89, is a key component in the initiation of many immunological defense mechanisms, such as the release of cytokines, superoxide, and other inflammatory mediators, as well as phagocytosis and antigen presentation.13,15 This gene is active in macrophages, monocytes, neutrophils, and eosinophils, and when it binds to IgA, it activates intracellular signaling that can result in the expression of inflammatory cytokines, such as tumor necrosis factor alpha and interleukin-1.16 In addition, previous clinical research has linked FcαR to a protective effect against pathogens by it increasing activity in IgA.13,17 This suggests that the upregulation of this gene's expression in patients with TMI may be part of a protective mechanism.
Our findings can be generalized to the broader TBI population. This is the first study to compare TBI patients with and without TMI that otherwise have similar presentation and injury severity. Our finding that hierarchical clustering could correctly predict 70% of patients' TBI groupings based on their gene expression changes suggest that expression of target genes may have utility as biomarkers that are diagnostic of TMI. Although this design allows for identification of gene activity specific to TMI, it is limited by a relatively small sample size, requiring future studies to validate and extend our findings. In addition, IPA analysis identified nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) as the most significant network (IPA network score of 49) in TMI+ patients. Activation of NF-κB is common in the brain during the acute stage post-TBI.18 Research has not investigated the relationship of NF-κB after meningeal injury; however, research on meningitis, an inflammatory disease of the meninges, reports that NF-κB largely regulates proinflammatory cytokines.19–21 NF-κB is closely associated with blood–brain barrier permeability as well22 and has been investigated as a treatment target for preventing secondary injury response from occurring in stroke patients.23 Despite the lack of research focused on meningeal injury, the hierarchical cluster and IPA analysis support that there may be a different mechanism occurring in patients with a TMI.
Advantages of this study include that patients were MRI imaged soon after what would previously have been called a “concussive event,” irrespective of the suspicion of TMI.
All patients experienced a trauma severe enough to warrant presentation to an emergency department for treatment of TBI. In addition, all patients had suspected mTBI determined by clinical evaluation and were excluded if there was evidence of injury other than TMI on the CT or MRI. Time to imaging was slightly longer for the TMI+ group. This may suggest a slight increase in the likelihood of detecting TMI with increasing severity of injury time.
Our findings likely have relevance to a broader population, given that previous research shows prevalence of TMI in the population screened with MRI was approximately 1 in 22 and is observed in concert with other intracranial injury.24 The existence of TMI after minor injury that does not result in acute medical evaluation, such as after sports concussion, has yet to be demonstrated. However, as is true of other forms of traumatic intracranial injury, it is reasonable to expect that injury to the meninges, and the related biological response examined here, may apply to all severities of TBI.
Whereas the clinical relevance to acute care or long-term outcome remains to be established, there is concern that meningeal injury could result in parenchymal injury. In support of this, a pre-clinical model of TBI found that meningeal injury can lead to secondary injury in the adjacent parenchyma,2 as a result of cytotoxic death and oxidative stress.2,25 In addition, recent pre-clinical findings on the presence of a lymphatic system in the meninges reveal the role of inflammatory cells that are able to have access to the meninges and the brain.1 The existence of these inflammatory cells within the brain supports the supposition that a disruption to the meninges during TBI may ignite an immune response that can be destructive if not sufficiently regulated. This pathway allows for easy access of activated immune cells to the meninges and surrounding parenchyma, thereby allowing these cells to transfer immune activity in the meninges to the parenchyma and promote secondary injury. This is supported by previous research finding gene expression changes in peripheral blood reflecting damage to central activities attributed to microglia communicating with peripheral immune cells post-TBI.26 Thus, identifying the relevant biological pathways associated with meningeal injury could provide an opportunity for treatment before a secondary injury response occurs.
Detection of meningeal injury is possible because of the contrast mechanism specific to FLAIR MRI used in imaging TMI. The preparation phase of the sequence nulls signal coming from spaces containing free fluid with a very long T1, similar to that of CSF, such as in the ventricles and sulci. This results in a T2-weigted image in which free fluid appears dark. Gadolinium-based contrast agents shorten T1. When contrast is extravasated into spaces containing free fluid, such as occurs with blood–brain barrier disruption after stroke,27,28 the FLAIR suppression fails and the free-fluid spaces appear bright. The null approach used in FLAIR dramatically increases the sensitivity to contrast extravasation, compared to that of conventional T1,29 as long as the space involved contains free fluid. With TMI, we presume that trauma results in the accumulation of fluid either in the dura or in a newly created space at the interface between the dura and arachnoid membrane. In more-severe TBI, injury to the meninges results in filling this potential “subdural” space with blood or xanthochromic fluid (i.e., a subdural hemorrhage or effusion). In patients with TMI, we suspect that the contrast agent is extravasated into these same fluid-filled spaces, T1 shortening from the contrast agent results in a failure of the FLAIR to suppress the water signal, and the meninges dramatically enhance when compared to pre-contrast FLAIR. TMI may be part of a continuum of severity shared with subdural effusion and hemorrhage.
Our findings suggest that meningeal enhancement on contrast-enhanced FLAIR imaging is a relevant phenotype of TBI with a biological profile evident in differential gene expression. The increased expression of inflammatory related genes, such as FcαR, suggests that TMI is related to inflammatory processes in the acute phase of TBI. This finding also suggests that peripheral immune cells may have differential expression that relates to TMI, and that diagnostics and pharmacological targets may be determined from validations of this line of research. Further research on the propagation of the secondary injury cascade subsequent to a positive post-contrast FLAIR for TMI could provide useful information for the identification of pharmacological targets that can improve clinical treatment for TBIs.
Acknowledgments
The authors are grateful to the patients and their families, for without them this research would not be possible. The authors acknowledge the staff at Johns Hopkins Suburban Hospital, MedStar Washington Hospital Center, and the National Institute of Neurological Disorders and Stroke (NINDS) Acute Stroke Team for their assistance during this study. Support for this work included Department of Defense in the Center for Neuroscience and Regenerative Medicine (CNRM), the NINDS, and the National Institute of Nursing Research (NINR).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Louveau A., Smirnov I., Keyes T.J., Eccles J.D., Rouhani S.J., Peske J.D., Derecki N.C., Castle D., Mandell J.W., Lee K.S., Harris T.H., and Kipnis J. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roth T.L., Nayak D., Atanasijevic T., Koretsky A.P., Latour L.L., and McGavern D.B. (2014). Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michael D.B., Byers D.M., and Irwin L.N. (2005). Gene expression following traumatic brain injury in humans: analysis by microarray. J. Clin. Neurosci. 12, 284–290 [DOI] [PubMed] [Google Scholar]
- 4.Frugier T., Crombie D., Conquest A., Tjhong F., Taylor C., Kulkarni T., McLean C., and Pébay A. (2011). Modulation of LPA receptor expression in the human brain following neurotrauma. Cell. Mol. Neurobiol. 31, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grond-Ginsbach C., Hummel M., Wiest T., Horstmann S., Pfleger K., Hergenhahn M., Hollstein M., Mansmann U., Grau A.J., and Wagner S. (2008). Gene expression in human peripheral blood mononuclear cells upon acute ischemic stroke. J. Neurol. 255, 723–731 [DOI] [PubMed] [Google Scholar]
- 6.Hernadez-Ontiveros D.G., Tajiri N., Acosta S., Giunta B., Tan J., and Borlongan C.V. (2013). Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 4, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schroeder A., Mueller O., Stocker S., Salowsky R., Leiber M., Gassmann M., Lightfoot S., Menzel W., Granzow M., and Ragg T. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill M., Windemuth R., Steele R., and Green S.M. (2005). A comparison of the Glasgow Coma Scale score to simplified alternative scores for the prediction of traumatic brain injury outcomes. Ann. Emerg. Med. 45, 37–42 [DOI] [PubMed] [Google Scholar]
- 9.Grmec S., and Gasparovic V. (2001). Comparison of APACHE II, MEES and Glasgow Coma Scale in patients with nontraumatic coma for prediction of mortality. Acute Physiology and Chronic Health Evaluation. Mainz Emergency Evaluation System. Crit. Care 5, 19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai C.L., Chu H., Peng G.S., Ma H.I., Cheng C.A., and Hueng D.Y. (2012). Preoperative APACHE II and GCS scores as predictors of outcomes in patients with malignant MCA infarction after decompressive hemicraniectomy. Neurol. India 60, 608–612 [DOI] [PubMed] [Google Scholar]
- 11.King P.R., Donnelly K.T., Donnelly J.P., Dunnam M., Warner G., Kittleson C.J., Bradshaw C.B., Alt M., and Meier S.T. (2012). Psychometric study of the Neurobehavioral Symptom Inventory. J. Rehabil. Res. Dev. 49, 879–888 [DOI] [PubMed] [Google Scholar]
- 12.Calvano S.E., Xiao W., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Miller-Graziano C., Moldawer L.L., Mindrinos M.N., Davis R.W., Tompkins R.G., and Lowry S.F. (2005). A network-based analysis of systemic inflammation in humans. Nature 437, 1032–1037 [DOI] [PubMed] [Google Scholar]
- 13.Ben Mkaddem S., Rossato E., Heming N., and Monteiro R.C. (2013). Anti-inflammatory role of the IgA Fc receptor (CD89): From autoimmunity to therapeutic perspectives. Autoimmun. Rev. 12, 666–669 [DOI] [PubMed] [Google Scholar]
- 14.Lu J., Marjon K.D., Marnell L.L., Wang R., Mold C., Du Clos T.W., and Sun P. (2011). Recognition and functional activation of the human IgA receptor (FcalphaRI) by C-reactive protein. Proc. Natl Acad. Sci. U. S. A. 108, 4974–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monteiro R.C. (2010). The role of IgA and IgA Fc receptors as anti-inflammatory agents. J. Clin. Immunol. 30, S61–S64 [DOI] [PubMed] [Google Scholar]
- 16.Shen L., Collins J.E., Schoenborn M.A., and Maliszewski C.R. (1994). Lipopolysaccharide and cytokine augmentation of human monocyte IgA receptor expression and function. J. Immunol. 152, 4080–4086 [PubMed] [Google Scholar]
- 17.Monteiro R.C., and van de Winkel J.G.J. (2003). IGA receptors. Annu. Rev. Immunol. 21, 177–204 [DOI] [PubMed] [Google Scholar]
- 18.Hang C.-H., Chen G., Shi J.-X., Zhang X., and Li J.-S. (2006). Cortical expression of nuclear factor κB after human brain contusion. Brain Res. 1109, 14–21 [DOI] [PubMed] [Google Scholar]
- 19.Deng G.-M., Liu Z.-Q., and Tarkowski A. (2001). Intracisternally localized bacterial DNA containing CpG motifs induces meningitis. J. Immunol. 167, 4616–4626 [DOI] [PubMed] [Google Scholar]
- 20.Kim K.S. (2003). Emerging molecular targets in the treatment of bacterial meningitis. Expert Opin. Ther. Targets 7, 141–152 [DOI] [PubMed] [Google Scholar]
- 21.Barichello T., Generoso J.S., Simões L.R., Elias S.G., and Quevedo J. (2013). Role of oxidative stress in the pathophysiology of pneumococcal meningitis. Oxid. Med. Cell. Longev. 2013, 371465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merrill J.E., and Murphy SP. (1997). Inflammatory events at the blood brain barrier: regulation of adhesion molecules, cytokines, and chemokines by reactive nitrogen and oxygen species. Brain Behav. Immun. 11, 245–263 [DOI] [PubMed] [Google Scholar]
- 23.Borlongan C.V., Glover L.E., Sanberg P.R., and Hess D.C. (2012). Permeating the blood brain barrier and abrogating the inflammation in stroke: implications for stroke therapy. Curr. Pharm. Des. 18, 3670–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S.C., Park S.-W., Ryoo I., Jung S.C., Yun T.J., Choi S.H., Kim J.H., and Sohn C.H. (2014). Contrast-enhanced FLAIR (fluid-attenuated inversion recovery) for evaluating mild traumatic brain injury. PLoS One 9, e102229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Werner C., and Engelhard K. (2007). Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99, 4–9 [DOI] [PubMed] [Google Scholar]
- 26.Woodcock T., and Morganti-Kossmann C. (2013). The role of markers of inflammation in traumatic brain injury. Front Neurol 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latour L.L., Kang D.W., Ezzeddine M.A., Chalela J.A., and Warach S. (2004). Early blood-brain barrier disruption in human focal brain ischemia. Ann. Neurol. 56, 468–477 [DOI] [PubMed] [Google Scholar]
- 28.Henning E.C., Latour L.L., and Warach S. (2008). Verification of enhancement of the CSF space, not parenchyma, in acute stroke patients with early blood-brain barrier disruption. J. Cereb. Blood Flow Metab. 28, 882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathews V.P., Caldemeyer K.S., Lowe M.J., Greenspan S.L., Weber D.M., and Ulmer J.L. (1999). Brain: gadolinium-enhanced fast fluid-attenuated inversion-recovery MR imaging. Radiology 211, 257–263 [DOI] [PubMed] [Google Scholar]