

Abstract
The complex Cladonia mediterranea belongs to the section Impexae and is formed by C. azorica, C. macaronesica and C. mediterranea. These species are basically distributed in the Mediterranean and Macaronesian Regions. In the present work the limits between the species of this complex are re-examined. To this end, the morphological characters were studied along with the secondary metabolites and the DNA sequences from three loci (ITS rDNA, IGS rDNA and rpb2). The morphological data were studied by principal component analysis (PCA), while the DNA sequences were analyzed using several approaches available to delimit species: genealogical concordance phylogenetic species recognition, species tree (BEAST* and spedeSTEM) and cohesion species recognition. In addition, the genealogical sorting index was used in order to assess the monophyly of the species. The different procedures used in our study turned out to be highly congruent with respect to the limits they establish, but these limits are not the ones separating the prior species. Either the morphological analysis or the different approaches to species delimitation indicate that C. mediterranea is a different species from C. macaronesica, while C. azorica and C. macaronesica, which are reduced to synonyms of C. portentosa, constitute a separate lineage.
Keywords: coalescence, Iberian Peninsula, integrative taxonomy, lichen forming fungi, Macaronesia, molecular systematic, species delimitation, taxonomy
INTRODUCTION
The development of molecular tools has brought about a more accurate species delimitation and a better understanding of the evolution of fungi. The definition of many species has changed. It is well-known that in many fungal groups a large number of morphological species hide cryptic species complexes (Bickford et al. 2007, Crespo & Lumbsch 2010). Despite the major methodological advances made in species delimitation, it still constitutes a challenge, since there does not exist a valid method that allows identification of independent evolutionary lineages in all the cases (Sites & Marshall 2003, Carstens et al. 2013). One of the most used criterion for species delimitation in fungi has been the Genealogical Concordance Phylogenetic Species Recognition (Taylor et al. 2000), that uses several unlinked loci and reciprocal monophyly to identify the species. In many cases this criterion has been very useful to distinguish divergent lineages (Kroken & Taylor 2001, Dettman et al. 2003, Ott et al. 2004, Fournier et al. 2005, Doyle et al. 2013, Morgado et al. 2013). Nevertheless, due to species divergence being a temporal process, this criterion can fail in cases of delimitation of closely related species that have diverged recently (Knowles & Carstens 2007). Some other facts such as hybridization, recombination and horizontal transfer can also cause the gene tree to be inconsistent with the species tree (Eckert & Carstens 2008). There are of course other procedures used for species delimitation in fungi (Wirtz et al. 2008, Gazis et al. 2011). One of them is Templeton’s (1989) cohesion species recognition, that does not require species monophyly (Weisrock & Larson 2006, Wirtz et al. 2008, 2012). This method evaluates two hypotheses for the evolutionary lineages to be considered as species. The first of them (H1) is that there is only one evolutionary lineage in the studied group; the second (H2) is that the evolutionary lineages are genetically or ecologically exchangeable. The rejection of both hypotheses along with the congruence of H2 with the lineages found in H1 permits to delimit the cohesion species (Templeton et al. 2000). Numerous methods based on coalescence have recently been combined with the species delimitation procedures (O’Meara et al. 2006, Pons et al. 2006, Liu et al. 2009, Ence & Carstens 2011, Yang & Rannala 2010). They have the advantage of taking into account the incomplete lineage sorting and not requiring the reciprocal monophyly (Carstens & Knowles 2007, Fujita et al. 2012). These methods have been already applied in several works on species delimitation in lichenized fungi (Leavitt et al. 2011, 2012, 2013, Parnmen et al. 2012).
An emergent approach that is gathering increasing approval is the one that uses diverse data and analysis types to trace the most significant evidence, which permits the establishing of boundaries among species (Padial & de la Riva 2010, Gazis et al. 2011, Gebiola et al. 2012). This is the procedure that some authors call ‘taxonomical integration’ (Wiens & Penkrot 2002, Dayrat 2005, Will et al. 2005, Padial et al. 2009). According to Carstens et al. (2013), several analytical methods must be used in order to delimit species within a group of organisms, since each of the extant methods takes prior assumptions that not always fit the available data or the speciation scenery under screening.
Cladonia comprises most species within the family Cladoniaceae. According to Stenroos et al. (2002), Cladonia is a monophyletic genus that encompasses all the species of the former genus Cladina (Ahti 1961, 1984, 2000), represented by about 36 species from all over the world (Ahti 2000). This group, commonly known as reindeer lichens, is characterized by a crustose evanescent primary thallus, densely branched podetia, ecorticated, arachnoid surface, and by the absence of scyphi and soredia (Ahti 1961, 1984). Furthermore, Stenroos et al. (2002) showed that Cladina is a monophyletic group (Group Cladinae) divided into two clades, one corresponding to the old section Impexae and the other to the sections Cladina and Tenues. However, some studies indicated that the Group Cladinae is not monophyletic (DePriest et al. 1999, 2000, Guo & Kashiwadani 2004) but the old section Impexae is monophyletic. The section Impexae is represented in Europe by C. azorica, C. macaronesica, C. mediterranea, C. portentosa and C. stellaris. The problematic C. mediterranea complex includes three of these species, viz. C. azorica, C. macaronesica and C. mediterranea. Cladonia azorica is reported to be widespread in Madeira, Azores, Ireland, England and Iceland (Ahti 1961, Ruoss 1989, James 2009, Ahti & Stenroos 2013), while C. macaronesica is known from the Canary Islands, Madeira and Azores (Ahti 1961). Cladonia mediterranea has the broadest distribution, from Portugal to Turkey, southwestern Britain and the Canary Islands (des Abbayes & Duvigneaud 1947, Ruoss 1989, James 2009, Hernández-Padrón & Pérez-Vargas 2009).
It is still unclear whether C. azorica, C. macaronesica and C. mediterranea represent independent species or not. Ahti (1977) synonymized C. azorica with C. macaronesica; but later on, he again recognized C. azorica (Ahti 1984), whereas Ruoss (1989) concluded that C. mediterranea and C. macaronesica were conspecific. However, in many floristic works C. macaronesica and C. mediterranea are still treated as separate species (Hafellner 1995, Flores Rodrígues & Aptroot 2005, Carvalho et al. 2008, Hernández-Padrón & Pérez-Vargas 2009, Gabriel 2012). Sicilia et al. (2009) refer to C. mediterranea group because of the high morphological variation they found, while they pointed out the necessity of molecular studies for clarifying the taxonomy of this complex. To date, analyses using DNA sequence data have not been carried out.
The aim of the present work is to study the species delimitation in the Cladonia mediterranea complex using different approaches and several data sources: DNA sequences from three loci (ITS rDNA, IGS rDNA and rpb2), morphological data and chemical data.
MATERIAL AND METHODS
Sampling
The specimens were collected during 2011 from the Canary Islands, Madeira, Azores and the coast of Portugal. To complete the sampling, specimens deposited at MACB and H were included. In all, 40 specimens of C. azorica, C. macaronesica, C. mediterranea and C. portentosa were selected (Table 1). We included C. portentosa, which is a common species in the Iberian Peninsula and Macaronesia, because of its morphological resemblance to the other three species (Ahti 1961, Ruoss 1989, Orange 1993, Burgaz & Ahti 2009, Sicilia et al. 2009, Ahti & Stenroos 2013). Two specimens of C. pycnoclada and two of C. confusa were also included, both South American members of the section Impexae; they have been considered by some authors to be close to C. azorica (des Abbayes 1946, Tavares 1952). Cladonia deformis, C. boryi and C. cenotea were chosen as outgroup species, according to the results of Stenroos et al. (2002).
Table 1.
Samples of Cladonia mediterranea complex used in this study with the GenBank accession numbers. The new sequences are in bold.
| Taxa | Voucher specimen | Chemistry1 | Code | GenBank numbers |
||
|---|---|---|---|---|---|---|
| IGS rDNA | ITS rDNA | rpb2 | ||||
| C. azorica | Azores Islands, São Miguel, Lago do Foco, Haikonen 26865 (H) | FUM, PERL, USN | 1AZO | KP941478 | KP941520 | – |
| Azores Islands, São Miguel, Serra de Aqua de Pau, Väre L1791 (H) | FUM, PERL, USN | 2AZO | – | KP941535 | – | |
| Madeira, Queimados, Pérez-Vargas (TFC) | FUM, PERL, USN | 1894 | KP941461 | KP941516 | KP941544 | |
| Canary Islands, La Palma, Los Sauces, Pérez-Vargas (TFC) | FUM, PERL, USN | 1866 | – | – | KP941547 | |
| Madeira, Pico Ruivo, Pérez-Vargas (TFC) | FUM, PERL, USN | 1898 | KP941463 | KP941525 | KP941545 | |
| Azores Islands, Terceira, Sierra de Santa Bárbara, Pérez de Paz (TFC) | FUM, PERL, USN | 1856 | KP941474 | KP941511 | – | |
| Azores Islands, Pico, Pérez de Paz (TFC) | FUM, PERL, USN | 1855 | – | – | KP941537 | |
| Madeira, Queimados, Pérez-Vargas (TFC) | FUM, PERL, USN | 1900 | KP941476 | KP941510 | – | |
| Madeira, Folhadal, Pérez-Vargas (TFC) | FUM, PERL | 1897 | KP941475 | KP941509 | KP941548 | |
| C. boryi | USA, Massachusett, Plymouth County, Burgaz (MACB) | USN, ZEO | 1904 | KP941495 | KP941532 | KP941563 |
| C. cenotea | Denmark, Vondrák 6965 (MACB) | SQUA | 1CENO | KP941497 | FN868596 | HM243221 |
| C. confusa | Brazil, Minas Gerais, Stenroos 5091 (TUR) | – | LK46 | KP941492 | AF458296 | KP941559 |
| Bolivia, Santa Cruz, Flakus & Plata 22689 (H) | – | CL271 | KP941491 | KP941536 | KP941560 | |
| C. deformis | USA, New Hampshire, Grafton County, Burgaz (MACB) | RHO, USN | 1905 | KP941496 | KP941533 | KP941570 |
| C. macaronesica | Canary Islands, La Gomera, Laguna Grande, Pérez-Vargas (TFC) | PERL, USN | 1849 | KP941470 | KP941503 | KP941538 |
| Canary Islands, La Gomera, Montaña de la Zarza, Pérez-Vargas (TFC) | PERL, USN | 1848 | KP941462 | KP941502 | – | |
| Canary Islands, Tenerife, Pico del Ingles, Pérez-Vargas (TFC) | PERL, USN | 1847 | KP941464 | KP941501 | KP941546 | |
| Canary Islands, Tenerife, El Pijaral, Pérez-Vargas (TFC) | PERL, USN | 1846 | KP941459 | KP941500 | – | |
| Canary Islands, Tenerife, El Bailadero, Pérez-Vargas (TFC) | PERL, USN | 1845 | KP941458 | KP941499 | KP941556 | |
| Canary Islands, Tenerife, El Bailadero, Pérez-Vargas (TFC) | PERL, USN | 1852 | KP941472 | KP941534 | KP941539 | |
| Canary Islands, La Gomera, Roque de la Zorcita, Hernandez-Padrón & Pérez de Paz (MACB) | PERL, USN | 1863 | KP941460 | KP941512 | – | |
| Canary Islands, La Gomera, Cumbres de Tajaqué, Pérez-Vargas (TFC) | PERL | 1853 | – | KP941505 | KP941543 | |
| Canary Islands, La Gomera, Laguna Grande, Pérez-Vargas (TFC) | PERL, USN | 1854 | KP941473 | – | KP941540 | |
| Canary Islands, Tenerife, El Bailadero, Pérez-Vargas (TFC) | PERL, USN | 1850 | KP941471 | KP941504 | KP941541 | |
| C. mediterranea | Balearic Islands, Ibiza, Sant Joseph de Sa Talaia, Pino-Bodas (MACB) | PERL, USN | 1MED | KP941489 | KP941524 | KP941569 |
| Portugal, Algarve, Maria Vinagre, Burgaz (MACB) | PERL, USN | 1861 | KP941483 | KP941522 | KP941551 | |
| Portugal, Beira Litoral, Figueira da Foz, Burgaz (MACB) | PERL, USN | 1862 | KP941484 | – | KP941552 | |
| Portugal, Beira Litoral, Vagos, Pino-Bodas (MACB) | PERL, USN | 1871 | – | KP941523 | KP941553 | |
| Portugal, Beira Litoral, Areao, Pino-Bodas (MACB) | PERL, USN | 1876 | KP941485 | KP941521 | – | |
| Portugal, Beira Litoral, Mira, Pino-Bodas (MACB) | PERL, USN | 1880 | KP941486 | KP941513 | KP941554 | |
| Portugal, Beira Litoral, Mira, Pino-Bodas (MACB) | PERL, USN | 1883 | KP941487 | KP941514 | KP941550 | |
| Canary Islands, Gran Canaria, El Palmital, Pérez-Vargas (TFC) | PERL, USN | 1895 | KP941488 | KP941517 | KP941555 | |
| Canary Islands, Gran Canaria, El Palmital, Pérez-Vargas (TFC) | PERL, USN | 1896 | KP941490 | KP941518 | KP941568 | |
| C. portentosa subsp. pacifica | USA, Alaska, Aleutian Islands, Talbot & Myers UNI 064-24 (H) | PERL, USN | CL308 | KP941479 | KP941528 | KP941557 |
| USA, Alaska, Aleutian Islands, Talbot WOS 025-24 (H) | PERL, USN | CL340 | KP941480 | KP941529 | KP941558 | |
| C. portentosa subsp. portentosa | Madeira, Folhadal, Pérez-Vargas (TFC) | PERL, USN | 1902 | KP941477 | KP941519 | KP941549 |
| Madeira, Queimados, Pérez-Vargas (TFC) | PERL | 1893 | – | KP941515 | KP941542 | |
| Portugal, Beira Litoral, Areao, Pino-Bodas (MACB) | PERL, USN | 1875 | KP941466 | KP941506 | – | |
| Portugal, Beira Litoral, Areao, Pino-Bodas (MACB) | PERL | 1878 | – | KP941507 | KP941564 | |
| Portugal, Beira Litoral, Canicrira, Pino-Bodas (MACB) | PERL, USN | 1884 | KP941469 | KP941508 | – | |
| United Kingdom, Scotland, Stenroos 6074 (H) | PERL, USN | CL92 | KP941482 | KP941530 | KP941565 | |
| United Kingdom, Scotland, Stenroos 6094 (H) | PERL, USN | CL97 | KP941481 | KP941531 | KP941567 | |
| United Kingdom, Scotland, Sanderson (MACB) | PERL, USN | 1/13 | KP941465 | KP941498 | KP941566 | |
| Spain, Valencia, Utiel, Burgaz (MACB) | PERL, USN | 8/13 | KP941467 | KP941526 | – | |
| Spain, Burgos, Pineda de la Sierra, Burgaz (MACB) | PERL, USN | 9/13 | KP941468 | KP941527 | – | |
| C. pycnoclada | Chile, Osorno, Feuerer 60257 (TUR) | – | AT509 | KP941494 | AF458297 | KP941561 |
| Chile, Osorno, Feuerer 60275 (TUR) | – | AT510 | KP941493 | AF458298 | KP941562 | |
1 FUM = fumarprotocetraric acid; PERL = perlatolic acid; RHO = rhodocladonic acid; SQUA = squamatic acid; USN = usnic acid; ZEO = zeorin.
Morphology and secondary metabolites
The morphological characters studied were selected on the basis of the original descriptions of the species (des Abbayes & Duvigneaud 1947, Ahti 1961, 1978) and other studies (Ruoss 1989, Burgaz & Ahti 2009). Fourteen quantitative morphological characters were measured (length of podetia, width of podetia, number of branches, dichotomous branches percentage, trichotomous branches percentage, tetrachotomous branches percentage, branching angles, length of internodes, length of last branch, thickness of podetia, thickness of medulla, thickness of stereome, number of open axils, number of closed axils). For each specimen, the measures were taken in one to three podetia, according to the available material. All the macroscopic characters were measured by means of a digital slide gauge (0.01 mm precision) under a binocular stereomicroscope (Olympus SZX9). Transverse sections of the podetia were made free-hand, and the microscopical measures were taken at 400× magnification using an Olympus CX41 microscope, in distilled water. The matrix containing the fourteen characters previously mentioned was analyzed using principal component analysis (PCA). This analysis was performed with the Canoco 4.5 program (ter Braak & Šmilauer 2002). The variables were centered and standardized before the PCA. The values for the first two components were plotted (Fig. 1). In Fig. 1a the data were coded according to the morphological identification. Using the same matrix the discriminatory descriptors were inferred from the lenght of the vector and its correlation with the respective axes, so Fig. 1b represents the correlation of the different morphological variables with the components.
Fig. 1.
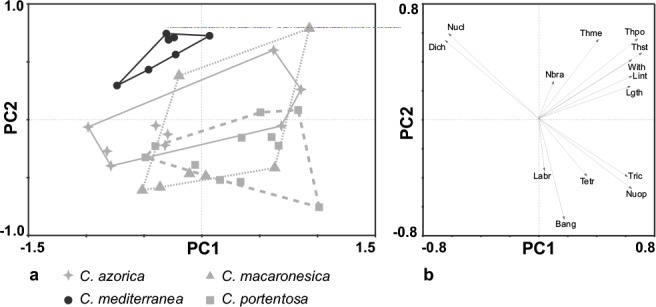
Results of PCA analysis. a. PCA scatterplot with all the standardized variables and species studied; b. vector plot on PC1 and PC2. The length of vectors show the importance of each character. Bang: branching angles; Dich: dichotomous branches percentage; Labr: length of last branch; Lgth: length of podetia; Lint: length of internodes; Nbra: number of branches; Nucl: number of closed axils; Nuop: number of open axils; Tetr: tetrachotomous branches percentage; Thme: thickness of medulla; Thpo: thickness of podetia; Thst: thickness of stereome; Tric: trichotomous branches percentage; With: width of podetia.
The secondary metabolites were studied in all the specimens using the solvents A (Toluene: dioxane: acetic acid) and B (Hexane: diethylether: formic acid) (White & James 1985).
DNA extraction, PCR and sequencing
Genomic DNA was extracted using DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer’s instructions. The DNA was eluted in the final step in 200 ml of elution buffer provided by the manufacturer. The following three nuclear loci were sequenced: ITS rDNA, rpb2 and IGS rDNA. The primers and PCR programs are described in Pino-Bodas et al. (2013). The amplifications were carried out with Ready-to-Go-PCR Beads (GE Healthcare Life Sciences, UK). PCR products were purified with ExoSAP-IT (USB Corporation, OH, USA). The sequencing was performed at Macrogen Europe service (www.macrogen.com), with the same primers used for the PCR.
Phylogenetic analyses
The consensus sequences from forward and reverse templates were assembled and edited in SequencherTM 4.1.4 program (Gene Codes Corporation, Inc, Ann Arbor, Michigan, USA). The sequences of each locus were manually aligned in BIO-EDIT 7.0 (Hall 1999). No ambiguous positions were found and all the positions of the alignments were included in the analyses. Each region was analyzed separately by Maximum Likelihood (ML) using RAxML 7.0.3 (Stamatakis et al. 2005), under the model GTRGAMMA. Fast bootstrap was run with 500 pseudoreplicates. The congruence among the different topologies inferred from the loci was tested following Hillis & Bull (1993): each clade with more than 75 % bootstrap support was scanned for conflict among loci. We considered the existence of a conflict whenever a clade was supported with a bootstrap (more than 75 %) in one locus, while it was not supported in another locus, and the individual sequences of this clade were part of another clade with bootstrap support 75 %. In the combined datasets, only the specimens with sequences at least for 2 genes were included. The combined dataset was analyzed by Maximum Parsimony (MP), ML and Bayesian analyses. MP analyses were performed in PAUP* v. 4.0.b.10 (Swofford 2003) using the heuristic searches with 1 000 random taxon-addition replicates, with TBR branch swapping and MulTrees option in effect, equally weighted characters. Gaps were treated as missing data. For the confidence analysis, the bootstrap (Felsentein 1985) was applied, with 1 000 replicates and heuristic searchs.
The ML analysis was performed in the same conditions as the single gene datasets but considering 5 partitions: ITS rDNA, IGS rDNA and each codon position of rpb2. The best fit substitution model for each region was calculated using MrModeltest 2.3 (Nylander 2004), with Akaike information criterion. The models selected and used in the Bayesian analysis were: GTR+G for IGS rDNA, SYM+G for ITS rDNA and SYM+I for rpb2. The Bayesian analysis was carried out using MrBayes 3.2 (Ronquist et al. 2012). The posterior probabilities were approximated by sampling trees using Markov Chain Monte Carlo (MCMC). The posterior probabilities of each branch were calculated by counting the frequency of trees visited during the MCMC analysis. Two simultaneous runs with 10 000 000 generations, each starting with a random tree and employing 4 simultaneous chains, were executed. Every 1 000th tree was saved into a file. The convergence was assessed checking that the average standard deviation of split frequencies was < 0.01. In addition, the compare and slide commands in AWTY (Nylander et al. 2008) were used. Afterwards, the 50 % majority-rule consensus tree was calculated after removing the first 2 500 000 generations (i.e. the first 2 500 trees) using the ‘burn in’ command.
Tests of monophyly and genealogical sorting index
In order to assess the hypotheses that C. azorica, C. macaronesica and C. portentosa were monophyletic, constraint trees were constructed. These alternatives topologies were supplied to RAxML to search the ‘best’ ML tree. The GTRGAMMA model was used. Shimodaira-Hasegawa test (SH; Shimodaira & Hasegawa 1999) and expected likelihood weight test (ELW; Strimmer & Rambaut 2002) were performed using the program TREE-PUZZLE 5.2 (Schmidt et al. 2002) with 1 000 replicates resampled using the RELL method.
The genealogical sorting index (GSI) was used to assess the level of genealogical exclusivity (Cummings et al. 2008) for C. azorica and C. macaronesica. The GSI value was not calculated for C. portentosa because we have more specimens of this species than of C. azorica and C. macaronesica, and the GSI has bias when unequal sampling size among the groups. The GSI was calculated for the ML tree of each locus and GSIT was calculated for the set of ML trees of the three loci. The significance was calculated using 10 000 permutations on the online platform at http://www.genealogicalsorting.org/. The GSI supports monophyly when this value is > 0.90 and the P-value < 0.001 according with Cummings et al. (2008) and Gazis et al. (2011).
Species tree
Two methods were used to calculate the species tree. First, the species tree under multispecies coalescent model was estimated using *BEAST implemented in BEAST (Drummond & Rambaut 2007), including only the specimens with sequence for the three loci. The specimens were assigned to species based on their morphology (C. azorica, C. macaronesica, C. mediterranea and C. portentosa). The model GTR+G was assigned to ITS and IGS partitions, and GTR+I to rpb2 partition, selecting birth-death speciation process with an uncorrelated relaxed lognormal clock (Drummond et al. 2006) and a constant size population. For the remaining priors the defect values were kept. The analysis was run for 50 000 000 generations, sampling every 1 000. The convergence was calculated with TRACER 1.5 (Rambaut & Drummond 2007). After discarding the first 10 000 000 generations the effective sample size for all the parameters of the evolutionary model reached values > 200. The tree was summarized with TREEANNOTATOR 1.7.5 (Rambaut & Drummond 2013) using maximum clade credibility tree option for the target tree type.
In the second method SpedeSTEM (Ence & Carstens 2011) was used to calculate the species tree. This method is based on coalescence that applies several loci gene trees to calculate the maximum likelihood species tree (Kubatko et al. 2009). This program allows not only to validate the species generated by other procedures, but also to delimit species with no a priori assignment of individuals. In this study only discovering analyses were made according to Satler et al. (2013). The ML gene trees were generated in PAUP (using the models estimated in MrModeltest), including C. mediterranea, C. macaronesica, C. portentosa, C. azorica and the outgroup. SpedeSTEM requires the specimens to be present in all the gene trees, and so only those can be studied for which it was possible to generate sequences for the three loci. SpedeSTEM needs a θ value for scaling the branch lengths in the species trees it produces. The θ value for each locus was calculated in DnaSP v. 5 (Librado & Rozas 2009), being θ = 0.04437 for IGS rDNA, θ = 0.04073 for ITS and θ = 0.02804 for rpb2. The analysis was repeated for several θ values: the average value of the three loci (θ = 0.03771), θ = 0.02, θ = 0.03 and θ = 0.04, to avoid the issues the program can have when calculating the likelihood for low θ values (Giarla et al. 2014).
Species delimitation by cohesion species recognition
Haplotypes networks under statistical parsimony with a confidential interval of 95 % were generated with TCS 1.21 (Clement et al. 2000) for each locus (ITS rDNA, IGS rDNA and rpb2). For the ITS rDNA analysis all the sequences of C. portentosa from GenBank (FR799166, FR799167, JQ695921, JQ695922, JQ695323) were included. Gaps were coded as missing data. The haplotypes were gathered manually in clades according to the rules of Templeton et al. (1987). This algorithm identifies clades united by mutational steps. The x-step clades are successively grouped in x+1-step clades and the final level of nested clades includes the complete network. The loops were resolved following the three criteria (frequency, topology and geographical) proposed by Pfenninger & Posada (2002). Table of contingence and Kruskal-Wallis analyses were done to test the null hypothesis (H2) of no significant association between haplotype variation and phenotypical variation. The quantitative variables with more contribution to separate the groups in the PCA analysis were analyzed by Kruskal-Wallis. The statistical analyses were performed in STATGRAPHICS 5.1 program. The clades 2-4 of ITS rDNA and 2-4 of rpb2 could not be included in the statistical analyses because they contained only one specimen. The pairwise fixation index FST (Weir & Cockerham 1984) was calculated with the program DnaSP. This value was used to assess whether gene flow exists among the cohesive species delimited.
RESULTS
Morphological analysis and secondary metabolites
Fig. 1 shows the results of PCA. The first two principal components PC1 and PC2 summarize 51.44 % of the total variance (29.93 % and 21.51 %, respectively). The analysis distinguished two groups (Fig. 1a). The first group contains all the specimens of Cladonia mediterranea (on the upper left area of the scatterplot) and the other group is formed by the rest of the species analyzed, C. azorica, C. macaronesica and C. portentosa (on the center of the scatterplot). The analysis shows a continuous morphological variation in the second group with a high degree of overlapping between the three species. The characters that most contribute to separate C. mediterranea from the other group were the dichotomous branching percentage and the number of closed axils (Fig. 1b).
The secondary metabolites found in each specimen are listed in Table 1. All the specimens of C. mediterranea and C. macaronesica contained perlatolic and usnic acids. One specimen of C. azorica lacked usnic acid, containing only fumarprotocetraric and perlatolic acids. The other specimens contained fumarprotocetraric, perlatolic and usnic acids. Three specimens of C. portentosa lacked usnic acid (C. portentosa subsp. portentosa f. subimpexae). The other specimens contained perlatolic and usnic acids.
Datasets and phylogenetic analyses
In this study 113 new sequences (39 from ITS rDNA, 40 from IGS rDNA and 34 from rpb2) have been generated, the GenBank accession numbers are listed in Table 1. The concatenated dataset contained 1 781 characters, 136 of which were parsimony-informative. The locus which contained more parsimony-informative positions was ITS rDNA with 50 positions, followed by IGS rDNA with 47 positions and rpb2 with 39 positions.
The separated analyses of ITS rDNA and IGS rDNA yielded trees with the same topology (not shown). Three well supported clades appeared. One clade contained all the samples of C. mediterranea, the second clade contained the samples of C. confusa and C. pycnoclada and the third included all the samples of C. azorica, C. macaronesica and C. portentosa. The ML analysis of rpb2 yielded a tree with 4 clades, one with all the samples of C. mediterranea, two clades containing samples of C. azorica, C. macaronesica and C. portentosa, and the fourth with the samples of C. confusa and C. pycnoclada. Only the C. confusa and C. pycnoclada clade was supported. No conflict among the loci was found and the datasets were combined. The MP analysis based on the concatenated dataset yielded trees of 357 steps long, CI = 0.8711 and RC = 0.9170. The ML analysis yielded a tree with a likelihood value of −LnL = 4431.65, while the mean likelihood of the Bayesian tree sampling was −LnL = 4620.16. The tree topology was the same in all the analyses, so only the 50 % consensus majority tree from Bayesian analysis is shown (Fig. 2). Three clades appeared in all the analyses. One of them contained all the specimens of C. mediterranea, the second the specimens of C. pycnoclada and C. confusa, and the third the specimens of C. azorica, C. macaronesica and C. portentosa. The C. mediterranea clade was basal to the group. This clade was well supported in MP and ML analyses but not in the Bayesian analysis (0.74 posterior probability). The clade formed by C. pycnoclada and C. confusa was supported in all the analyses, and the third clade had high support in all the analyses. In the ML and Bayesian analyses two unsupported subclades can be distinguished in the third clade. One of them was constituted by some samples of C. macaronesica and C. azorica and the other was formed by all the specimens of C. portentosa, some specimens of C. macaronesica and some specimens of C. azorica.
Fig. 2.
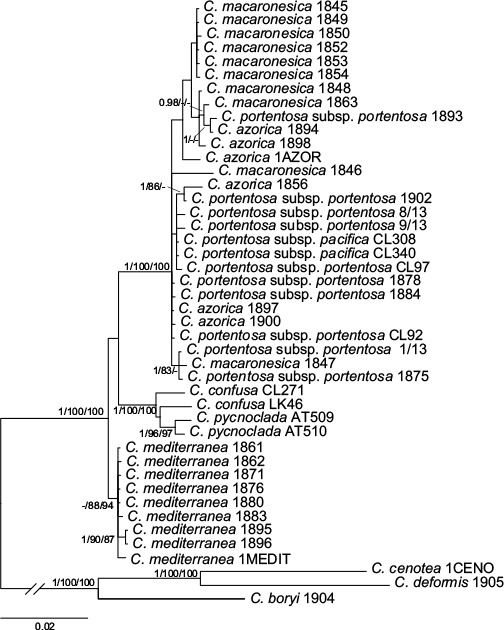
Phylogeny of Cladonia mediterranea complex. The 50 % consensus majority tree of the Bayesian analysis based on concatenated dataset. The values on the branches are the posterior probability from Bayesian analysis (≥ 0.95), bootstrap values from ML analysis (≥ 75 %) and bootstrap values from MP analysis (≥ 75 %).
Species trees
The results of the *BEAST analysis are shown in Fig. 3a. Cladonia mediterranea was well supported, C. azorica and C. portentosa form a clade but the node was not significantly supported with posterior probability. The clade clustering C. azorica, C. macaronesica and C. portentosa was significantly supported. The results from SpedeSTEM analyses were similar for different θ values. In all the cases the model that obtained most support was the one that considers 2 lineages in the ingroup (Fig. 3b). One of them was composed only by C. mediterranea and the other by C. macaronesica, C. portentosa and C. azorica. The probability of this model was wi = 0.99 for θ = 0.03771, wi = 1.0 for θ = 0.02, wi = 0.99 for θ = 0.03 and wi = 0.99 for θ = 0.04. The probability for alternative models was wi = 0.0.
Fig. 3.

a. Species tree inferred in *BEAST based on the concatenated dataset. The values on the branches correspond to the posterior probability; b. species tree estimated in spedeSTEM based on discovery approach.
Hypotheses and GSI
The SH and ELW significantly rejected the three hypotheses:
the monophyly of C. azorica (SH, P-values = 0.0090 and ELW P-value = 0.0009);
the monophyly of C. macaronesica (SH, P-values = 0.0290 and ELW P-value = 0.0423);
the monophyly of C. portentosa (SH, P-value = 0.0270 and ELW P-value = 0.0142).
The GSI test results are shown in the Table 2. The GSI values for C. azorica were similar among the different loci and the P-values rejected the monophyly in all the loci. The GSI values for C. macaronesica were low in ITS rDNA and rpb2, and not significant. However, the GSI value of IGS rDNA was 0.5806 and significant. The GSIT rejected the exclusive ancestry for both species.
Table 2.
Genealogical sorting index and probability values under the null hypothesis that the samples labeled as putative species are monophyletic.
| Species | ITS rDNA |
IGS rDNA |
rpb2 |
GSIT |
||||
|---|---|---|---|---|---|---|---|---|
| GSI | P-value | GSI | P-value | GSI | P-value | GSIt | P-value | |
| C. azorica | 0.1429 | 0.1798 | 0.2114 | 0.0659 | 0.2614 | 0.0463 | 0.1429 | 0.1782 |
| C. macaronesica | 0.1556 | 0.1376 | 0.5806 | 1e-04* | 0.1795 | 0.3308 | 0.1556 | 0.1343 |
* denotes significant result.
Networks and nested clade analyses
A total of fifteen haplotypes of ITS rDNA were identified, connected in a single network (Fig 4a). Two haplotypes were unique for C. mediterranea, two were unique for C. macaronesica, two were unique for C. azorica and five were unique for C. portentosa. The other four haplotypes were shared by samples of different species (C. macaronesica, C. portentosa and C. azorica). The IGS rDNA network analysis yielded a total of six haplotypes connected into a single network (Fig. 4b). All the samples of C. mediterranea were represented in one unique haplotype, one haplotype was unique for C. azorica, and one was unique for C. portentosa. The other three haplotypes were shared by samples of C. macaronesica and C. azorica; C. macaronesica and C. portentosa; or C. macaronesica, C. portentosa and C. azorica. The rpb2 network analysis yielded nine haplotypes connected into a single network (Fig. 4c), four of them were unique for C. mediterranea, one for C. macaronesica and one was unique for C. azorica. The other three haplotypes were shared by samples of different species.
Fig. 4.
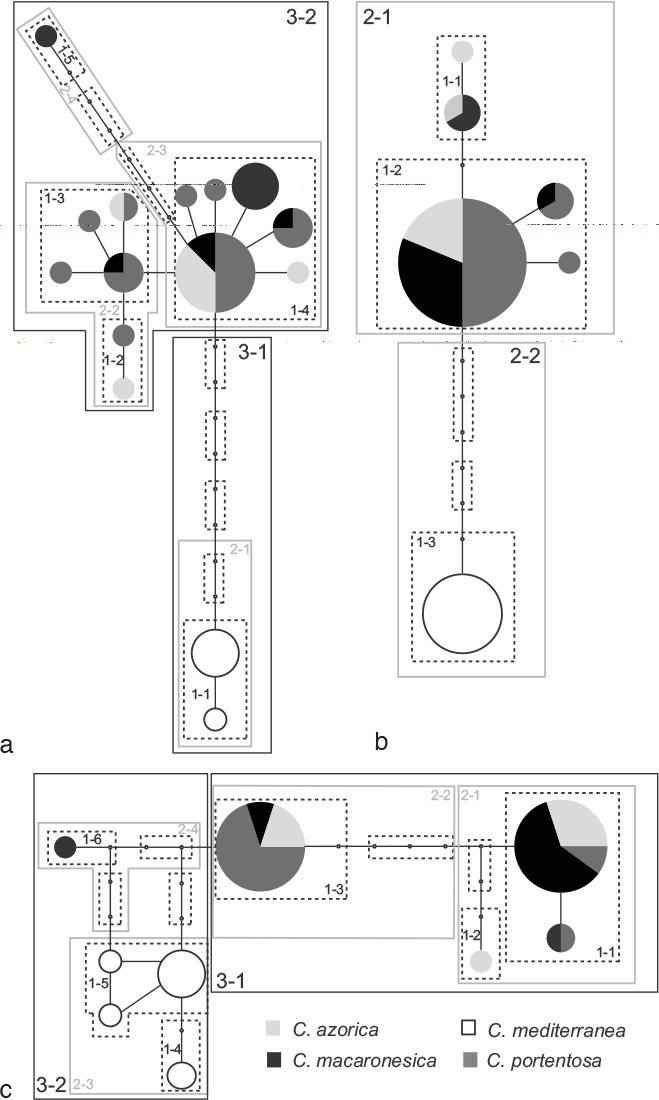
Haplotype networks at 95 % of probability of C. mediterranea complex based on a. ITS rDNA; b. IGS rDNA; c. rpb2. The circles represent the haplotypes and the size is proportional to haplotype frequency. The small circles represent missing haplotypes. The discontinuous lines outline the 1-step nested clades, the grey lines outline the 2-step nested clades and the black lines outline the 3-step nested clades.
The nested clade analyses generated five 1-step clades, four 2-step clades and two 3-step clades for ITS rDNA; for IGS rDNA, three 1-step clades and two 2-step clades were generated; and for rpb2 six 1-step clades, four 2-step clades and two 3-step clades were generated. All the specimens of 2-2 clade from IGS rDNA and 3-1 clade from ITS were identified as C. mediterranea, while the 3-2 clade of rpb2 contained all the specimens of C. mediterranea and one of C. macaronesica. The specimens of C. macaronesica, C. portentosa and C. azorica appeared together in the 3-2 clade of ITS rDNA, 2-1 clade of IGS rDNA and 3-1 clade of rpb2. The specimens grouped together in the 2-1 clade of IGS rDNA, 3-2 clade of ITS rDNA and 3-1 clade of rpb2 are from Macaronesia, North America and Europe while the specimens of the clades 2-2 of IGS rDNA and 3-2 of rpb2 are from the Canary Islands and the Iberian Peninsula.
Table 3 and 4 summarize the results of the contingency table analyses. In the analyses of the 3 networks (ITS rDNA, IGS rDNA and rpb2) significant differences in characters were observed (Table 3). These characters include medulla (loose/compact), branching pattern (isotomic/anisotomic/subisotomic) and algal layer (continuous/discontinuous). These differences were observed between the 3-step clades of ITS rDNA and rpb2 and 2-step clades in IGS rDNA. No significant differences in the presence/absence of fumarprotocetraric acid among these clades were found. The results of the contingency table analyses at 2-step level are presented in Table 4. In ITS rDNA, most of the significant differences were detected among the 2-1 clade and the other clades; in rpb2 significant differences were detected among the 2-3 clade and the other clades; and in IGS rDNA significant differences were detected among the 1-3 clade and the other clades.
Table 3.
Contingency test results for association of haplotype clades and phenotypical characters at highest step clade level for each locus.
| Comparation level | Character | P-value |
|---|---|---|
| ITS rDNA | ||
| 3-step | Presence / lack of fumarprotocetraric acid | 0.193 |
| 3-step | Isotomic / anisotomic or subisotomic pattern | 0.001* |
| 3-step | Compact / loose medulla | 0.000* |
| 3-step | Continuous / discontinuous algal layer | 0.000* |
| IGS rDNA | ||
| 2-step | Presence / lack of fumarprotocetraric acid | 0.211 |
| 2-step | Isotomic / anisotomic or subisotomic pattern | 0.007* |
| 2-step | Compact / loose medulla | 0.000* |
| 2-step | Continuous / discontinuous algal layer | 0.000* |
| rpb2 | ||
| 3-step | Presence / lack of fumarprotocetraric acid | 0.111 |
| 3-step | Isotomic / anisotomic or subisotomic pattern | 0.004* |
| 3-step | Compact / loose medulla | 0.000* |
| 3-step | Continuous / discontinuous algal layer | 0.000* |
* denotes significant result.
Table 4.
Contingency test results for association of haplotype clades and phenotypical characters.
| Comparation level | Character | P-value |
|---|---|---|
| ITS rDNA | ||
| 2-1 to 2-2 | Presence / lack of fumarprotocetraric acid | 0.429 |
| 2-1 to 2-3 | Presence / lack of fumarprotocetraric acid | 0.171 |
| 2-2 to 2-3 | Presence / lack of fumarprotocetraric acid | 0.412 |
| 2-1 to 2-2 | Compact / loose medulla | 0.001* |
| 2-1 to 2-3 | Compact / loose medulla | 0.000* |
| 2-2 to 2-3 | Compact / loose medulla | 0.704 |
| 2-1 to 2-2 | Continuous / discontinuous algal layer | 0.007* |
| 2-1 to 2-3 | Continuous / discontinuous algal layer | 0.000* |
| 2-2 to 2-3 | Continuous / discontinuous algal layer | 0.296 |
| 2-1 to 2-2 | Isotomic / anisotomic or subisotomic pattern | 0.001* |
| 2-1 to 2-3 | Isotomic / anisotomic or subisotomic pattern | 0.002* |
| 2-2 to 2-3 | Isotomic / anisotomic or subisotomic pattern | 0.406 |
| IGS rDNA | ||
| 1-1 to 1-2 | Presence / lack of fumarprotocetraric acid | 0.179 |
| 1-1 to 1-3 | Presence / lack of fumarprotocetraric acid | 0.091 |
| 1-2 to 1-3 | Presence / lack of fumarprotocetraric acid | 0.348 |
| 1-1 to 1-2 | Compact / loose medulla | 0.662 |
| 1-1 to 1-3 | Compact / loose medulla | 0.002* |
| 1-2 to 1-3 | Compact / loose medulla | 0.487 |
| 1-1 to 1-2 | Continuous / discontinuous algal layer | 0.450 |
| 1-1 to 1-3 | Continuous / discontinuous algal layer | 0.018* |
| 1-2 to 1-3 | Continuous / discontinuous algal layer | 0.000* |
| 1-1 to 1-2 | Isotomic / anisotomic or subisotomic pattern | 0.134 |
| 1-1 to 1-3 | Isotomic / anisotomic or subisotomic pattern | 0.160 |
| 1-2 to 1-3 | Isotomic / anisotomic or subisotomic pattern | 0.001* |
| rpb2 | ||
| 2-1 to 2-2 | Presence / lack of fumarprotocetraric acid | 0.166 |
| 2-1 to 2-3 | Presence / lack of fumarprotocetraric acid | 0.054 |
| 2-2 to 2-3 | Presence / lack of fumarprotocetraric acid | 1.000 |
| 2-1 to 2-2 | Compact / loose medulla | 0.045* |
| 2-1 to 2-3 | Compact / loose medulla | 0.000* |
| 2-2 to 2-3 | Compact / loose medulla | 0.022* |
| 2-1 to 2-2 | Continuous / discontinuous algal layer | 0.191 |
| 2-1 to 2-3 | Continuous / discontinuous algal layer | 0.000* |
| 2-2 to 2-3 | Continuous / discontinuous algal layer | 0.006* |
| 2-1 to 2-2 | Isotomic / anisotomic or subisotomic pattern | 0.208 |
| 2-1 to 2-3 | Isotomic / anisotomic or subisotomic pattern | 0.015* |
| 2-2 to 2-3 | Isotomic / anisotomic or subisotomic pattern | 0.003* |
* denotes significant result.
Table 5 shows the Kruskal-Wallis results. Significant differences were obtained for all of the characters among the 3-step clades in ITS rDNA and rpb2 and the 2-step clades in IGS rDNA. However, there were not significant differences among all the 2-step clades (see Tukey test, Table 6). No significant differences were found among the 2-step clades in rpb2 for dichotomous branching rate and trichotomous branching rate.
Table 5.
Kruskal-Wallis results for association of haplotype clades and phenotypical characters.
| Comparation level | Character | Estatistic | P-value |
|---|---|---|---|
| ITS rDNA | |||
| 3-step | Dichotomous branching rate (%) | 5.53597 | 0.0186* |
| 3-step | Trichotomous branching rate (%) | 3.90411 | 0.0481* |
| 3-step | Closed axils rate (%) | 12.4813 | 0.0004* |
| 2-step | Dichotomous branching rate (%) | 8.26747 | 0.0407* |
| 2-step | Trichotomous branching rate (%) | 9.39649 | 0.0244* |
| 2-step | Closed axils rate (%) | 12.5187 | 0.0058* |
| IGS rDNA | |||
| 2-step | Dichotomous branching rate (%) | 10.6712 | 0.0010* |
| 2-step | Trichotomous branching rate (%) | 7.5518 | 0.0059* |
| 2-step | Closed axils rate (%) | 11.8636 | 0.0057* |
| 1-step | Dichotomous branching rate (%) | 10.9286 | 0.0042* |
| 1-step | Trichotomous branching rate (%) | 7.64197 | 0.0219* |
| 1-step | Closed axils rate (%) | 13.5391 | 0.0011* |
| rpb2 | |||
| 3-step | Dichotomous branching rate (%) | 5.16176 | 0.02308* |
| 3-step | Trichotomous branching rate (%) | 4.64772 | 0.03109* |
| 3-step | Closed axils rate (%) | 11.268 | 0.00078* |
| 2-step | Dichotomous branching rate (%) | 5.63538 | 0.1307 |
| 2-step | Trichotomous branching rate (%) | 6.65459 | 0.0837 |
| 2-step | Closed axils rate (%) | 14.1365 | 0.00272* |
* denotes significant result.
Table 6.
Tukey’s multiple comparison test for significant results of the Kruskal-Wallis analyses.
| Dichotomic | Trichotomic | Closed axil | |
|---|---|---|---|
| ITS rDNA | |||
| 2-1 to 2-2 | * | ns | * |
| 2-1 to 2-3 | ns | ns | * |
| 2-1 to 2-4 | ns | ns | ns |
| 2-2 to 2-3 | ns | ns | ns |
| 2-2 to 2-4 | ns | ns | ns |
| 2-3 to 2-4 | ns | ns | ns |
| IGS rDNA | |||
| 1-1 to 1-2 | ns | ns | ns |
| 1-1 to 1-3 | * | * | * |
| 1-2 to 1-3 | ns | ns | ns |
| rpb2 | |||
| 2-1 to 2-2 | – | – | ns |
| 2-1 to 2-3 | – | – | ns |
| 2-1 to 2-4 | – | – | * |
| 2-2 to 2-3 | – | – | * |
| 2-2 to 2-4 | – | – | ns |
| 2-3 to 2-4 | – | – | ns |
ns = not significant; * = significant with 95 % of probability.
The FST values between the 3-step clades of ITS rDNA and rpb2 and the 2-step clades of IGS rDNA are shown in Table 7. In all the comparations the values were high. The lowest value was between the clades appearing in rpb2.
Table 7.
Pairwise FST for each clade defined in the networks.
| Locus | Comparisons | FST |
|---|---|---|
| ITS rDNA | 3-1 to 3-2 step clade | 0.87659 |
| IGS rDNA | 2-1 to 2-2 step clade | 0.93114 |
| rpb2 | 3-1 to 3-2 step clade | 0.69796 |
DISCUSSION
This work addresses the species delimitation in the C. mediterranea complex using two data sources: phenotypical data (morphological and chemical) and DNA sequences from three nuclear genes. The DNA data were analyzed by different methods often used for species delimitation (gene trees, species trees, haplotype networks) and they were highly congruent. When the analyses performed with different type of data show congruent results (as in this case), the concordant inferred boundaries likely correspond to existing biological entities. According to our results, the most probable scenario is the one that comprises two species.
The analyses of the morphological data reveal that C. mediterranea is different from the remaining species. The genealogical phylogenetic species recognition (GPSR) was congruent with the results of the analysis of the morphological data (Fig. 2). Cladonia mediterranea formed a monophyletic clade well supported in MP and ML analyses, but not in the Bayesian analysis. The hypotheses tests (SH and EWL) significantly rejected the alternative topologies, in which C. azorica, C. macaronesica and C. portentosa were divided into independent monophyletic groups. Since the incomplete lineage sorting could be responsible for the lack of monophyly of C. azorica, C. macaronesica and C. portentosa, we applied the GSI test to evaluate the degree of genealogic divergence. The monophyly of C. azorica and C. macaronesica was not supported by this test. The species trees generated by means of *BEAST and SpedeSTEM gave rise to two well-supported species (Fig. 3). These analyses are congruent with the gene trees and the morphological analysis, leading to consider C. azorica, C. macaronesica and C. portentosa as a unique species, and C. mediterranea as a different one. The cohesion species recognition requires, in addition to rejecting the two null hypothesis, that the groups delimited during the evaluation of H1 be congruent with H2 hypothesis (Templeton et al. 2000). This congruence happens at 3-step clade level in ITS rDNA and rpb2 and at 2-step clade level in IGS, since at an inferior level (2-step clade level in ITS and rpb2 and 1-step clade level in IGS) significant results were obtained, but not among all the clades. The morphological differences occur between the clades that contain samples of C. mediterranea and the remaining clades, while there are no significant differences between the clades that contain the samples of C. azorica, C. macaronesica and C. portentosa (2-2, 2-3 and 2-4 in ITS rDNA, 2-1, 2-2, 2-4 in rpb2 and 1-1 and 1-2 in IGS rDNA). Strong evidence for the fact that C. mediterranea is a different species from C. macaronesica, is that all the samples of C. mediterranea are confined to a unique clade in all the haplotype networks. In addition, C. mediterranea shows high levels of genetic differentiations, according with the FST values.
The analyses of the morphological data and also numerous analyses based on the DNA sequences are consistent, indicating that C. mediterranea is an independent evolutionary lineage and C. azorica, C. macaronesica and C. portentosa are conspecific. Thus our results reject the taxonomical proposal that C. mediterranea and C. macaronesica are conspecific (Ruoss 1989). This author studied the branching pattern and the characteristics of the algal layer and concluded that C. macaronesica and C. mediterranea were the same species. The diagnostic characters used to distinguish these species were the following: length of the internodes are longer in C. mediterranea than in C. macaronesica; the algal layer is continuous in C. mediterranea and discontinuous in C. macaronesica; a compact medulla present in C. mediterranea and a lax medulla in C. macaronesica; the axils are frequently closed in C. mediterranea and generally open in C. macaronesica (Ahti 1961). The PCA analyses carried out in this work show that the most relevant variables to distinguish C. mediterranea from the remaining species are the percentage of dichotomous branching and the number of closed axils. According to Ruoss (1989) C. mediterranea had more closed axils than C. macaronesica. However, we think that the internodal length does not contribute to the separation of C. mediterranea from the remaining samples, since C. portentosa has internodes of similar length or even with greater variation (Burgaz & Ahti 2009). Burgaz & Martínez (2008) found that the podetial wall is thicker in C. mediterranea than in C. portentosa; however, in our analysis this character had a scant contribution to distinguish C. mediterranea from the other species. The morphological characters that distinguish C. mediterranea are: the presence of a continuous algal layer, the presence of a compact medulla, the prevalence of isotomy, with dichotomous branching and closed axils. These characters are the originally used ones to describe the species (des Abbayes & Duvigneaud 1947).
The boundaries among C. azorica, C. macaronesica and C. portentosa were not supported by any of the analyses carried out in this work. Cladonia azorica was distinguised from C. macaronesica mainly by the presence of fumarprotocetraric acid and by having a greater number of trichotomous branching, although the dichotomous pattern is also prevailing in this species (Ahti 1961). But our analyses did not show a correlation between the presence of fumarprotocetraric acid and a greater number of trichotomous branching. In previous works based on the study of the morphological variation, the species status of C. azorica and C. macaronesica had already been questioned (Ahti 1977). Although no previous study has suggested that C. portentosa is conspecific with the latter, Orange (1993) pointed out that in Britain it was impossible to distinguish C. azorica from C. portentosa only by means of morphological characters. The morphological similarities of C. macaronesica and C. azorica with C. portentosa are clear, even with C. mediterranea (Fig. 5), and the identification keys and floristic works usually point out the possible confusion of C. portentosa with these other species (James 2009, Sicilia et al. 2009). But C. portentosa is generally distinguished by the prevailing trichotomous branching and an anisotomous pattern, where a main axis is clear. Nonetheless, C. portentosa is a very variable species, either morphologically or chemically (des Abbayes 1939, Ahti 1961, 1978, Burgaz & Martínez 2008). Within this taxon several forms and subspecies have been described. Cladonia portentosa subsp. pacifica, growing in western North America, is more slender and deflexed than C. portentosa subsp. portentosa and shows a greater number of dichotomous branches (Ahti 1978, Brodo & Ahti 1996). Cladonia portentosa subsp. pacifica f. decolorans is a chemotype that lacks usnic acid, turning to a greyish shade (Brodo & Ahti 1996). Cladonia portentosa subsp. portentosa f. subimpexa also lacks usnic acid (Ahti 1978). The ITS rDNA sequences of C. portentosa subsp. pacifica and C. portentosa subsp. portentosa were recently compared and it was found that there was no genetical difference between them (Smith et al. 2012). Our analyses confirm these results. The two specimens of C. portentosa subsp. pacifica here included share a haplotype with some of C. portentosa subsp. portentosa samples in each of the 3 loci.
Fig. 5.

Photographs of the four species studied, showing the general configurations of podetia a. Cladonia azorica (Haikonen 26865, H); b. C. macaronesica (Pérez-Vargas s.n., TFC 10602); c. C. mediterranea (Burgaz s.n., MACB 61559); d. C. portentosa (Stenroos 6074, H).
In other species of the Group Cladinae (Stenroos et al. 2002) similar results have been found. This is the case of C. arbuscula, for which several subspecies were defined on the basis of the morphological and chemical variation. However, much of this variation is not correlated with the genetic variation (Piercey-Normore et al. 2010). The authors attribute the high variation within this species to environmental agents such as lighting, humidity, nutrients and thallus age. The warm temperatures throughout the year in Macaronesia, which causes a continuous development of the podetia, could be the environmental agent that determines C. portentosa to develop a prevailing dichotomous branching, instead of trichotomous. Ahti (1961) had already pointed out that in southern Europe (Portugal) C. portentosa tended to produce dichotomous branching, being easily mistaken for C. mediterranea, with which it often coexists (Burgaz & Ahti 2009).
Our results indicate that C. confusa and C. pycnoclada are related, while in the phylogeny submitted by Stenroos et al. (2002) C. confusa appeared closely related to C. terra-novae and C. portentosa. However, in our analyses C. confusa is not monophyletic, which could reveal a lack of genetic homogeneity of this species. Further studies, based on a wide range of sampling, should be made to confirm this observation.
TAXONOMY
In this section we present formally the taxonomical changes.
Cladonia portentosa (Dufour) Coem., Bull. Acad, Roy. Sci. Belgique, ser. 2, 19: 49. 1865.
Basionym. Cenomyce portentosa Dufour, Ann. Gen. Sci. Phys. 8: 69. 1821.
Type. FRANCE, Landes, Saint-Sever-sur-Adour, 1818, J.-M. Dufour (PC-Herb. Desmazières lectotype, Ahti, Ann. Bot. Fenn. 15: 8. 1978).
= Cladonia azorica Ahti, Ann. Bot. Soc. Zool.-Bot. Fenn. Vanamo 32, 1: 36. 1961.
Type. PORTUGAL, Azores, Pico, 25 Sept. 1961, A.G. da Cunha & L. Sobrinho (LISU holotype).
= Cladonia macaronesica Ahti, Ann. Bot. Soc. Zool.-Bot. Fenn. Vanamo 32, 1: 37. 1961.
Type. PORTUGAL, Madeira, Entre as Queimadas e o Caldeirão Verde, 1951, C.N. Tavares 4583 (LISU holotype).
Acknowledgments
This study was supported by Caja Canarias-Bancacívica project 31195/2011. R. P-B was funded by Marie Curie IEF-program (PIEF-GA-2013-625653). A.R. B. thanks the project CGL2013-41839-P (Ministry of Economy & Competitiveness, Spain).
REFERENCES
- Abbayes H des. 1939. Revision monographique des Cladonia du sous-genre Cladina (Lichen). Bulletin de la Société Scientifique de Bretagne 16 (hors sér. 2): 1–156. [Google Scholar]
- Abbayes H des. 1946. Les Cladonia (Lichens) des Iles Açores. Portugaliae Acta Biologica 1: 243–254. [Google Scholar]
- Abbayes H des, Duvigneaud P. 1947. Un nouveau lichen méditerranéo-atlantique: Cladonia mediterranea Duvign. et des Abb. Revue Bryologique et Lichénologique 16: 95–104. [Google Scholar]
- Ahti T. 1961. Taxonomic studies on reindeer lichens (Cladonia, subgenus Cladina). Annales Botanici Societatis Zoologicae Botanicae Fennicae Vanamo 32: 1–160. [Google Scholar]
- Ahti T. 1977. Cladonia Wigg. subg. Cladina (Nyl) Leight. In: Poelt J, Vezda A. (eds), Bestimmungsschlüssel europäischer Flechten. Ergänzungsheft 1: 45–59. Cramer, Germany. [Google Scholar]
- Ahti T. 1978. Nomenclatural and taxonomic remarks on European species of Cladonia. Annales Botanici Fennici 15: 7–14. [Google Scholar]
- Ahti T. 1984. The status of Cladina as a genus segregated from Cladonia. Nova Hedwigia 79: 25–61. [Google Scholar]
- Ahti T. 2000. Cladoniaceae. Flora Neotropica Monograph 78: 1–363. [Google Scholar]
- Ahti T, Stenroos S. 2013. Cladoniaceae. In: Ahti T, Stenroos S, Moberg R. (eds), Nordic Lichen Flora Vol. 5: 1–117. Uppsala, Museum of Evolution, Uppsala University, Sweden. [Google Scholar]
- Bickford D, Lohman DJ, Sodhi NS, et al. 2007. Cryptic species as a window on diversity and conservation. Trends in Ecology & Evolution 22: 148–155. [DOI] [PubMed] [Google Scholar]
- Braak CJF ter, Šmilauer P. 2002. Reference manual and Canodraw for Windows user’s guide: software for canonical community ordination (Version 4.5). Microcomputer Power, Ithaca, New York. [Google Scholar]
- Brodo IM, Ahti T. 1996. Lichens and lichenicolous fungi of the Queen Charlotte Islands, British Columbia, Canada. 2. The Cladoniaceae. Canadian Journal of Botany 74: 1147–1180. [Google Scholar]
- Burgaz AR, Ahti T. 2009. Cladoniaceae. Flora liquenológica Ibérica Vol. 4 Sociedad Española de Liquenología, Spain. [Google Scholar]
- Burgaz AR, Martínez I. 2008. El género Cladonia en la península Ibérica. Supergrupo Crustaceae. Botanica Complutensis 32: 21–36. [Google Scholar]
- Carstens BC, Knowles LL. 2007. Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: An example from Melanoplus grasshoppers. Systematic Biology 56: 400–411. [DOI] [PubMed] [Google Scholar]
- Carstens BC, Pelletier TA, Reid NM, et al. 2013. How to fail at species delimitation. Molecular Ecology 22: 4369–4383. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Figueira R, Jones MP. 2008. List of lichens and lichenicolous fungi (Fungi). In: Borges PAV, Abreu C, Aguiar AMF, et al. (eds), A list of terrestrial fungi, flora and fauna of Madeira and Sevagens archipelagos: 105–122. Direcção Regional do Ambiente da Madeira and Universidade dos Açores, Portugal. [Google Scholar]
- Clement M, Posadas D, Crandall K. 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1660. [DOI] [PubMed] [Google Scholar]
- Crespo A, Lumbsch HT. 2010. Cryptic species in lichen-forming fungi. IMA Fungus 1: 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings MP, Nell MC, Shaw KL. 2008. A genealogical approach to quantifying lineage divergence. Evolution 62: 2411–2422. [DOI] [PubMed] [Google Scholar]
- Dayrat B. 2005. Towards integrative taxonomy. Biological Journal of the Linnean Society 85: 407–415. [Google Scholar]
- DePriest PT, Piercey-Normore M, Sikaroodi M, et al. 1999. Phylogenetic analyses of Cladonia and Cladina (lichen-forming Ascomycota). In: XVI International Botanical Congress, Abstract book: 325 St. Louis, USA. [Google Scholar]
- DePriest PT, Piercey-Normore M, Sikaroodi M, et al. 2000. Phylogenetic relationships among sections of Cladonia and Cladina. In: Progress and problems in lichenology at the turn of the millenium: the fourth IAL symposium, Abstract book: 14. University of Barcelona, Spain. [Google Scholar]
- Dettman JR, Jacobson DJ, Taylor JW. 2003. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 57: 2703–2720. [DOI] [PubMed] [Google Scholar]
- Doyle VP, Oudemans PV, Rehner SA, et al. 2013. Habitat and host indicate lineage identity in Colletotrichum gloeosporioides s.l. from wild and agricultural landscapes in North America. PLoS ONE 8, 5: e62394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ho SY, Phillips MJ, et al. 2006. Relaxed phylogenetics and dating with confidence. PLoS Biology 4: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology 7: e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert AJ, Carstens BC. 2008. Does gene flow destroy phylogenetic signal? The performance of three methods for estimating species phylogenies in the presence of gene flow. Molecular Phylogenetics and Evolution 49: 832–842. [DOI] [PubMed] [Google Scholar]
- Ence DD, Carstens BC. 2011. SpedeSTEM: a rapid and accurate method for species delimitation. Molecular Ecology Resources 11: 473–480. [DOI] [PubMed] [Google Scholar]
- Felsentein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- Flores Rodrígues AF, Aptroot A. 2005. New data and corrections to the list of lichens and lichenicolous fungi from the Azores. In: Borges PAV, Cunha R, Gabriel R, et al. (eds), A list of terrestrial fauna (Mollusca and Arthropoda) and flora (Bryophyta, Pteridophyta and Spermatophyta) from the Azores: 231–247. Direccção Regional do Ambiente, Universidade dos Acçores, Portugal. [Google Scholar]
- Fournier E, Giraud T, Albertini C, et al. 2005. Partition of the Botrytis cinerea complex in France using multiple gene genealogies. Mycologia 97: 1251–1267. [DOI] [PubMed] [Google Scholar]
- Fujita MK, Leaché AD, Burbrink FT, et al. 2012. Coalescent-based species delimitation in an integrative taxonomy. Trends in Ecology & Evolution 27: 480–488. [DOI] [PubMed] [Google Scholar]
- Gabriel R. 2012. Base de dados da Biodiversidade dos Açores. Líquenes. http://www.azoresbioportal.angra.uac.pt/pesquisa.php?sstr=1〈=pt. [Google Scholar]
- Gazis R, Rehner S, Chaverri P. 2011. Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Molecular Ecology 20: 3001–3013. [DOI] [PubMed] [Google Scholar]
- Gebiola M, Gómez-Zurita J, Monti MM, et al. 2012. Integration of molecular, ecological, morphological and endosymbiont data for species delimitation within the Pnigalio soemius complex (Hymenoptera: Eulophidae). Molecular Ecology 21: 1190–1208. [DOI] [PubMed] [Google Scholar]
- Giarla TS, Voss RS, Jansa SA. 2014. Hidden diversity in the Andes: Comparison of species delimitation methods in montane marsupials. Molecular Phylogenetics and Evolution 70: 137–151. [DOI] [PubMed] [Google Scholar]
- Guo S, Kashiwadani H. 2004. Recent study on the phylogeny of the genus Cladonia (s.lat.) with the emphasis on the integrative biology. In: Akiyama S. (ed), Proceedings of the 5th and 6th symposia on collection building and natural history studies in Asia and the Pacific Rim. National Science Museum Monographs 24, Japan: 207–225. [Google Scholar]
- Hafellner J. 1995. A new checklist of lichens and lichenicolous fungi of Insular Laurimacaronesia including a lichenological bibliography for the area. Fritschiana 5: 1–132. [Google Scholar]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98. [Google Scholar]
- Hernández-Padrón CE, Pérez-Vargas I. 2009. Lichenes, lichenicolous fungi. In: Arechavaleta M, Rodríguez S, Zurita N, et al. (eds), Lista de especies silvestres de Canarias. Hongos, plantas y animales terrestres. Consejería de Medio Ambiente y Ordenación Territorial: 71–105. Gobierno de Canarias, Spain. [Google Scholar]
- Hillis DM, Bull JJ. 1993. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analyses. Systematic Biology 42: 182–192. [Google Scholar]
- James PW. 2009. Cladonia. In: Smith CW, Aptroot A, Coppins BJ, et al. (eds), The lichens of Great Britain and Ireland: 309–338. Natural History Museum Publications, UK. [Google Scholar]
- Knowles LL, Carstens BC. 2007. Delimiting species without monophyletic gene trees. Systematic Biology 56: 887–895. [DOI] [PubMed] [Google Scholar]
- Kroken S, Taylor JW. 2001. A gene genealogical approach to recognize phylogenetic species boundaries in the lichenized fungus Letharia. Mycologia 93: 38–53. [Google Scholar]
- Kubatko LS, Carstens BC, Knowles LL. 2009. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics 25: 971–973. [DOI] [PubMed] [Google Scholar]
- Leavitt SD, Esslinger TL, Spribille T, et al. 2013. Multilocus phylogeny of the lichen-forming fungal genus Melanohalea (Parmeliaceae, Ascomycota): Insights on diversity, distributions, and a comparison of species tree and concatenated topologies. Molecular Phylogenetics and Evolution 66: 138–152. [DOI] [PubMed] [Google Scholar]
- Leavitt SD, Fankhauser JD, Leavitt DH, et al. 2011. Complex patterns of speciation in cosmopolitan “rock posy” lichens – discovering and delimiting cryptic fungal species in the lichen-forming Rhizoplaca melanophthalma species-complex (Lecanoraceae, Ascomycota). Molecular Phylogenetics and Evolution 59: 587–602. [DOI] [PubMed] [Google Scholar]
- Leavitt SD, Johnson LA, Goward T, et al. 2012. Species delimitation in taxonomically difficult lichen-forming fungi: an example from morphologically and chemically diverse Xanthoparmelia (Parmeliaceae) in North America. Molecular Phylogenetics and Evolution 60: 317–332. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452 [DOI] [PubMed] [Google Scholar]
- Liu L, Yu L, Kubatko L, et al. 2009. Coalescent methods for estimating phylogenetic trees. Molecular Phylogenetics and Evolution 53: 320–328. [DOI] [PubMed] [Google Scholar]
- Morgado LN, Noordeloos ME, Lamoreux Y, et al. 2013. Multi-gene phylogenetic analyses reveal species limits, phylogeographic patterns, and evolutionary histories of key morphological traits in Entoloma (Agaricales, Basidiomycota). Persoonia 31: 159–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander JAA. 2004. MrModelTest 2.1. Program distributed by the author. Uppsala, Evolutionary Biology Centre, Uppsala University. [Google Scholar]
- Nylander JAA, Wilgenbusch DL, Warren JC, et al. 2008. AWTY (Are we there yet?): A system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24: 581–583. [DOI] [PubMed] [Google Scholar]
- O’Meara BC, Ané C, Sanderson MJ, et al. 2006. Testing for different rates of continuous trait evolution using likelihood. Evolution 60: 922–933. [PubMed] [Google Scholar]
- Orange A. 1993. Cladonia azorica in the British Isles. Lichenologist 25: 105–114. [Google Scholar]
- Ott S, Brinkmann M, Wirtz N, et al. 2004. Mitochondrial and nuclear ribosomal DNA data do not support the separation of the Antarctic lichen Umbilicaria kappenii and Umbilicaria antartica as distinct species. Lichenologist 36: 227–234. [Google Scholar]
- Padial JM, Castroviejo-Fisher S, Köhler J, et al. 2009. Deciphering the products of evolution at the species level: the need for an integrative taxonomy. Zoologica Scripta 38: 431–447. [Google Scholar]
- Padial JM, Riva I de la. 2010. A response to recent proposals for integrative taxonomy. Biological Journal of the Linnean Society 101: 747–756. [Google Scholar]
- Parnmen S, Rangsiruji A, Mongkolsuk P, et al. 2012. Using phylogenetic and coalescent methods to understand the species diversity in the Cladia aggregata complex (Ascomycota, Lecanorales). PLoS ONE 7: e52245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger M, Posada D. 2002. Phylogeographic history of the land snail Candidula unifasciata (Helicellinae, Stylommatophora): fragmentation, corridor migration, and secondary contact. Evolution 56: 1776–1788. [DOI] [PubMed] [Google Scholar]
- Piercey-Normore MD, Ahti T, Goward T. 2010. Phylogenetic and haplotype analyses of four segregates within Cladonia arbuscula sl. Botany 88: 397–408. [Google Scholar]
- Pino-Bodas R, Ahti T, Stenroos S, et al. 2013. Multilocus approach to species recognition in the Cladonia humilis complex (Cladoniaceae, Ascomycota). American Journal of Botany 100: 664–678. [DOI] [PubMed] [Google Scholar]
- Pons J, Barraclough TG, Gomez-Zurita J, et al. 2006. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology 55: 595–609. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond A. 2007. Tracer v1.5. http://beast.bio.ed.ac.uk/Tracer. [Google Scholar]
- Rambaut A, Drummond AJ. 2013. TreeAnnotator v1.7.0. Available from: http://beast.bio.ed.ac.uk. [Google Scholar]
- Ronquist F, Teslenko M, Mark P, et al. 2012. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoss E. 1989. Verzweigung als Unterscheidungsmerkmal bei Rentierflechten (Cladonia subg. Cladina). Herzogia 8: 125–136. [Google Scholar]
- Satler JD, Carstens BC, Hedin M. 2013. Multilocus species delimitation in a complex of morphologically conserved trapdoor spiders (Mygalomorphae, Antrodiaetidae, Aliatypus). Systematic Biology 62: 805–823. [DOI] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, et al. 2002. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18: 502–504. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. 1999. Multiple comparisons of log-likehoods with applications to phylogenetic inference. Molecular Biology and Evolution 16: 1114–1116. [Google Scholar]
- Sicilia D, Hernández-Padrón C, Burgaz AR. 2009. The genus Cladonia in Garajonay National Park, La Gomera, Canary Islands. Cryptogamie, Mycologie 30: 305–316. [Google Scholar]
- Sites JJW, Marshall JC. 2003. Delimiting species: a Renaissance issue in systematic biology. Trends in Ecology & Evolution 18: 462–470. [Google Scholar]
- Smith RJ, Arvidson R, Bono G, et al. 2012. Rare inland reindeer lichens at Mima Mounds in southwest Washington State. North American Fungi 7: 1–25. [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. 2005. RAxML-III: A fast program for maximum likelihood- based inference of large phylogenetic trees. Bioinformatics 21: 456–463. [DOI] [PubMed] [Google Scholar]
- Stenroos S, Hyvönen J, Myllys L, et al. 2002. Phylogeny of the genus Cladonia s.lat. (Cladoniaceae, Ascomycetes) inferred from molecular, morphological, and chemical data. Cladistics 18: 237–278. [DOI] [PubMed] [Google Scholar]
- Strimmer K, Rambaut A. 2002. Inferring confidence sets of possibly misspecified gene trees. Proceedings of the Royal Society B 269: 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. 2003. PAUP*: Phylogenetic analysis using parsimony (*and other methods), version 4.0b10. Sinauer, Sunderland, Massachusetts, USA. [Google Scholar]
- Tavares CN. 1952. Contributions to the lichen flora of Macaronesia I. Lichens from Madeira. Portugaliae Acta Biologica 3: 308–391. [Google Scholar]
- Taylor JW, Jacobson DJ, Kroken S, et al. 2000. Phylogenetic species recognition and species concepts in fungi. Fungal Genetics and Biology 31: 21–32. [DOI] [PubMed] [Google Scholar]
- Templeton AR. 1989. The meaning of species and speciation: A genetic perspective. In: Otte D, Endler JA. (eds), Speciation and its consequences: 3–27. Sunderland, Sinauer Associates, USA. [Google Scholar]
- Templeton AR, Maskas SD, Cruzan MB. 2000. Gene trees: A powerful tool for exploring the evolutionary biology of species and speciation. Plant Species Biology 15: 211–222. [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Weisrock DW, Larson A. 2006. Testing hypotheses of speciation in the Plethodon jordani species complex with allozymes and mitochondrial DNA sequences. Biological Journal of the Linnean Society 89: 25–51. [Google Scholar]
- White FJ, James PW. 1985. A new guide to microchemical techniques for the identification of lichen substances. British Lichen Society Bulletin 75 (supplement): 1–41. [Google Scholar]
- Wiens JJ, Penkrot TA. 2002. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Systematic Biology 51: 69–91. [DOI] [PubMed] [Google Scholar]
- Will KG, Mishler BD, Wheeler QD. 2005. The perils of DNA barcoding and the need for integrative taxonomy. Systematic Biology 54: 844–851. [DOI] [PubMed] [Google Scholar]
- Wirtz N, Printzen C, Lumbsch HT. 2008. The delimitation of Antarctic and bipolar species of neuropogonoid Usnea (Ascomycota, Lecanorales): a cohesion approach of species recognition for the Usnea perpusilla complex. Mycological Research 112: 472–484. [DOI] [PubMed] [Google Scholar]
- Wirtz N, Printzen C, Lumbsch HT. 2012. Using haplotype networks, estimation of gene flow and phenotypic characters to understand species delimitation in fungi of a predominantly Antarctic Usnea group (Ascomycota, Parmeliaceae). Organisms Diversity & Evolution 12: 17–37. [Google Scholar]
- Yang Z, Rannala B. 2010. Bayesian species delimitation using multilocus sequence data. Proceedings of the National Academy of Sciences, USA 107: 9264–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]