

Abstract
Background:
Laquinimod 0.6 mg is a once-daily, oral, disease-modifying therapy in development for the treatment of multiple sclerosis (MS) that was investigated in two double-blind, placebo-controlled, phase 3 trials: ALLEGRO and BRAVO.
Methods:
Data from these studies were pooled to assess the safety profile of laquinimod versus placebo. Adverse events (AEs), laboratory value changes, and potential risks identified in preclinical studies were evaluated in participants in ALLEGRO and BRAVO treated with at least one dose of laquinimod or matching placebo (1:1 random assignment).
Results:
In total, 1988 patients received at least one dose of study drug (laquinimod: n = 983 [mean ± SD duration, 639 ± 190 days]; placebo: n = 1005 [mean ± SD duration, 627 ± 198 days]). Early terminations due to AEs were infrequent (laquinimod: 6.4%; placebo: 4.7%). Death was reported in four patients (laquinimod: n = 1; placebo: n = 3). Rates of serious AEs (including malignancies, infections, and cardiovascular AEs) were similar between groups. The most common AEs identified with laquinimod use were back and neck pain and appendicitis. Laquinimod was also associated with asymptomatic changes in liver enzyme levels, fibrinogen levels, and hematologic parameters that followed a consistent temporal pattern: mild, nonprogressive, and occurring within 90 days of treatment initiation, then stabilizing or reverting to baseline levels during continued treatment.
Conclusions:
Data from these pivotal laquinimod studies demonstrate a safety profile comprising benign or manageable AEs and asymptomatic laboratory findings with a clear temporal pattern. Potential risks noted in preclinical studies were not observed.
Laquinimod is a once-daily, oral, quinoline-3-carboxamide, disease-modifying therapy in development for the treatment of relapsing-remitting multiple sclerosis (RRMS) and progressive MS. It is a small molecule that passively crosses the intact blood-brain barrier, allowing direct modulation of pathologic processes related to MS in the central nervous system.1 Pharmacokinetically, laquinimod has a predictable and linear pharmacokinetic profile characterized by high plasma protein binding (>98%), high oral bio-availability (80%–90%), and a long half-life (~80 hours). The drug is metabolized predominantly by cytochrome P450 3A4 and is a strong inducer of cytochrome P450 1A2.
Laquinimod is a synthetic compound chemically related to roquinimex (linomide). It is the result of a structure-activity relationship screening program aimed at identifying a new, pharmacologically modified substance active in MS animal models that, compared with roquinimex, would have a superior safety profile. Roquinimex demonstrated clinical efficacy in MS in phase 2 studies.2,3 However, serious cardiopulmonary toxicities (including myocardial infarction, pericarditis, and pleuritis) that occurred during the first months of phase 3 trials led to early termination of these trials.2
Preclinical studies with laquinimod showed that laquinimod is not immunosuppressive4–6; phenotypical and functional measures of peripheral blood mono-nuclear cells (PBMCs) were assessed in a prospective longitudinal analysis comparing laquinimod- and placebo-treated cohorts. These data demonstrated that there were no significant changes in the distribution and function of PBMCs in patients receiving 2 years or more of continuous laquinimod therapy. The major populations of circulating PBMCs did not change and retained their capacity to respond to immunologic stimuli.7–9
Laquinimod is neither mutagenic nor clastogenic in in vitro and in vivo assays. Treatment with laquinimod resulted in the formation of micronuclei in vitro and in vivo through an aneugenic mechanism with broad safety margins (99-fold) above the clinical dose of 0.6 mg/day in humans. In a 2-year rat carcinogenicity study, an increase in the incidence of uterine adenocarcinomas and oral cavity tumors was observed in female rats. The mechanisms proposed for these tumors are either species or model specific, and the tumors are, therefore, not considered relevant to humans. Studies in rats have shown reproductive toxicity, including teratogenicity (urogenital malformations) at doses similar to the clinical dose of 0.6 mg/day, and a slight delay in puberty and reduced fertility in rat offspring exposed to laquinimod in utero at higher doses. In monkeys, there were no treatment-related malformations at any tested dose. The relevance to humans of these rat findings is not known but cannot be excluded. Therefore, exposure to laquinimod during pregnancy must be avoided and a stringent contraception approach applied in all laquinimod clinical trials.
A double-blind, placebo-controlled, 36-week, phase 2b trial (LAQ5062) showed that laquinimod 0.6 mg/day had a significant effect on magnetic resonance imaging–monitored disease activity, with no significant effect for the treatment arm using 0.3 mg/day.10 Based on the results, an open-label study with laquinimod 0.6 mg/day is ongoing. Two double-blind, placebo-controlled, 24-month, phase 3 trials were completed with laquinimod 0.6 mg: Assessment of Oral Laquinimod in Preventing Progression in Multiple Sclerosis (ALLEGRO)11 and BRAVO.12 Although the ALLEGRO study met its primary endpoint and the BRAVO study did not, the efficacy profile stemming from the individual studies was similar and attested to a new and important clinical profile. This profile is characterized by a pronounced, robust effect on slowing of disability progression (34% and 46% reduction in 3- and 6-month confirmed disability progression, respectively, in the pooled ALLEGRO and BRAVO analyses13), larger than predicted by the effect on relapse rate (21% in the pooled analysis), together with a noted effect on reduction in brain atrophy (30% in the pooled analysis). Such a profile suggests a distinct mechanism of action on underlying MS pathology that is only minimally related to an anti-inflammatory effect.
Reported herein is the clinical safety profile of laquinimod 0.6 mg/day in patients with RRMS, as demonstrated by pooled safety results from the ALLEGRO and BRAVO clinical studies.
Methods
Clinical Studies and Patient Population
The ALLEGRO and BRAVO trial protocols were approved by local ethics committees/institutional review boards, including committees on human experimentation, as required. The studies are registered at ClinicalTrials.gov (ALLEGRO: NCT00509145; BRAVO: NCT00605215). Patients provided written informed consent before participating in these studies.
The ALLEGRO and BRAVO studies were phase 3, randomized, double-blind, placebo-controlled, parallel-group studies lasting 2 years each that were designed to assess the efficacy and safety of oral, once-daily laquinimod 0.6 mg in patients with RRMS.11,12 The BRAVO study included an active treatment arm, intramuscular interferon beta-1a (IFNβ-1a) (Avonex; Biogen, Cambridge, MA), which is not reported in this analysis. Detailed patient inclusion and exclusion criteria, randomization and blinding procedures, and other study procedures were comparable and are described in detail elsewhere.11,12
The two studies enrolled similar cohorts of patients with RRMS, in whom diagnosis was based on the 2005 revised McDonald criteria,14 with a relapsing-remitting course of at least 6 months' duration before screening, an age of 18 to 55 years, an Expanded Disability Status Scale score of 0 to 5.5, and at least one documented relapse in the 12 months before screening, two or more documented relapses in the 24 months before screening, or one documented relapse 12 to 24 months before screening with at least 1 gadolinium-enhancing lesion in the previous year. Patients were excluded if they had progressive forms of MS or an unstable neurologic condition or other clinically significant medical or surgical condition that would preclude safe and complete study participation.
Safety monitoring guidelines and stopping criteria addressed liver enzyme level elevation, thrombotic events, and systemic inflammation events, as well as laboratory parameters and pregnancy.
Study assessments were performed at screening; baseline; months 1, 2, and 3; and every 3 months thereafter until month 24 and included recording of adverse events (AEs) and concomitant medications, measurement of vital signs, physical examination, electrocardiography, and laboratory tests.
Analysis Methods
Patient characteristics at baseline were similar in the ALLEGRO and BRAVO studies. In both studies, a similar pattern of common AEs and similar ratios of AEs between the laquinimod and placebo arms were observed. Based on these considerations, it was decided that data from the two studies were appropriate for a pooled analysis of all patients who received at least one dose of either laquinimod 0.6 mg or placebo to allow a more comprehensive interpretation of available data.
Tolerability was analyzed based on proportions of patients who discontinued the study 1) for any reason or 2) due to an AE. Common AEs were defined as occurring in at least 5% of patients in either treatment group. Serious AEs (SAEs) that occurred in at least two patients are presented.
In laboratory analyses, changes in liver function test results were assessed as shifts from normal levels at baseline to abnormal levels categorized as greater than 1 to 3× the upper limit of normal (ULN), greater than 3 to 5×ULN, greater than 5 to 8×ULN, and greater than 8×ULN. Changes in hemoglobin levels were evaluated by shifting from the normal Common Terminology Criteria for Adverse Events (CTCAE, v.4.) category at baseline to higher CTCAE categories. Changes in platelet count were assessed as shifts from a normal level at baseline to a low postbaseline level (<100 × 109/L). Changes in C-reactive protein (CRP) levels, fibrinogen levels, and white blood cell (WBC) counts were assessed by shifts to abnormally high levels (CRP, >10 mg/mL; fibrinogen, >4 g/L; WBC, >10 × 109/L). Characterization of the magnitude and temporal pattern of changes in the levels of fibrinogen, WBCs, CRP, hemoglobin, and platelets after laquinimod administration were also assessed by the group means of values and the temporal course.
Potential risks identified in preclinical studies (eg, teratogenicity and carcinogenicity), as well as AEs associated with roquinimex, were investigated.
Results
In total, 1988 of 1990 randomized patients with RRMS received at least one dose of study drug and composed the safety population (laquinimod: n = 983; placebo: n = 1005) (Figure 1), with mean ± SD exposure of 639 ± 190 days (range, 1–764 days) for laquinimod and 627 ± 198 days (range, 1–796 days) for placebo. The treatment arms were balanced with respect to baseline demographic and disease characteristics (Table 1). Consistent with the MS population at large,15 most patients in the ALLEGRO and BRAVO studies were female (69%), 30 to 49 years old (66%), and of white race (97%).
Figure 1.
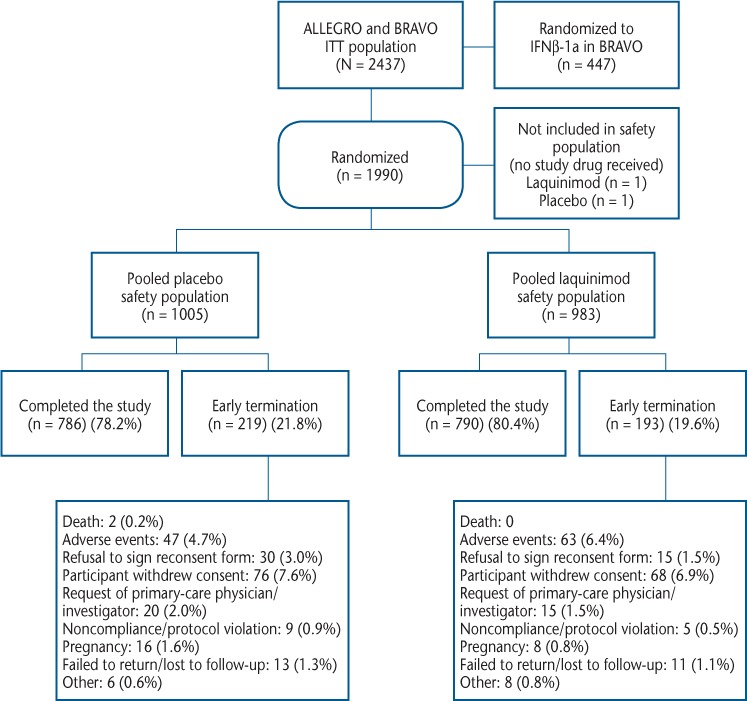
Patient disposition
IFNβ-1a, interferon beta-1a; ITT, intention to treat.
Table 1.
Patient demographic and disease characteristics at baseline: pooled from the phase 3 ALLEGRO and BRAVO studies

Overall, 1576 patients completed the 24-month studies (laquinimod: 80.4%; placebo: 78.2%) (Figure 1). Total exposure to laquinimod was 1720.6 patient-years and to placebo was 1726.2 patient-years. The most frequent reason for discontinuation in both treatment groups was withdrawal of consent (laquinimod: 6.9%; placebo: 7.6%). Early termination due to AEs occurred infrequently (laquinimod: 6.4%; placebo: 4.7%). Adverse events leading to early termination of two or more patients that occurred more frequently in the laquinimod group were abdominal pain, headache, diarrhea, and elevated liver enzyme levels (Supplementary Table 1 (211.7KB, pdf) , published in the online version of this article at ijmsc.org).
Serious AEs
Death was reported in three patients receiving placebo and one patient receiving laquinimod. A single laquinimod-treated patient in the BRAVO trial died of sepsis 3 weeks after early termination of the study; this was a 44-year-old cachectic man with severe MS (baseline Expanded Disability Status Scale score of 5.5) who was a Chernobyl survivor with severe debilitation after this radiation exposure. Eight months into the study, the patient presented weakness and diarrhea, for which no diagnosis could be established, and he terminated the study early. The patient died 3 weeks later after refusing hospitalization. Autopsy revealed that the cause of death was sepsis, with bilateral purulent pneumonia with microabscesses (no bacterial origin was confirmed), and the case was assessed by the study investigators as not being related to the study drug.
Two placebo-treated patients in the ALLEGRO trial died on-study: a 52-year-old man committed suicide, and a 33-year-old man died of injuries sustained in a train accident. In addition, a 40-year-old placebo-treated woman was discontinued from the ALLEGRO study due to pneumonia and later died of complications of this infection.
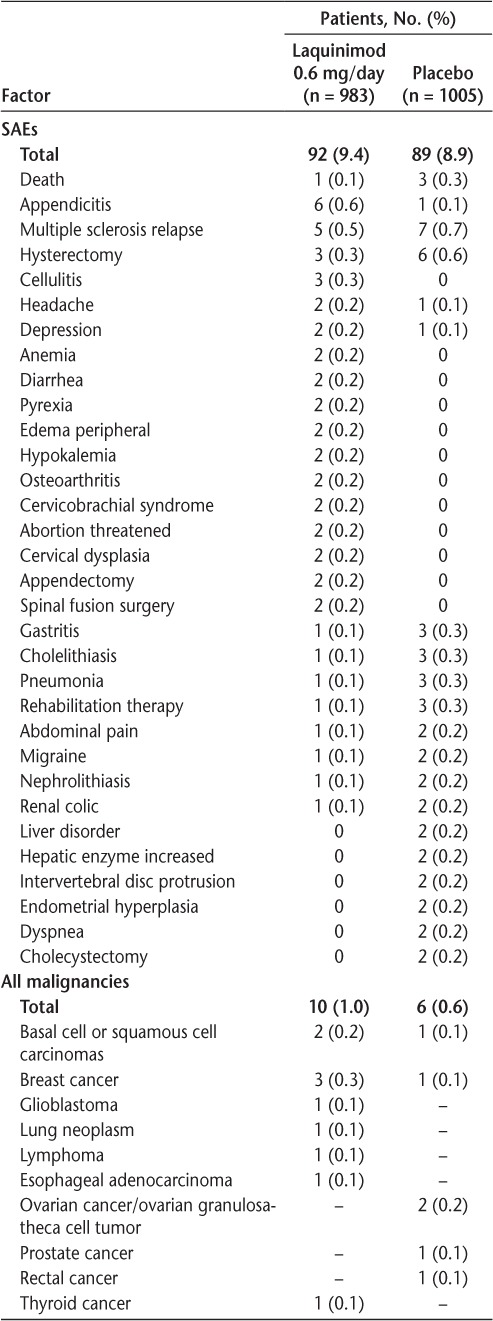
The overall frequency of SAEs was similar in the laquinimod- and placebo-treated groups (9.4% and 8.9%, respectively), and the types of SAEs reported for the two groups were similar (Table 2). An increased frequency of appendicitis was documented in laquinimod-treated patients (laquinimod: 0.6%; placebo: 0.1%). This finding was not associated with atypical histologic features or with any detectable pattern based on treatment duration, sex, age, or geography. Incidences of malignancies, infections, and cardiovascular events were similar between laquinimod- and placebo-treated patients.
Table 2.
Serious adverse events (SAEs) reported by two or more patients (either treatment group) and all malignancies
Adverse Events
A common AE that was reported more frequently for laquinimod-treated patients than for placebo-treated patients was back pain (laquinimod: 13.6%; placebo: 8.2%) (Table 3). The grouped events “back and neck pain” were reported in 15.6% and 9.4% of laquinimod- and placebo-treated patients, respectively. Events usually occurred during the first 3 months of treatment and were generally of mild severity but occasionally occurred at a later time point, were of longer duration, or required symptomatic treatment.
Table 3.
Common a adverse events

Headache was reported in 18.2% and 15.1% of laquinimod- and placebo-treated patients, respectively. Abdominal pain was reported in 5.0% and 2.6% of laquinimod- and placebo-treated patients, respectively. A higher frequency of alanine aminotransferase (ALT) increase (reported as an AE: 5.9% vs. 2.7%) was documented in laquinimod-treated patients; the issue of elevated liver enzyme levels is further addressed in the “Laboratory Measures” section that follows.
Laboratory Measures
Treatment with laquinimod was associated with some laboratory changes, including elevated liver enzyme levels, hematologic changes (decreased hemoglobin level/anemia, decreased platelet count, and increased WBC count), and elevated fibrinogen levels.
Liver Enzyme Levels
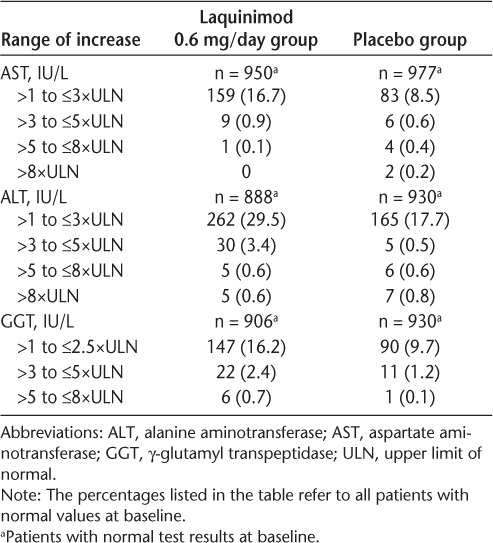
A higher incidence of liver enzyme level elevations was observed with laquinimod therapy compared with placebo use (Table 4). Most liver enzyme elevations were shifts from normal levels to between greater than 1×ULN and less than or equal to 3×ULN. Moderate increases (to ≤5×ULN) were much less frequent and occurred for ALT only (laquinimod: 3.4%; placebo: 0.5%). Shifts to greater than 5×ULN were rare and were reported with similar incidence in the laquinimod and placebo groups. Liver enzyme level elevations were mostly mild, asymptomatic, and reversible, generally occurring within 6 months of initiation of treatment and returning to baseline within 3 months under continued treatment. Hy's law criteria16 were not met, there was no concomitant elevation of bilirubin levels, and there were no cases of liver failure.
Table 4.
Shift from normal test results at baseline to the highest values for ALT, AST, and GGT
Hematologic Analysis
Hemoglobin Level. A mild decrease in hemoglobin level was noted early after initiation of treatment and was not progressive (Supplementary Figure 1 (211.7KB, pdf) ). Post-baseline hemoglobin level shifts were mostly to CTCAE grade 1 (hemoglobin level less than the lower limit of normal to 10.0 g/dL, defined as asymptomatic or mildly symptomatic anemia), reported for 44% of laquinimod-treated patients and 28% of placebo-treated patients. Postbaseline decreases to higher CTCAE grades were infrequent and occurred with similar or lower incidence in the laquinimod group relative to the placebo group. Hemoglobin level decreases were usually transient during continued laquinimod therapy and did not require treatment. There were no concurrent decreases in platelet or WBC counts in patients with decreased hemoglobin levels.
Total WBC Count. A small increase in the group mean total WBC count (within the reference range), consistent across all WBC subtypes, was observed within the first 2 months of initiation of laquinimod treatment, remaining stable thereafter. In the subgroup of patients with WBC counts greater than the ULN, mean total WBC counts in laquinimod- and placebo-treated patients were similar, ranging from 11.6 to 12.0 × 109/L in the laquinimod group versus 11.5 to 12.3 × 109/L in the placebo group, only slightly higher than the normal threshold (10.5 × 109/L) and without associated clinical manifestations (Supplementary Figure 2 (211.7KB, pdf) ).
Platelet Count. A mild, nonprogressive platelet count decrease occurred early after initiation of treatment in laquinimod-treated patients (Supplementary Figure 3 (211.7KB, pdf) ). Platelet count decreases were mild and asymptomatic, with no evidence of an increased bleeding risk or an association with any other clinical manifestations. Shifts from normal platelet values at baseline to a lower level (<100 × 109/L) occurred in six patients (0.6%) in the laquinimod group and two (0.2%) in the placebo group. Except for one patient, in all cases the low platelet count was a single measurement or occurred in patients with a stable, low platelet count that was recorded at baseline. The single laquinimod-treated patient with more profound thrombocytopenia was ultimately diagnosed as having idiopathic thrombocytopenic purpura (asymptomatic and responsive to corticosteroid treatment).
Fibrinogen Level
A small increase in the group mean level of fibrinogen (within the reference range) was documented in laquinimod-treated patients. Generally, this increase was documented after the first month of treatment and was followed by a return to baseline level without further increase after 6 months of treatment (Supplementary Figure 4 (211.7KB, pdf) ). The frequency of patients with above-normal fibrinogen levels was increased in the laquinimod-treated group (laquinimod: 43%; placebo: 34%), but the mean fibrinogen level over time in the subgroup of patients with an elevated fibrinogen level was similar to that in the placebo group, ranging from 4.7 to 4.9 g/L in the laquinimod group versus 4.4 to 4.6 g/L in the placebo group, slightly above the normal threshold (4.3 g/L). The maximal fibrinogen level did not exceed 2.5×ULN, and there were no associated clinical manifestations.
CRP Level
No differences in the proportion of patients with elevated CRP levels were documented in laquinimod- versus placebo-treated patients at any time during the study (laquinimod: 16.5%; placebo: 17.8%). In addition, there was no difference between laquinimod and placebo in group mean CRP level change over time (Supplementary Figure 5 (211.7KB, pdf) ).
Vital Signs and Electrocardiographic Outcomes
No trend was observed in changes from baseline over time for any vital sign, and no significant differences between treatment groups were observed in mean values for any electrocardiographic parameter.
Potential Risk Outcomes
Malignancy
There was no evidence that laquinimod 0.6 mg increased the risk of cancer. Malignant tumors were reported in ten patients in the laquinimod group (1.0%) and in six patients in the placebo group (0.6%) (Table 2). Oral and uterine tumors, the malignancy types documented in preclinical studies, were not reported during the clinical studies. The most frequently reported malignancy was breast cancer (laquinimod: n = 3; placebo: n = 1), followed by skin cancers (basal and squamous cell carcinoma) (laquinimod: n = 2; placebo: n = 1). Ovarian tumors were reported in the placebo group (n = 2) but not in the laquinimod group. Other reported malignancies were single reports of various cancers (laquinimod: glioblastoma, lung, lymphoma, esophageal, and thyroid cancers; placebo: prostate and rectal cancers). No rare malignancies, atypical malignancy patterns, or malignancies suggestive of immunosuppression were observed in either trial.
Among the reported malignancies, five occurred very early (<6 months) after treatment initiation. According to World Health Organization convention, tumor promotion by a medication is a process that occurs over a prolonged period (ie, years), and a medicine should not be considered to have promoted a malignancy unless it had been taken for at least 6 months.
Teratogenicity
Thirty pregnancies were reported in the ALLEGRO and BRAVO studies: 12 in laquinimod-treated patients and 18 in placebo-treated patients. No malformations were reported in newborns of laquinimod-treated patients. For pregnancies that occurred during placebo treatment, one patient with cryptorchism and cardiac septal defect and one patient with hypospadias were reported.
AEs Associated with Roquinimex
No increases in the incidence of myocardial infarction, pericarditis, or pleuritis were noted during laquinimod treatment relative to placebo treatment.
Discussion
The safety profile of laquinimod described herein is based on data from 1988 patients with MS (laquinimod: n = 983; placebo: n = 1005) participating in two double-blind studies, ALLEGRO and BRAVO, who received up to 24 months of laquinimod treatment. Adherence to the study was high: approximately 80% of laquinimod- and placebo-treated patients completed the studies, without systematic or informative censoring of patients in the intention-to-treat population. Notably, laquinimod, administered orally once daily, has no unblinding features, and, thus, the reported AEs are unlikely to have been biased. This is in contrast to many other MS treatments, which have frequent unblinding effects to patients or health-care providers, which may bias the reporting of parameters.
Based on these data, the safety profile of laquinimod is consistent with that reported in the phase 2 laquinimod study10 and characterized by AEs and laboratory findings that are mostly mild, transient, easily monitored, or readily diagnosed. Appendicitis, reported in 0.6% of laquinimod-treated patients and 0.1% of placebo-treated patients, was not associated with atypical histologic features or with any detectable pattern based on treatment duration, sex, age, or geography. The relationship between laquinimod and appendicitis is unclear, but appendicitis should be considered if typical symptoms are present. The most common AE noted with increased incidence with laquinimod was back and neck pain, which typically occurred in the first 3 months of treatment, as reflected by decreased AE reporting and less use of anti-inflammatory and antirheumatic medications thereafter. These AEs were usually of mild severity, and no specific pattern was identified that may suggest a potential mechanism.
Liver-related SAEs were reported with similar incidence and types in the laquinimod and placebo groups. Treatment with laquinimod is associated with mostly mild (≤3×ULN), asymptomatic, and reversible liver enzyme level elevations that generally occurred within 6 months after initiation of treatment, returning to baseline within 3 months under continued treatment. No patient met Hy's law16 or had concomitant bilirubin level elevations. Altogether, 11 patients in each treatment group terminated early due to liver function test result elevations (placebo: 5.5%; laquinimod: 3.3%). Of those, results in more patients in the placebo group versus the laquinimod group (ten [5%] vs. 5 [1.5%]) did not return to baseline. Transient elevations in liver enzyme levels have been noted in the previous phase 2 laquinimod study.10
Most approved RRMS treatments have been associated with elevations in liver enzyme levels. Hepatic injury, including elevated serum hepatic enzyme levels, has been reported with intramuscular IFNβ-1a in postmarketing surveillance,17 and severe elevations in hepatic transaminase levels are likewise listed as a common adverse effect for subcutaneous IFNβ-1a.18 Similarly, for subcutaneous IFNβ-1b, increased ALT and AST levels, as well as increased bilirubin and γ-glutamyl transpeptidase levels, although considered rare, are of enough consequence to require monitoring.19 Abnormal liver function test results are labeled as common adverse reactions for glatiramer acetate,20 and liver enzyme level elevations up to fivefold the ULN have been reported more frequently with fingolimod treatment than with placebo use.21 Likewise, an increased incidence of elevations in hepatic transaminase levels, seen primarily during the first 6 months of treatment, is known for dimethyl fumarate and teriflunomide.22,23
Other laboratory findings with laquinimod included increased fibrinogen levels and mild hematologic changes. Increased fibrinogen levels were demonstrated in 34% of placebo-treated patients and 43% of laquinimod-treated patients. The magnitudes of the elevations were small and are not considered to be clinically meaningful. Specifically, no clinical manifestations were noted in either group (placebo or laquinimod). Hematologic changes were mild and asymptomatic, including a decrease in hemoglobin levels, an increase in WBC counts, and a decrease in platelet counts. An interesting stereotypical temporal pattern was noted for the laboratory value changes, with onset during the first 60 to 90 days of treatment followed by stabilization (hematologic) or a return to baseline levels (fibrinogen). This pattern suggests an initial induction followed by adaptive responses.
Death occurred in three placebo-treated patients and one patient treated with laquinimod, and, except for the aforementioned case of appendicitis, SAEs were of similar incidence and type. There was no increase in infections, malignancies, or cardiovascular events in laquinimod-treated patients relative to placebo users.
Malignancies that were the focal point of interest in preclinical data (oral and uterine tumors in rats) were not observed in the ALLEGRO and BRAVO trials. No malignancies suggestive of immunosuppression (eg, Kaposi's sarcoma or unusual lymphoproliferative disorders) were seen. No atypical malignancies and no unusual shift in the expected cancer frequency were observed. Thus, not unexpected for this population comprising mostly women aged 30 to 49 years, the most frequently reported malignancy was breast cancer; other reported invasive cancers were individual cases of different types. The three cases of breast cancer were clinically typical, diagnosed at early stages, and without unusual histologic patterns. In two patients, diagnosis occurred within 6 months of initiation of the drug, supporting the notion that undiagnosed malignancy was present before entering the study. Altogether, there is no evidence of increased cancer risk in these 2-year studies.
No pregnancy of a laquinimod-treated patient resulted in fetal malformations, and no fetal defects were reported in cases of induced abortion. Nevertheless, these small numbers preclude conclusions, so the relevance of animal teratogenicity findings to humans remains unknown, and exposure to laquinimod during pregnancy must be avoided.
No signal of concern was identified regarding serious toxicities associated with laquinimod's predecessor compound, roquinimex (namely, myocardial infarction, pericarditis, and pleuritis). Such toxicities were noted in the first months of treatment with this compound and were not seen with an increased incidence during laquinimod treatment compared with placebo use during 24 months of treatment. Notably, this safety database is approximately threefold larger in terms of treated patients, and approximately 20-fold larger in terms of drug exposure, compared with that for roquinimex (946 patients, 330 patient-years of exposure), indicating that the safety profile of laquinimod differs substantially from that of roquinimex.
In conclusion, pooled results of the placebo-controlled ALLEGRO and BRAVO trials for the assessment of laquinimod 0.6 mg once daily demonstrate a safety profile characterized by events that are mostly mild, transient, easily monitored, or readily diagnosed and treated. Laboratory findings have a temporal pattern and are asymptomatic. The potential risks noted in the pre-clinical studies or related to the predecessor compound, roquinimex, were not observed.
PracticePoints
Laquinimod is a once-daily oral treatment currently in development for relapsing-remitting and progressive MS, with an emerging safety profile comprising manageable or benign adverse effects that are mostly mild, transient, easily monitored, or readily diagnosed.
Identified laquinimod-related events include generally mild back and neck pain during early treatment, and appendicitis reported more frequently in laquinimod-treated patients than in placebo-treated patients. The relationship between laquinimod and appendicitis is unclear, without a pattern to suggest a causative mechanism, but appendicitis should be considered if typical symptoms are present.
All other identified safety-related events observed during laquinimod treatment are laboratory abnormalities. Changes in laboratory values follow a stereotypical temporal pattern, with mild, nonprogressive changes that occur in the first months of treatment and then stabilize or revert to baseline levels during continued treatment and are not accompanied by clinical symptoms.
The incidence of malignancies, infections, and cardiovascular events is similar in laquinimodand placebo-treated patients, and potential risks noted in the preclinical studies or related to the predecessor compound, roquinimex, were not observed in the clinical setting.
Supplementary Material
Footnotes
Financial Disclosures: Dr. Sørensen has served on scientific advisory boards for Biogen, Merck Serono, Novartis, Genmab, Teva Pharmaceutical Industries Ltd, Elan, and GlaxoSmithKline; has been on steering committees or independent data monitoring boards in clinical trials sponsored by Merck Serono, Genmab, Teva Pharmaceutical Industries Ltd, GlaxoSmithKline, and Bayer Schering and has received travel funding for these activities; has served as Editor-in-Chief of the European Journal of Neurology and is an editorial board member for Therapeutic Advances in Neurological Disorders and Multiple Sclerosis; has received speaker honoraria from Biogen, Merck Serono, Teva Pharmaceutical Industries Ltd, Bayer Schering, Sanofi-Aventis, and Novartis; and has received research support from Biogen, Bayer Schering, Merck Serono, Teva Pharmaceutical Industries Ltd, Baxter, Sanofi-Aventis, BioMS, Novartis, Bayer, RoFAR, Roche, Genzyme, the Danish Multiple Sclerosis Society, the Danish Medical Research Council, and the European Union Sixth Framework Programme: Life Sciences, Genomics and Biotechnology for Health. Dr. Comi has received consulting fees for participation on advisory boards, consultancy, and speaker activities from Novartis, Teva Pharmaceutical Industries Ltd, Sanofi-Aventis, Merck Serono, and Bayer Schering; and lecture fees from Novartis and Teva Pharmaceutical Industries Ltd. Dr. Vollmer has received payment for participation on advisory boards, lectures, and consultancy from Biogen, Consortium of Multiple Sclerosis Centers, Daiichi Sankyo, Elan Pharma, Eli Lilly and Co, Global Prairie, Guidepoint Global, Hoffman-LaRoche, Medical Logix, MSDx, Prime Education, Projects in Knowledge, Teva Pharmaceutical Industries Ltd, Xenoport, Esai Pharmaceuticals, Schering-Plough Biopharma, Acorda, and Novartis; and has received research support from Biogen, Teva Pharmaceutical Industries Ltd, Lilly Research Laboratories, Genzyme, Ono Pharmaceuticals, and Elan Pharmaceuticals. Dr. Montalban has received honoraria for speaking and travel expenses to scientific meetings; was a steering member or participated in advisory boards in corporate-sponsored clinical trials or has had consulting agreements with Bayer Schering Pharma, Biogen, EMD Merck Serono, Genentech, Genzyme, Novartis, Sanofi-Aventis, and Teva Pharmaceutical Industries Ltd. The University Hospital Basel, as the employer of Dr. Kappos, has received and dedicated to research support fees for board membership, consultancy or speaking, or grants in the past 3 years from Actelion, Advancell, Allozyne, Bayer, Bayhill, Biogen Idec, BioMarin, CSL Behring, Eli Lilly EU, Genmab, GeNeuro SA, Gianni Rubatto Foundation, Glenmark, Merck Serono, MediciNova, Mitsubishi Pharma, Novartis, Novartis Research Foundation, Novo Nordisk, Peptimmune, Roche, Roche Research Foundation, Santhera, Sanofi-Aventis, Swiss MS Society, Swiss National Research Foundation, Teva Pharmaceutical Industries Ltd, UCB, and Wyeth. Dr. Dadon, Dr. Gorfine, Dr. Margalit, Mr. Sasson, Dr. Rubinchick, and Dr. Knappertz are employed by and receive travel/meeting expenses from Teva Pharmaceutical Industries Ltd. Mr. Sasson and Dr. Knappertz own stock in Teva Pharmaceutical Industries Ltd, and Dr. Knappertz is a coholder of patents with Teva Pharmaceutical Industries Ltd.
References
- 1. Brück W, Wegner C.. Insight into the mechanism of laquinimod action. J Neurol Sci. 2011; 306: 173– 179. [DOI] [PubMed] [Google Scholar]
- 2. Noseworthy JH, Wolinsky JS, Lublin FD, . et al.; North American Linomide Investigators Linomide in relapsing and secondary progressive MS, part I: trial design and clinical results. Neurology. 2000; 54: 1726– 1733. [DOI] [PubMed] [Google Scholar]
- 3. Wolinsky JS, Narayana PA, Noseworthy JH, . et al; MRI Analysis Center of the University of Texas-Houston, Health Science Center, and the North American Linomide Investigators Linomide in relapsing and secondary progressive MS, part II: MRI results. Neurology. 2000; 54: 1734– 1741. [DOI] [PubMed] [Google Scholar]
- 4. Brunmark C, Runstrom A, Ohlsson L, . et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002; 130: 163– 172. [DOI] [PubMed] [Google Scholar]
- 5. Yang JS, Xu LY, Xiao BG, Hedlund G, Link H.. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J Neuroimmunol. 2004; 156: 3– 9. [DOI] [PubMed] [Google Scholar]
- 6. Lund BT, Kelland EE, Hayardeny L, Barilan O, Gilmore W, Weiner LP.. Assessment of changes in immune measures of multiple sclerosis patients treated with laquinimod. J Neuroimmunol. 2013; 263: 108– 115. [DOI] [PubMed] [Google Scholar]
- 7. Stasiolek M, Linker RA, Hayardeny L, Bar Ilan O, Gold R.. Immune parameters of patients treated with laquinimod, a novel oral therapy for the treatment of multiple sclerosis: results from a double-blind placebo-controlled study. Immun Inflamm Dis. 2015; 3: 45– 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lund BT, Kelland EE, Hayardeny L, Barilan O, Gilmore W, Weiner LP.. Assessment of changes in immune measures of multiple sclerosis patients treated with laquinimod. J Neuroimmunol. 2013; 263: 108– 115. [DOI] [PubMed] [Google Scholar]
- 9. Jolivel V, Luessi F, Masri J, . et al. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain. 2013; 136( pt 4): 1048– 1066. [DOI] [PubMed] [Google Scholar]
- 10. Comi G, Pulizzi A, Rovaris M, . et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomized, double-blind, placebo-controlled phase IIb study. Lancet 2008; 371: 2085– 2092. [DOI] [PubMed] [Google Scholar]
- 11. Comi G, Jeffery D, Kappos L, . et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis. N Engl J Med. 2012; 366: 1000– 1009. [DOI] [PubMed] [Google Scholar]
- 12. Vollmer T, Sorensen PS, Selmaj K, . et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol. 2014; 261: 773– 783. [DOI] [PubMed] [Google Scholar]
- 13. Comi G, Ladkani D, Vollmer T, Sormani MP, Sidi Y, Knappertz V.. Mediation of the effect of laquinimod on disability progression in relapsing-remitting multiple sclerosis. Paper presented at: 2014 Annual Meeting of the American Academy of Neurology; April 26–May 3, 2014; Philadelphia, PA. [Google Scholar]
- 14. Polman CH, Reingold SC, Edan G, . et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria.” Ann Neurol. 2005; 58: 840– 846. [DOI] [PubMed] [Google Scholar]
- 15. National Multiple Sclerosis Society. . Who gets MS (epidemiology). http://www.nationalmssociety.org/What-is-MS/Who-Gets-MS. Accessed 2014.
- 16. Reuben A. Hy's law. Hepatology. 2004; 39: 574– 578. [DOI] [PubMed] [Google Scholar]
- 17. Avonex [package insert]. Cambridge, MA: Biogen Idec Inc; 2014. [Google Scholar]
- 18. Rebif [package insert]. Rockland, MA: EMD Serono Inc; 2014. [Google Scholar]
- 19. Betaseron [package insert]. Whippany, NJ: Bayer Healthcare Pharmaceuticals Inc; 2014. [Google Scholar]
- 20. Copaxone [summary of product characteristics]. West Yorkshire, UK: Teva Pharmaceuticals Ltd; 2015. [Google Scholar]
- 21. Gilenya [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2014. [Google Scholar]
- 22. Gold R, Kappos L, Arnold DL, . et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012; 367: 1098– 1107. [DOI] [PubMed] [Google Scholar]
- 23. O'Connor P, Wolinsky JS, Confavreux C, . et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011; 365: 1293– 1303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.