

Abstract
Mine tailings from copper mines are considered as one of the sources of highly hazardous acid mine drainage (AMD) due to bio-oxidation of its sulfidic constituents. This study was designed to understand microbial community composition and potential for acid generation using samples from mine tailings of Malanjkhand copper project (MCP), India through 16S rRNA gene based amplicon sequencing approach (targeting V4 region). Three tailings samples (T1, T2 and T3) with varied physiochemical properties selected for the study revealed distinct microbial assemblages. Sample (T3) with most extreme nature (pH < 2.0) harbored Proteobacteria, Actinobacteria, Chloroflexi while the samples (T1 and T3) with slightly moderate nature (pH < 4.0 and > 3.0) exhibited abundance of Proteobacteria, Fimicutes, Actinobacteria and/or Nitrospirae. Metagenomic sequences are available under the BioProject ID PRJNA361456.
Keywords: Mine tailings, Metagenomics, Next generation sequencing, Microbial community, Acid mine drainage
| Specifications | |
|---|---|
| Organism/cell line/tissue | Mine tailings sediment |
| Sex | Not applicable |
| Sequencer or array type | Ion Torrent S5 Platform |
| Data format | Raw data: FASTQ file |
| Experimental factors | Environmental samples |
| Experimental features | 16S rRNA gene amplicon (V4 region) sequencing using Ion Torrent platform and diversity analysis using QIIME 1.9.0 |
| Consent | Not applicable |
| Sample source location | Tailing dam of Malanjkhand copper project, Balaghat district, M.P., India. T1 (N21°59.105″ E080°42.423″), T2 (N21° 59.096″ E080° 42.427″) and T3 (N21° 59.915″ E 080°42.081″) |
1. Direct link to deposited data
2. Experimental design, materials and methods
Mine tailings are considered as a major source of acid mine drainage (AMD) due to the microbial bio-oxidation of sulfidic constituents [1]. Mine tailings are characterized with low –pH, − organic matters and other nutrients, as well as high concentrations of toxic metals and sulfate which pose a severe environmental threat [2], [3]. Several studies have been conducted worldwide to understand microbial community composition within mine tailings and their metabolic role in biogeochemical cycles of acid generation using either clone library/cultivation or pyrosequencing based approach [3], [4], [5], [6], [7], [8], [9]. In recent years, advances in semiconductor based next generation sequencing technology has shown high promises to gain better insights into the microbial community structure and dynamics in natural ecosystems. Numerous studies have been conducted to understand the microbial community structure and function of AMD ecosystem [10], [11], [12], [13], [14], [15], [16]. However, dynamics of microbial community composition from mine tailings (source) to the generation of AMD (product) remains less explored. This study was designed to target unexplored mine tailings of Malanjkhand copper project (MCP), Asia's biggest copper mine, situated in Balaghat district of Madhya Pradesh, India to analyze in-depth microbial community composition and their potential role in acid generation.
2.1. Sampling, DNA extraction and sequencing
Tailing sediments (T1 and T2) were collected from tailing dam and its seepage point (T3) of MCP during April 2014 (T1 and T3) and September 2016 (T2). Samples were collected in sterile container and stored at 4 °C, till further analysis. Metagenome was extracted from 0.25 g of each sample using Power soil DNA isolation kit (MoBio laboratories) according to the manufacturer protocol; quantified in Qubit 3.0 fluorometer (Invitrogen, Thermo Fisher Scientific) and V4 region of 16S rRNA gene was amplified with V4 specific primers [17]. Each forward primer was tagged with 10-12 bp barcode for multiplexing during sequencing run. PCR was performed in 25 μl of reaction using Amplitaq Gold 360 Master Mix (Invitrogen, Thermo Fisher Scientific), 40 pico mole each fusion primers and 10–50 ng of template DNA. PCR amplified products (V4 amplicons libraries) were extracted using E gel base system (Invitrogen, Thermo Fisher Scientific). Purified amplicons were processed and sequenced in Ion S5 sequencer.
2.2. Bioinformatic processing of 16S rRNA gene sequence and diversity analysis
Total 0.97 million raw reads were obtained after sequencing. These reads were analyzed in QIIME pipeline [18]. Quality filtering was performed using split_libraries.py to remove primers, sequences with homopolymers run of > 6 bp and read length beyond the range of 230-300 bp. Minimum 3 primer mismatches were allowed in this step due to degeneracy of primer set. The output files from three samples were merged together and pick_closed_reference_otus.py was used to pick OTUs, assign taxonomy and create OTU table against a SILVA 119 reference database (www.arb-silva.de/documentation/release-119). Number of OTUs was obtained using alpha_diversity.py. Agglomerative hierarchical clustering on the abundance of major genera (cumulative abundance > 0.5%) were performed with weighted pair group average (WPGA) clustering method using Bray- Curtis dissimilarity distance matrix in XLSTAT 2014 software.
Out of 0.7 million operational reads (obtained after quality filtering), 0.58 million reads were assigned to taxonomy using closed reference method. Alpha diversity indicator revealed that T1 was highly diverse (OTUs 4885) followed by T3 (OTUs 3776) and T2 (OTUs 211). Sample (T3) with most extreme nature (pH < 2.0) harbored Proteobacteria, Actinobacteria and Chloroflexi while the samples (T1 and T2) with slightly moderate nature (pH < 4.0 and > 3.0) exhibited abundance of Proteobacteria, Fimicutes, Actinobacteria and/or Nitrospirae. Archaeabacterial phyla, Euryarchaeota and Thaumarchaeota were abundant in the extreme sample (T3) and accounted for 7.2% of total assigned reads. (Fig. 1).
Fig. 1.
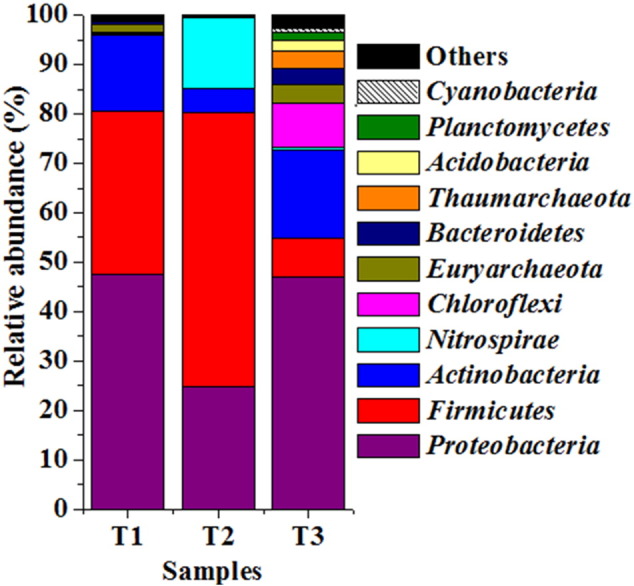
Distribution of taxa at phylum level across the samples (cumulative abundance > 0.5%).
T1 and T2 samples showed commonality with presence of genera Sulfobacillus and Alicyclobacillus. T1 showed specific abundance of Halomonas, Corynebacterium, Staphylococcus, Rhizobium, Acinetobacter and Stenotrophomonas while T2 harbored Leptospirillum, Acidithiobacillus, Acidiferrobacter and Acidiphilium. In contrast, T3 sample showed distinct abundance of Ralstonia, Metallibacterium, Acidovorax, Rhodanobacter, uncultured-Chloroflexi, -Chitinophagaceae, -Thermoplasmatales, -Thaumarcheota and Thiobacillus. Hierarchical clustering on abundance of these genera across the samples was also evident in Fig. 2, indicating distinct patterns of assemblage among the organisms with common metabolic/biogeochemical functions. Detection of major genera such as Sulfobacillus, Leptospirillum, Acidithiobacillus, Acidiferrobacter, Metallibacterium, Acidiphilium and Alicyclobacillus in the tailings samples indicates the intrinsic acid generation potential of the microbial community. Observed variation in community composition among the samples could be attributed to the prevalent physicochemical conditions and their role in shaping the community structure. This study provides an insight into the microbial community structure and their relation to biogeochemistry of copper mine tailings.
Fig. 2.

Weighted pair average group based hierarchical clustering with Bray Curtis dissimilarity distance matrix of major abundant genera (cumulative abundance > 0.5%) across the samples.
Acknowledgements
This work was financially supported by the Department of Biotechnology (DBT), Govt. of India (BT/PR 7533/BCE/8/959/2013). Next generation facility (with Ion S5) is funded by the Indian Institute of Technology (IIT) Kharagpur (IIT/SRIC/BT/ODM/2015-16/141) through SGBSI challenge grant. Abhishek Gupta (DBT-JRF) is a recipient of DBT-JRF (DBT/2014/IITKH/113). Financial support to Avishek Dutta and Jayeeta Sarkar from IIT Kharagpur (IIT/ACAD(PGS&R)/F.II/2/14/BS/91R01 and IIT/ACAD(PGS&R)/F.II/2/13BT91P01) institutional fellowship is acknowledged. Dhiraj Paul was recipient of UGC research fellowship serial no.2120930743 and Ref. No.20-12/2009(ii)EU-IV. Authors are thankful to Malanjkhand copper project, Balaghat district, M.P. India for providing samples.
References
- 1.Schippers A., Breuker A., Blazejak A., Bosecker K., Kock D., Wright T.L. The biogeochemistry and microbiology of sulfidic mine waste and bioleaching dumps and heaps, and novel Fe (II)-oxidizing bacteria. Hydrometallurgy. 2010;104(3):342–350. [Google Scholar]
- 2.Mendez M.O., Neilson J.W., Maier R.M. Characterization of a bacterial community in an abandoned semiarid lead-zinc mine tailing site. Appl. Environ. Microbiol. 2008;74(12):3899–3907. doi: 10.1128/AEM.02883-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan G.L., Shu W.S., Hallberg K.B., Li F., Lan C.Y., Zhou W.H., Huang L.N. Culturable and molecular phylogenetic diversity of microorganisms in an open-dumped, extremely acidic Pb/Zn mine tailings. Extremophiles. 2008;12(5):657–664. doi: 10.1007/s00792-008-0171-9. [DOI] [PubMed] [Google Scholar]
- 4.Kock D., Schippers A. Geomicrobiological investigation of two different mine waste tailings generating acid mine drainage. Hydrometallurgy. 2006;83(1):167–175. [Google Scholar]
- 5.Diaby N., Dold B., Pfeifer H.R., Holliger C., Johnson D.B., Hallberg K.B. Microbial communities in a porphyry copper tailings impoundment and their impact on the geochemical dynamics of the mine waste. Environ. Microbiol. 2007;9(2):298–307. doi: 10.1111/j.1462-2920.2006.01138.x. [DOI] [PubMed] [Google Scholar]
- 6.Kock D., Schippers A. Quantitative microbial community analysis of three different sulfidic mine tailing dumps generating acid mine drainage. Appl. Environ. Microbiol. 2008;74(16):5211–5219. doi: 10.1128/AEM.00649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang L.N., Zhou W.H., Hallberg K.B., Wan C.Y., Li J., Shu W.S. Spatial and temporal analysis of the microbial community in the tailings of a Pb-Zn mine generating acidic drainage. Appl. Environ. Microbiol. 2011;77(15):5540–5544. doi: 10.1128/AEM.02458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L.X., Li J.T., Chen Y.T., Huang L.N., Hua Z.S., Hu M., Shu W.S. Shifts in microbial community composition and function in the acidification of lead/zinc mine tailings. Environ. Microbiol. 2013;15(9):2431–2444. doi: 10.1111/1462-2920.12114. [DOI] [PubMed] [Google Scholar]
- 9.Dha P.K., Sar P. Microbial communities in uranium mine tailings and mine water sediment from Jaduguda U mine, India: a culture independent analysis. J. Environ. Sci. Health A. 2014 May 49;6 doi: 10.1080/10934529.2014.865458. [DOI] [PubMed] [Google Scholar]
- 10.Hallberg K.B., Johnson D.B. Biodiversity of acidophilic prokaryotes. Adv. Appl. Microbiol. 2001;49:37–84. doi: 10.1016/s0065-2164(01)49009-5. [DOI] [PubMed] [Google Scholar]
- 11.Baker B.J., Banfield J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 2003;44(2):139–152. doi: 10.1016/S0168-6496(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 12.Tan G.L., Shu W.S., Hallberg K.B., Li F., Lan C.Y., Huang L.N. Cultivation-dependent and cultivation-independent characterization of the microbial community in acid mine drainage associated with acidic Pb/Zn mine tailings at Lechang, Guangdong, China. FEMS Microbiol. Ecol. 2007;59(1):118–126. doi: 10.1111/j.1574-6941.2006.00216.x. [DOI] [PubMed] [Google Scholar]
- 13.González-Toril E., Llobet-Brossa E., Casamayor E.O., Amann R., Amils R. Microbial ecology of an extreme acidic environment, the Tinto River. Appl. Environ. Microbiol. 2003;69(8):4853–4865. doi: 10.1128/AEM.69.8.4853-4865.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Druschel G.K., Baker B.J., Gihring T.M., Banfield J.F. Acid mine drainage biogeochemistry at Iron Mountain, California. Geochem. Trans. 2004;5(2):13. doi: 10.1186/1467-4866-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertin P.N., Heinrich-Salmeron A., Pelletier E., Goulhen-Chollet F., Arsène-Ploetze F., Gallien S., Bonnefoy V. Metabolic diversity among main microorganisms inside an arsenic-rich ecosystem revealed by meta-and proteo-genomics. ISME J. 2011;5(11):1735–1747. doi: 10.1038/ismej.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuang J.L., Huang L.N., Chen L.X., Hua Z.S., Li S.J., Hu M., Shu W.S. Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J. 2013;7(5):1038–1050. doi: 10.1038/ismej.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bates S.T., Berg-Lyons D., Caporaso J.G., Walters W.A., Knight R., Fierer N. Examining the global distribution of dominant archaeal populations in soil. ISME J. 2011;5(5):908–917. doi: 10.1038/ismej.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Huttley G.A. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]