

The cobalt–nitrogen catalyst shows high activity for selective reduction of nitro compounds with H2 and other hydrogen donors.
Keywords: Non-noble heterogeneous catalyst, Nitro compounds, Hydrogenation, Transfer hydrogenation, Reductive coupling reaction
Abstract
Replacement of precious noble metal catalysts with low-cost, non-noble heterogeneous catalysts for chemoselective reduction and reductive coupling of nitro compounds holds tremendous promise for the clean synthesis of nitrogen-containing chemicals. We report a robust cobalt–nitrogen/carbon (Co–Nx/C-800-AT) catalyst for the reduction and reductive coupling of nitro compounds into amines and their derivates. The Co–Nx/C-800-AT catalyst was prepared by the pyrolysis of cobalt phthalocyanine–silica colloid composites and the subsequent removal of silica template and cobalt nanoparticles. The Co–Nx/C-800-AT catalyst showed extremely high activity, chemoselectivity, and stability toward the reduction of nitro compounds with H2, affording full conversion and >97% selectivity in water after 1.5 hours at 110°C and under a H2 pressure of 3.5 bar for all cases. The hydrogenation of nitrobenzene over the Co–Nx/C-800-AT catalyst can even be smoothly performed under very mild conditions (40°C and a H2 pressure of 1 bar) with an aniline yield of 98.7%. Moreover, the Co–Nx/C-800-AT catalyst has high activity toward the transfer hydrogenation of nitrobenzene into aniline and the reductive coupling of nitrobenzene into other derivates with high yields. These processes were carried out in an environmentally friendly manner without base and ligands.
INTRODUCTION
Selective reduction of nitro compounds into amines is one of the most important chemical reactions in synthetic organic chemistry (1, 2). Amines with an annual production of more than 4 million metric tons (3) have been widely used for the production of bulk and fine chemicals, such as dyes, agrochemicals, pharmaceuticals, pesticides, and polymers (4). In the past, a noncatalytic process was used for the reduction of nitro compounds using stoichiometric reducing agents (5), an approach that demonstrated some drawbacks, such as the release of wastes and the high cost of reducing agents. Therefore, considerable attention has shifted to the catalytic reduction of nitro compounds into amines because of the compatibility of environmentally friendly catalytic methods with industrial processes (6, 7).
The catalytic reduction of nitro compounds with hydrogen or hydrogen donors has been mainly performed over noble metal catalysts (8, 9). Unfortunately, most catalysts do not meet the dual requirements of activity and selectivity (10). On the one hand, catalysts with a high selectivity require a high reaction temperature or a high H2 pressure because of their low intrinsic activity. On the other hand, catalysts with high intrinsic activity suffer from poor chemoselectivity. Thus, modification of metal catalysts with well-chosen additives, such as Pt/C-H3PO2-VO(acac)2 and Pt–Pb/CaCO3-FeCl2-nBu4NCl, has been used to improve the chemoselectivity at the expense of the activity (10). In recent years, some new kinds of noble metal catalysts, such as Au and Pt with specific structures, were observed to be active for the chemoselective hydrogenation of nitroarenes with H2 (11–13). For example, Wei and co-workers (13) reported that FeOx-supported Pt catalysts (Pt/FeOx) with activity associated with a single atom or a pseudo–single atom showed high activity and chemoselectivity toward the hydrogenation of nitroarenes at 40°C and under a H2 pressure of 3 bar in toluene. Besides H2, some hydrogen donors, such as hydrazine, sodium borohydride, alcohols, and formic acid, were also used for the catalytic transfer hydrogenation (CTH) of nitro compounds (14). A CTH process does not need high pressurized H2 and elaborate experimental setups, and most reactions were performed over noble metal catalysts with difficult-to-obtain and environmentally unfriendly ligands or base as additives (15, 16).
Despite notable achievements over noble metal catalysts in recent years, there remains significant room for the design of inexpensive and active non-noble metal catalysts for the chemoselective reduction of nitro compounds. In recent years, carbon-supported cobalt or iron oxides from the pyrolysis of metal-phenanthroline complexes were used for the chemoselective hydrogenation of nitroarenes (17, 18) but required relatively demanding conditions, such as long reaction times (12 to 24 hours) and high H2 pressures (50 bar) in a mixed solvent consisting of water and tetrahydrofuran. Therefore, there is still a great need to develop highly efficient and environmentally benign methods to selectively transform nitro compounds into amines using either H2 or hydrogen donors. It is also highly desirable to perform the reductive coupling of nitro compounds into other nitrogen-containing derivatives, which avoids the isolation and purification of intermediates.
Recently, catalysts based on nitrogen-doped carbon materials have received worldwide attention, because the incorporation of nitrogen atoms can enhance their chemical, electrical, and functional properties (19, 20). This kind of catalyst can be easily prepared by a one-pot annealing process using suitable precursors containing nitrogen, carbon, and transition metals. Here, cobalt phthalocyanine (fig. S1) was selected as the precursor to prepare mesoporous nitrogen-doped carbon-based cobalt catalysts for the reduction of nitro compounds. The cobalt–nitrogen (Co–Nx) complex was identified to be the active site, which was generated by the acid washing of the Co nanoparticles and silica after pyrolysis of the cobalt phthalocyanine/silica composite. The Co–Nx/C-800-AT catalyst was active for the chemoselective reduction of nitro compounds with H2 or other hydrogen donors under mild conditions. In addition, it was also effective for the one-pot synthesis of imines, N-phenylformamide, and benzimidazole from nitro compounds.
RESULTS
Catalyst preparation
The catalyst preparation procedure is schematically illustrated in Fig. 1. Silica was used as the hard template to enlarge the surface area and porosity of the catalysts. Typically, Co/N-C-SiO2-800 was generated by the pyrolysis of the cobalt phthalocyanine/silica composite at 800°C under a N2 atmosphere. Then, Co nanoparticles and silica were simultaneously removed with 10 weight % (wt %) HF, and the resulting catalyst was denoted as Co–Nx/C-800-AT. For comparison, the Co/N-C-SiO2-800 powder was soaked with NaOH to remove silica to give a catalyst abbreviated as Co/N-C-800-BT.
Fig. 1. Schematic illustration for the preparation of the Co–Nx/C-800-AT catalyst.
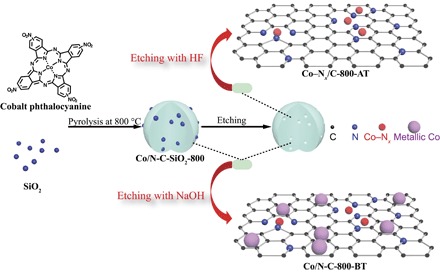
Thermogravimetric analysis (TGA) of the cobalt phthalocyanine/silica composite was performed under a N2 atmosphere (fig. S2). The weight loss before 200°C was attributed to the loss of the physically absorbed water. According to previous work on the TGA behavior of the cobalt phthalocyanine (21, 22), the weight loss between 200°C and 440°C was due to the initial release of axially coordinated molecules with the Co2+ center, such as water, followed by the release of nitro groups and the loss of phenyl groups. During this step, the polymerization of the phthalocyanine unit was completed. The decomposition of the cobalt phthalocyanine–like polymer was responsible for the weight loss between 430°C and 630°C. During this step, some of the fragments might form a Co–N4 moiety or a Co–N2 moiety, and some metallic Co at the subnanometer scale might also form (22, 23). At a pyrolysis temperature higher than 650°C, the weight loss was sharper, indicating that Co–N bonds were cleaved seriously, and cobalt nanoparticles were formed, as evidenced from transmission electron microscope (TEM) images. When the pyrolysis temperature was beyond 820°C, the weight loss was further enhanced, likely due to the further breakage of the carbon framework at high reaction temperatures. From the TGA, four representative pyrolysis temperatures (400°C, 600°C, 800°C, and 900°C) were adopted for preparation of the catalysts.
Solid ultraviolet-visible (UV-Vis) spectra were used to probe the structural changes of the cobalt phthalocyanine after pyrolysis (fig. S3). The peak intensity of the sample after pyrolysis at 200°C was almost the same as that of the initial cobalt phthalocyanine/silica composite, suggesting that cobalt phthalocyanine was stable at 200°C. At 400°C, the intensity of absorption peak at 650 nm became much weaker, suggesting that some Co–N4 moieties of the phthalocyanine ring decomposed to other fragments, such as Co–N2 moieties. A few Co–N4 moieties still presented at 600°C. The characteristic peak of the Co–N4 moiety disappeared at 800°C, suggesting the complete decomposition of Co–N4 moieties.
Figure S4 shows the TEM images of Co/N-C-SiO2-X samples. There were no Co nanoparticles in the TEM images of Co/N-C-SiO2-400 and Co/N-C-SiO2-600. However, Co nanoparticles were clearly observed in the TEM images of Co/N-C-SiO2-800 and Co/N-C-SiO2-900. The average size of the Co nanoparticles was 17.6 nm (200 particles measured in 20 TEM images) for Co/N-C-SiO2-800 and 28.1 nm for Co/N-C-SiO2-900 (fig. S5), suggesting that a higher pyrolysis temperature resulted in the growth of the Co nanoparticles. As shown in Fig. 2 (A and B), no Co nanoparticles were observed in the TEM images of the Co–Nx/C-800-AT and Co–Nx/C-900-AT, and pores were clearly observed, suggesting that Co nanoparticles and silica were successfully washed off by 10 wt % HF. The enlarged TEM images (scale, 20 nm) (fig. S6) also revealed that no Co nanoparticles were present in the C–-Nx/C-800-AT and Co–Nx/C-900-AT catalysts, further confirming that Co nanoparticles were fully washed off by 10 wt % HF. Nitrogen adsorption isotherms of Co–Nx/C-800-AT displayed a type IV curve and an H1-type hysteresis loop, characteristic of a mesoporous structure. The average pore size was determined to be 3.6 nm, according to the Barrett-Joyner-Halenda method. The Brunauer-Emmett-Teller (BET) surface area and pore volume were determined to be 530 m2/g and 0.52 cm3/g, respectively. Co nanoparticles were observed in Co/N-C-SiO2-800 (Fig. 2C) and Co/N-C-800-BT (Fig. 2D). In addition, the porous structure was also observed in Fig. 2D, suggesting that NaOH can only wash off silica from Co/N-C-SiO2-800. Table S1 summarizes the cobalt weight % of the samples. The weight % of cobalt in Co/N-C-800-BT was 12.3%, whereas it was only 0.25% in the Co–Nx/C-800-AT catalyst. This means that about 98 wt % cobalt was presented in the form of metallic Co nanoparticles, which can be washed off by 10 wt % HF. The state of Co in the Co–Nx/C-800-AT catalyst was present in the form of Co–Nx, as will be discussed later. Other researchers have also prepared other kinds of Co–Nx/C catalysts for electrochemical reactions (24, 25). The method they used for the preparation of Co–Nx/C catalysts was similar to ours (as shown in Fig. 1), using different kinds of precursors, and the weight % of Co in the Co–Nx/C catalyst was low (0.14%) after washing off the Co nanoparticles (24, 25).
Fig. 2. TEM images of the samples.

(A) Co–Nx/C-800-AT, (B) Co–Nx/C-900-AT, (C) Co/N-C-SiO2-800, and (D) Co/N-C-800-BT.
X-ray powder diffraction (XRD) patterns of the samples were also measured. No XRD peaks of Co nanoparticles were observed in Co/N-C-SiO2-400 and Co/N-C-SiO2-600 (fig. S7), which is consistent with the TEM results. XRD patterns of Co/N-C-SiO2-800 (Fig. 3) and Co/N-C-SiO2-900 (fig. S6) showed three peaks at 2θ = 44.2°, 51.5°, and 75.9°, corresponding to the (111), (200), and (220) planes of metallic Co nanoparticles [Joint Committee on Powder Diffraction Standards (JCPDS) no. 15-0806], respectively (24). As shown in Fig. 3, XRD peaks of metallic Co nanoparticles were also observed in Co/N-C-800-BT, suggesting that Co nanoparticles remained after the removal of the silica in Co/N-C-SiO2-800 by NaOH. However, metallic Co peaks and the silica peak at 2θ around 22° disappeared in Co–Nx/C-800-AT and Co–Nx/C-900-AT (Fig. 3), revealing that the treatment of Co/N-C-SiO2-800 and Co/N-C-SiO2-900 by HF could simultaneously wash off both Co nanoparticles and silica. It is worth noting that a new and weak peak at 2θ around 43.7° was observed in Co–Nx/C-800-AT and Co–Nx/C-900-AT, which is the characteristic peak for Co–Nx (JCPDS no. 41-0943) (24, 25). Although the Co–Nx species has recently been believed to be the active sites for electrocatalytic reactions (24, 25), it has no specific structure. Cobalt phthalocyanine (Co2+ coordinating with four nitrogen atoms, as shown in fig. S1) was inactive for the hydrogenation of nitro compounds (Table 1, entry 2). However, the Co–Nx/C-800-AT catalyst showed high catalytic activity for this reaction. Meanwhile, Co–N4 moieties (Co2+ coordinating with four nitrogen atoms in the phthalocyanine ring, as shown in fig. S1) disappeared at a pyrolysis temperature of ≥800°C, as shown in UV-Vis spectra (fig. S3). Thus, we can conclude that the Co valence state in the Co–Nx/C-800-AT catalyst should most probably not be Co2+, which coordinates to N atoms. In addition, no Co nanoparticles were observed in the TEM image of the Co–Nx/C-800-AT catalyst. Thus, we speculate that metallic subnanometer-scale Co stabilized by nitrogen atoms is most probably the state of Co in the Co–Nx/C-800-AT catalyst (26).
Fig. 3. XRD patterns of the samples.
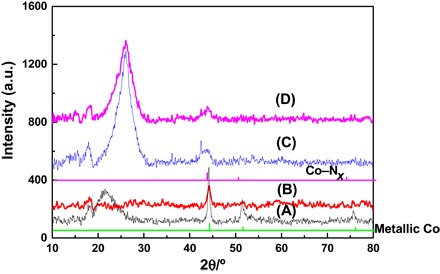
(A) Co/N-C-SiO2-800, (B) Co/N-C-800-BT, (C) Co–Nx/C-800-AT, and (D) Co–Nx/C-900-AT. a.u., arbitrary units.
Table 1. The results of nitrobenzene hydrogenation by H2 over different catalysts.
The reaction conditions are as follows: nitrobenzene, 1 mmol; H2O, 15 ml; temperature, 110°C; H2 pressure, 3.5 bar; 1.5 hours.
| Entry | Catalyst | Catalyst amount (mg) | Molar ratio of nitrobenzene to Co | Conversion (%) | Selectivity (%) |
| 1 | Co–Nx/C-600-AT | 40 | 409 | 9.0 | >99 |
| 2 | Cobalt phthalocyanine | 40 | 18 | — | — |
| 3 | Co–Nx/C-800-AT | 40 | 589 | 100 | >99 |
| 4 | Co–Nx/C-900-AT | 40 | 818 | 70.8 | >99 |
| 5 | N-C-800-AT | 40 | — | — | — |
| 6* | Co–Nx/C-800-AT | 40 | 589 | 47.1 | >99 |
| 7* | Co/N-C-800-BT | 40 | 14 | 54 | >99 |
*The reaction time was 0.5 hour.
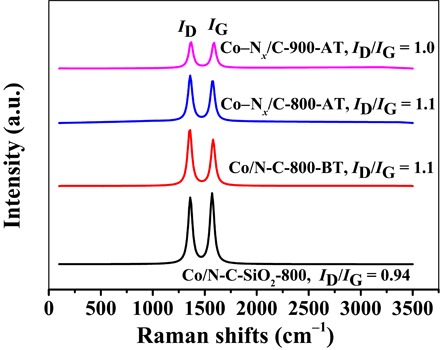
In addition, the graphite peak at 2θ = 26.4° (JCPDS no. 01-0646), which emerges in the XRD patterns of Co–Nx/C-800-AT and Co–Nx/C-900-AT, is due to the increase in the carbon content after the removal of Co nanoparticles and silica. This peak, with an interlayered spacing of 3.42 Å, was assigned to a turbostratic ordering of the carbon and nitrogen atoms in the graphite layers (27). Raman spectra of the four representative catalysts display a defect (D) band at 1360 cm−1 and a graphite (G) band at 1580 cm−1 (Fig. 4), corresponding to the disordered graphitic carbon and the graphitization degree, respectively (28). After the removal of silica, the intensity ratio of ID/IG increased from 0.94 in Co/N-C-SiO2-800 to 1.1 for both Co/N-C-800-BT and Co–Nx/C-800-AT, which indicated that the crystallographic defects in the graphitic carbon increased after the removal of silica. The intensity ratio of ID/IG in Co–Nx/C-800-AT was higher than that in Co–Nx/C-900-AT, suggesting that there were fewer defects in Co–Nx/C-900-AT. Less nitrogen, which can cause the distortion of the sp2 carbon lattice of graphene, was present in Co–Nx/C-900-AT (table S1) (24).
Fig. 4. Raman spectra of the samples.
X-ray photoelectron spectroscopy (XPS) experiments were performed to probe the valence state of Co in the three representative catalysts at 800°C (Fig. 5). The Co 2p3/2 peaks with binding energies (BEs) of 780.2 and 780.7 eV were observed for Co/N-C-SiO2-800 and Co/N-C-800-BT, respectively. The BE of the Co 2p3/2 peak around 780 eV can be assigned to cobalt oxides (29), which was due to the surface oxidation of metallic Co in Co/N-C-SiO2-800 and Co/N-C-800-BT during storage in the air (29). The Co 2p peak intensity of Co/N-C-800-BT was much stronger than that of Co/N-C-SiO2-800 because of the removal of silica. However, the Co 2p peak of Co–Nx/C-800-AT was too weak to be visible because of the removal of Co nanoparticles by HF. The higher resolution of the Co 2p spectra of Co–Nx/C-800-AT and Co–Nx/C-900-AT (fig. S8) showed that Co 2p3/2 peaks with a BE of around 781.2 eV were observed, characteristic of Co–Nx (30, 31). Figure S9 shows the high-resolution C 1s XPS spectra of Co/N-C-800-BT and Co–Nx/C-800-AT, which were deconvoluted into four peaks: 284.5 eV (C–C or C=C), 285.1 eV (C–N), 286.0 eV (C–O), and 288.3 eV (C–N=C) (32). The C 1s peak intensity of Co/N-C-800-BT was slightly weaker than that of Co–Nx/C-800-AT because of the presence of Co nanoparticles in Co/N-C-800-BT. As regards the N 1s XPS spectra (fig. S10), three kinds of nitrogen, including pyridinic N (398.4 eV), pyrrolic N (400.3 eV), and graphitic N (401.0 eV), were observed in both Co/N-C-800-BT and Co–Nx/C-800-AT catalysts (32). The peak intensity of N 1s of Co–Nx/C-800-AT was also slightly stronger than that of Co/N-C-800-BT.
Fig. 5. XPS spectra of the samples.
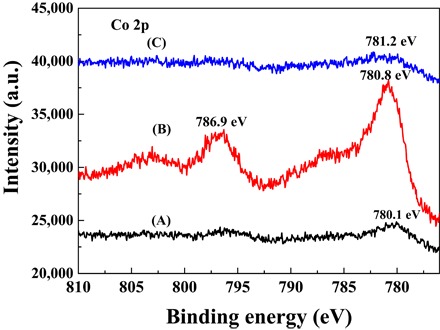
(A) Co/N-C-SiO2-800, (B) Co/N-C-800-BT, and (C) Co–Nx/C-800-AT.
Catalyst screening
In a preliminary study, screening of catalysts for the hydrogenation of nitrobenzene with H2 was studied. As presented in Table 1, the pyrolysis temperature showed a great effect on the catalytic activity of the Co–Nx/C-T-AT catalysts. Co–Nx/C-400-AT gave no conversion, and the conversion was only 9% over the Co–Nx/C-600-AT catalyst (Table 1, entry 1), suggesting that Co–N4 and Co–N2 moieties in Co–Nx/C-400-AT and Co–Nx/C-600-AT were not active for nitrobenzene hydrogenation. This was also confirmed by the use of cobalt phthalocyanine (Co–N4 moiety), which gave no conversion (Table 1, entry 2). Co–Nx/C-800-AT exhibited the highest catalytic activity with respect to 100% conversion, whereas Co–Nx/C-900-AT produced a lower conversion of 70.8% under identical conditions (Table 1, entry 3 versus entry 4). It is worth noting that aniline selectivity was >99% for all cases. As a control experiment, no conversion was observed over the metal-free N-C-800-AT catalyst (Table 1, entry 5), suggesting that Co was the active site for nitrobenzene hydrogenation. A Co–Nx species should be the active site for nitrobenzene hydrogenation from the results in entries 1 to 4. The lower nitrobenzene conversion over Co–Nx/C-900-AT as compared with Co–Nx/C-800-AT was due to the lower content of Co–Nx when the same amount of catalyst was used (table S1). This means that Co–Nx in the Co–Nx/C-800-AT was not stable, and it could decompose at a pyrolysis temperature of more than 800°C. A similar phenomenon was also reported by Zhang and co-workers (19). They also prepared a Co-N-C catalyst for the aerobic oxidative cross-coupling of primary and secondary alcohols to directly produce α,β-unsaturated ketones. They observed that the Co-N-C catalyst at a pyrolysis temperature of 800°C produced α,β-unsaturated ketone with a yield of 71%, whereas the yield decreased to 27% over the same amount of the Co-N-C catalyst at a pyrolysis temperature of 880°C. In addition, Co/N-C-800-BT was also used for this reaction. Co/N-C-800-BT produced a similar nitrobenzene conversion to Co–Nx/C-800-AT, but the Co weight content in Co/N-C-800-BT was 48 times higher than that in Co–Nx/C-800-AT (Table 1, entry 6 versus entry 7). These results once again revealed that the Co–Nx complex was highly active for the hydrogenation of nitrobenzene, showing much higher activity than Co nanoparticles in the Co/N-C-800-BT catalyst.
Hydrogenation of nitrobenzene with H2 under various conditions
Nitrobenzene hydrogenation was then performed under different conditions over the Co–Nx/C-800-AT catalyst. High conversion (91 to 100%) and high aniline selectivity (>99%) were produced in all tested solvents after 1.5 hours at 110°C and under a H2 pressure of 3.5 bar (Table 2, entries 1 to 7). Ethanol and water are highly desirable solvents because they are cheap, nontoxic, and environmentally benign. The Co–Nx/C-800-AT catalyst even showed high activity at 110°C and under a H2 pressure of 1 bar, affording 100% conversion and >99% high selectivity after 6 hours (Table 2, entry 8). The high catalytic activity of Co–Nx/C-800-AT inspired us to perform the reaction even under milder conditions (40°C and a H2 pressure of 1 bar). A high conversion of 98.7% and a selectivity of >99% were still obtained within 18 hours (Table 2, entry 9). Although some reports have claimed that the reduction of nitrobenzene could be performed even at room temperature with NaBH4 as the reducing agent (33), no other heterogeneous non-noble metal catalysts have been reported to be highly active toward the hydrogenation of nitrobenzene at a low H2 pressure of 1 bar and a low temperature of 40°C. Note that the mild reaction conditions (40°C and atmospheric H2 pressure) enable the use of common glass reactors, demonstrating a promising potential in industrial applications from an economical, environmental, and safety viewpoint. For comparison, the commercially available 5 wt % Pd/C catalyst was also used for the hydrogenation of nitrobenzene at 40°C and under a H2 pressure of 1 bar. The 5 wt % Pd/C catalysts showed lower catalytic activity with a nitrobenzene conversion of 68% with the same mole of Pd to the Co in 40 mg of Co–Nx/C-800-AT (Table 2, entry 10 versus entry 9). These results once again revealed that the Co–Nx/C-800-AT catalyst was highly active toward the hydrogenation of nitro compounds.
Table 2. The results of nitrobenzene hydrogenation with H2 under different conditions.
The reaction conditions are as follows: nitrobenzene, 1 mmol; Co–Nx/C-800-AT catalyst, 40 mg; and solvent, 15 ml.
| Entry | Solvent | Catalyst | H2 pressure (bar) | Temperature (°C) | Time (hours) | Conversion (%) | Selectivity (%) |
| 1 | H2O | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 100 | >99 |
| 2 | THF | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 95 | >99 |
| 3 | CH3CN | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 96 | >99 |
| 4 | EtOH | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 100 | >99 |
| 5 | Isopropanol | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 98 | >99 |
| 6 | Toluene | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 99 | >99 |
| 7 | Ethyl acetate | Co–Nx/C-800-AT | 3.5 | 110 | 1.5 | 91 | >99 |
| 8* | H2O | Co–Nx/C-800-AT | 1 | 110 | 6 | 100 | >99 |
| 9* | H2O | Co–Nx/C-800-AT | 1 | 40 | 18 | 98.7 | >99 |
| 10*† | H2O | Pd/C | 1 | 40 | 18 | 68 | >99 |
*Nitrobenzene (0.5 mmol) was used.
†The same molar of Pd to Co in 40 mg of Co–Nx/C-800-AT.
Mechanism and kinetic study
Two pathways are widely accepted for nitrobenzene hydrogenation (fig. S11) (34). Nitrobenzene-to-aniline conversion occurs via the hydroxyl amine intermediate (direct pathway; fig. S11, path A) or via the azoxybenzene intermediate (indirect pathway; fig. S11, path B). In our reaction, no intermediates were observed by gas chromatography (GC) at 110°C during the reaction process (fig. S12). Thus, we tried to observe the intermediates at a lower reaction temperature. A new and weak peak was observed in the GC spectrum at 60°C and under a H2 pressure of 50 bar (fig. S12), which was identified to be phenylhydroxylamine by comparison with the authentic phenylhydroxylamine and GC–mass spectrometry (MS). As shown in figs. S13 and S14, nitrobenzene conversion gradually increased with an increase in reaction temperature from 30°C to 60°C, whereas the molar percentage of phenylhydroxylamine increased from 30°C to 40°C and then decreased from 40°C to 60°C. These results indicate that phenylhydroxylamine becomes less stable at higher temperatures. Hydrogenation of azobenzene (a stable intermediate in path B) over the Co–Nx/C-800-AT catalyst did not produce aniline under the reaction conditions for the hydrogenation of nitrobenzene; thus, it indicated that hydrogenation of nitrobenzene over the Co–Nx/C-800-AT catalyst did not proceed via a “condensation way” but instead followed the “direct way” mechanism with phenylhydroxylamine as the intermediate.
A kinetic study was performed at 30°C, 40°C, and 50°C under a H2 pressure of 50 bar. Under this pressure, the H2 concentration can be considered constant (35). Thus, nitrobenzene hydrogenation could be considered as a pseudo–first-order reaction. Figure S15 shows a plot of ln(Ct/C0) versus time at 30°C, 40°C, and 50°C. The reaction rate constant k was determined to be 0.0040, 0.0059, and 0.012 min−1 for 30°C, 40°C, and 50°C, respectively. According to the Arrhenius plot, the activation energy of nitrobenzene hydrogenation with H2 over the Co–Nx/C-800-AT catalyst was calculated to be 44.8 kJ·mol−1, which was much lower than for that over the noble metal Au/ZrO2 catalyst (67.2 kJ·mol−1) (35).
Substrate scope
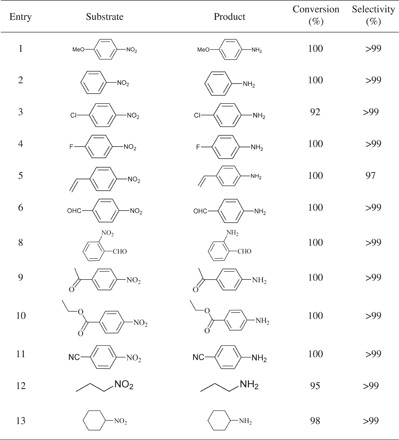
Besides the high activity of the Co–Nx/C-800-AT catalyst, the chemoselective reduction of nitro compounds is also of great importance. As shown in Table 3, the Co–Nx/C-800-AT catalyst showed high chemoselectivity toward the hydrogenation of substituted nitroarenes in the presence of other reducible groups (halogen, alkene, aldehyde, ketone, ester, and nitrile groups) (Table 3, entries 1 to 11). Moreover, the Co–Nx/C-800-AT catalyst also showed high activity toward nonactivated aliphatic and cyclic nitro compounds (Table 3, entries 12 and 13). In all cases, high conversions (92 to 100%) and high selectivities (≥97%) were produced.
Table 3. Chemoselective hydrogenation of different nitro compounds.
The reaction conditions are as follows: substrate, 1 mmol; Co–Nx/C-800-AT catalyst, 40 mg; H2O, 15 ml; H2 pressure, 3.5 bar; 1.5 hours.
Transfer hydrogenation and reductive coupling of nitro compounds
The excellent catalytic performance of Co–Nx/C-800-AT toward nitrobenzene hydrogenation with H2 inspired us to study the CTH of nitrobenzene with readily available hydrogen donors. CO and H2O can serve as a substitute for H2 via water gas shift reaction (CO + H2O↔H2 + CO2) (36). Although CO is cheaper than H2, there were a few examples on the reduction of nitro compounds with CO/H2O (37, 38). Excitingly, the Co–Nx/C-800-AT catalyst was active toward the reduction of nitrobenzene into aniline in H2O under a CO pressure of 5 bar, affording full conversion and a selectivity of >99% after 6 hours at 110°C (Table 4, entry 1).
Table 4. The results of transfer hydrogenation and reductive coupling of nitrobenzene.
Reaction was performed under a N2 pressure of 20 bar except entry 1. In all cases, nitrobenzene (1 mmol) and the Co–Nx/C-800-AT (40 mg) catalyst were used.
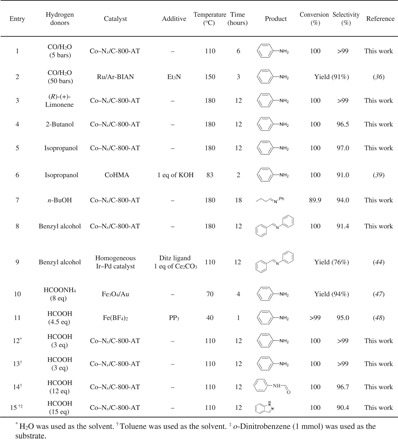
The reason for the excellent transfer hydrogenation of nitrobenzene with CO in water was that the Co–Nx/C-800-AT can effectively promote the water gas shift reaction (CO + H2O↔H2 + CO2), and the produced H2 was used in situ for the hydrogenation of nitrobenzene. We also conducted a control experiment on the reduction of nitrobenzene with CO in toluene. Toluene was freshly distilled to remove any water. As expected, no conversion was observed in this case, confirming that the transfer hydrogenation of nitrobenzene with CO/H2O over the Co–Nx/C-800-AT occurred via the water gas shift reaction. The Co–Nx/C-800-AT catalyst even exhibited much higher activity than some noble metal catalysts (38). For example, homogeneous ruthenium bis(arylimino)acenaphthene (Ru/Ar-BIAN) with 2 eq triethylamine (Et3N) produced aniline with a similar yield of 98% under harsh conditions (150°C and a CO pressure of 50 bar; Table 4, entry 2) (38). The naturally abundant (R)-(+)-limonene was studied as a representative hydrogen donor of the hydrocarbon compounds for the CTH of nitrobenzene. The reaction produced excellent results (Table 4, entry 3). There have been no other reports on the use of (R)-(+)-limonene as a hydrogen donor for this reaction. Alcohol, as one of the most common hydrogen donors, was also used for the CTH of nitrobenzene. We observed that the products were dependent on the type of alcohol. Aniline was produced in high yields (>95%) with secondary alcohols, such as isopropanol and 2-butanol, as the hydrogen donors after 12 hours at 180°C without any additives (Table 4, entries 4 and 5). Some other methods have also been reported for the CTH of nitrobenzene with isopropanol, but a base was generally required as additive (15, 39, 40). For example, 1 eq of KOH was required for the CTH of nitroarenes with isopropanol over cobalt(II)-substituted hexagonal mesoporous aluminophosphate molecular sieves (CoHMA) (Table 4, entry 6) (39). Obviously, our method is preferable because no additive was needed.
Imines were the main products via tandem reaction when primary alcohols were used as hydrogen donors (Table 4, entries 7 and 8). Synthesis of imines included the CTH of nitrobenzene to aniline and the subsequent condensation of aniline with the in situ formed aldehydes (fig. S16). However, the CTH of nitrobenzene with secondary alcohols could not generate the corresponding imines, possibly because of the steric hindrance of ketones from the dehydrogenation of secondary alcohols that block the subsequent condensation reaction. Imines are important chemicals with multiple applications, which are commonly synthesized from amines (41, 42). Very few examples of the synthesis of imines via the reductive coupling of nitro compounds with alcohols have been documented (43, 44). For example, Zanardi and co-workers (44) used a homogeneous Ir–Pd bimetallic catalyst with the 1,2,4-trimethyltriazolyldiylidene (ditz) ligand and Ce2CO3 (1 eq) as the additive for the reductive coupling of nitrobenzene with benzyl alcohol, affording a 76% yield of N-benzylideneaniline after 20 hours at 110°C (Table 4, entry 9). Compared with the method by Zanardi et al. (44), our method is attractive, because a higher N-benzylideneaniline yield of 91.4% was produced over the non-noble metal catalyst without base or ligands.
One of the distinct advantages of the Co–Nx/C-800-AT catalyst is that it is stable under acidic conditions. Compared with other hydrogen donors, formic acid has received much more interest (45) because it is a renewable product produced from biomass. In previous work, the CTH of nitro compounds with formic acid was mainly performed over noble metal catalysts with base as the additive (46, 47). For example, Fe3O4/Au catalyzed the hydrogenation of nitrobenzene to yield 92% aniline in ethanol with 8 eq of HCOONH4 (Table 4, entry 10) (47). Apparently, the use of non-noble catalysts without base would be attractive. To date, there is only one example of a base-free reduction of nitrobenzene with formic acid over a non-noble metal catalyst (Table 4, entry 10) (48). The homogeneous catalyst Fe(BF4)2·6H2O, together with the tetraphos ligand tris[(2-diphenylphosphino)-ethyl]phosphine (PP3), afforded >99% nitrobenzene conversion and 95% aniline yield in ethanol with 4.5 eq of HCOOH (Table 4, entry 11). Although this method did not use base as additive, the use of the difficult-to-prepare and expensive ligands with 4 mol % homogeneous catalyst resulted in a system through which it is difficult to recycle the catalyst and to purify the product. To our delight, nitrobenzene was quantitatively converted into aniline in water with 3 eq of HCOOH over the Co–Nx/C-800-AT catalyst after 12 hours at 110°C in water or toluene without any additive (Table 4, entries 12 and 13). Furthermore, this reaction has high atom efficiency because a stoichiometric amount of formic acid (3 eq) was used. Note that no base was required in our method. According to the references, the electronegative nitrogen atoms should act as a base to capture the H+ from formic acid to generate NH+. Then, it was activated by Co sites to generate the active Co-hydride, which was then used for the transfer hydrogenation of nitrobenzene (49). For example, Wang and co-workers (49) reported that the Pd/TiO2@CN catalyst for the transfer hydrogenation of vanillin with formic acid could produce >99.5% conversion of vanillin after 4 hours at 150°C. However, the Pd/TiO2 catalyst can only provide a very low conversion of 9.7% under the same conditions. These results also confirmed that the nitrogen atom acted as a base to promote the transfer hydrogenation with formic acid. Thus, both the nitrogen atoms and Co sites in the Co–Nx/C-800-AT were important in the transfer hydrogenation of nitrobenzene with formic acid as the hydrogen donor. Further increasing the amount of formic acid to 12 eq, N-phenylformamide was produced in a high yield of 96.7% via two consecutive steps: the CTH of nitrobenzene to aniline and the subsequent condensation of aniline with formic acid (fig. S17 and Table 4, entry 14). The high selectivity of N-phenylformamide is due to the fact that the carbonyl group was inert. The high efficiency of Co–Nx/C-800-AT toward the transformation of nitrobenzene into N-phenylformamide inspired us to use o-dinitrobenzene as the starting material to synthesize benzimidazole, which involves the reduction of one nitro group into one formamide group and the reduction of one nitro group into one amino group, as well as the final condensation of the amino group with the formamide group (fig. S18). Benzimidazole was produced in a high yield of 90.4% (Table 4, entry 15).
Stability of the Co–Nx/C-800-AT catalysts
Finally, the stability of Co–Nx/C-800-AT was investigated. Hydrogenation of nitrobenzene by H2 was used as the model reaction. After the reaction, the Co–Nx/C-800-AT catalyst was washed with ethanol and water, respectively. To avoid catalyst weight loss, we recovered the Co–Nx/C-800-AT catalyst by centrifugation. The moist catalyst was directly used for the next run under identical conditions. The reactivity was fully restored for up to eight runs (table S2). The reaction solution was analyzed by inductively coupled plasma (ICP)–atomic emission spectroscopy, and the Co content was below the limit. All these results suggested that Co–Nx/C-800-AT was highly stable.
DISCUSSION
In summary, a highly efficient method has been developed for the reduction and reductive coupling of nitro compounds into amines and their derivatives over a non-noble Co–Nx/C-800-AT catalyst. The Co–Nx/C-800-AT catalyst was generated by a practical and scalable two-step method via the pyrolysis of a cobalt phthalocyanine/silica composite and the subsequent leaching of Co nanoparticles and silica by 10 wt % HF. As revealed by different physical characterization techniques, the pyrolysis temperature markedly affected the structure of the Co–Nx/C-X-AT catalysts, and a Co–Nx complex was identified to be an active site in Co–Nx/C-800-AT and Co–Nx/C-900-AT. Co–Nx/C-800-AT exhibited an extremely high catalytic activity toward the reduction of nitro compounds. Under mild conditions (40°C and a H2 pressure of 1 bar), full nitrobenzene conversion and high aniline selectivity (>99%) were achieved in water. Co–Nx/C-800-AT also showed high catalytic activity and chemoselectivity to substituted nitroarenes with other reducible groups and nonactive aliphatic and cyclic nitro compounds. In addition, Co–Nx/C-800-AT also exhibited high catalytic activity toward the transfer hydrogenation of nitro compounds with some hydrogen donors without base or ligands as additives. Some significant nitrogen-containing compounds, including imines, formamide, and benzimidazole, were also produced from nitrobenzene via tandem reactions. The Co–Nx/C-800-AT catalyst exhibited excellent stability. We consider that, after acid treatment, the Co–Nx site was embedded in a C–N composite and was stabilized by nitrogen atoms. Thus, the nonprecious metal catalyst Co–Nx/C-800-AT is a promising potential alternative to noble metal catalysts for the industrial synthesis of amines and their derivates from nitro compounds because of the high activity, high selectivity, high stability, low cost, and environmental benignancy of the reaction. Furthermore, there is room to enlarge the scope of the Co–Nx/C catalyst for the green synthesis of a broad spectrum of fine chemicals.
MATERIALS AND METHODS
Catalyst preparation and characterization
Typically, colloidal silica (2.5 g, 40 wt %) and cobalt phthalocyanine (1.0 g) were added to a mixed solvent consisting of ethanol and N,N′-dimethylformamide (v/v, 3:1) and stirred at 25°C for 1 hour. After the evaporation of the solvents, the cobalt phthalocyanine/silica composite was pyrolyzed at 800°C (or 400°C, 600°C, or 900°C) for 2 hours under a N2 atmosphere with a ramp rate of 3°C min−1. The as-made powder (abbreviated as Co/N-C-SiO2-800) was treated with 10 wt % HF at 25°C for 12 hours to remove silica and Co nanoparticles, and the as-prepared catalyst was denoted as Co–Nx/C-800-AT.
For comparison, Co/N-C-SiO2-800 was also treated with 1 M NaOH at 90°C for 12 hours to remove silica, which was abbreviated as Co/N-C-800-BT. The metal-free mesoporous nitrogen-doped carbon (N/C-800) was prepared using cobalt-free phthalocyanine/silica composite. The methods used for catalyst characterization are provided in the Supplementary Materials.
Hydrogenation of nitro compounds with H2
Typically, Co–Nx/C-800-AT (40 mg), nitrobenzene (1 mmol), and H2O (15 ml) were charged in a 60-ml autoclave. After removal of air, the autoclave was charged with a H2 pressure of 3.5 bar at room temperature and then it was heated from room temperature to 110°C within 5 min and kept at 110°C for 1.5 hours with magnetic stirring at 1000 rpm. After cooling to room temperature, ethyl acetate was used to extract the organic chemicals from water. To ensure that the organic chemicals were fully extracted from water, we used 15 ml of ethyl acetate each time to repeat the extraction six times, and 4-chlorotoluene was then added as an internal standard. Products were identified by comparison of the retention time with the authentic chemicals and further confirmed by GC-MS (Agilent 7890A GC/5973 MS, HP-5 column). The content of each compound was quantitatively analyzed by GC via interpolation from calibration curves.
For the reactions in organic solvents, the internal standard (4-chlorotoluene) was added into the reaction mixture and then the reaction mixture was diluted with ethyl acetate to a certain volume. Then, the reaction mixture was analyzed by GC after filtration.
Transfer hydrogenation and reductive coupling of nitro compounds
For alcohols or hydrocarbons as hydrogen donors, they were also used as solvents. The procedure was almost the same as described above, but the reactor was purged with a N2 pressure of 10 bar. For formic acid as the hydrogen donor, the reaction was performed in water or toluene under a N2 pressure of 10 bar. For CO/H2O as the hydrogen donor, the reaction was performed in water under a CO pressure of 5 bar.
Supplementary Material
Acknowledgments
Funding: The project was supported by the National Natural Science Foundation of China (no. 21203252 & 21373275). Author contributions: P.Z. performed the catalyst preparation, characterizations (TGA, solid UV-Vis spectra, Raman spectra, and BET), and hydrogenation of nitro compounds with H2 as the reducing agent. L.J. participated in the catalyst preparation and characterizations (TEM, XRD, and ICP) and performed the transfer hydrogenation and reductive coupling of nitrobenzene. F.W. analyzed all reaction products by GC-MS. K.D. and K.L. helped in revising the manuscript. Z.Z. conceived the idea, designed the study, analyzed the data, and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1601945/DC1
fig. S1. Structure of cobalt phthalocyanine.
fig. S2. TGA of the cobalt phthalocyanine/silica composite under a N2 atmosphere.
fig. S3. Solid UV-Vis spectra of the samples after the pyrolysis of the cobalt phthalocyanine/silica composite at different temperatures.
fig. S4. TEM images of the Co/N-C-SiO2-X samples.
fig. S5. Particle size distribution of Co nanoparticles.
fig. S6. Higher-resolution TEM images of the Co/N-C-AT-X samples.
fig. S7. XRD patterns of the samples.
fig. S8. Higher-resolution Co 2p XPS spectra.
fig. S9. Higher-resolution C 1s XPS spectra.
fig. S10. Higher-resolution N 1s XPS spectra.
fig. S11. Reaction pathways of the reduction of nitrobenzene.
fig. S12. Time course of the molar percentage of each compound during the hydrogenation of the nitrobenzene process.
fig. S13. GC analysis of the hydrogenation of nitrobenzene over the Co/Nx-C-800-AT catalyst at low reaction temperatures.
fig. S14. Molar percentage of the samples at three different temperatures.
fig. S15. Plot of ln(Ct/C0) versus time for the reduction of nitrobenzene over the Co–Nx/C-800-AT catalyst at different temperatures.
fig. S16. Tandem reaction of nitrobenzene with primary amines to produce imines.
fig. S17. Reductive N-formylation of nitrobenzene to N-phenylformamide by formic acid.
fig. S18. Synthesis of benzimidazole with o-dinitrobenzene and formic acid.
table S1. The content of Co and N in the as-prepared catalysts.
table S2. Recycling results for the Co–Nx/C-800-AT catalyst.
REFERENCES AND NOTES
- 1.S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis (Wiley, 2001). [Google Scholar]
- 2.Adams J. P., Paterson J. R., Nitro and related compounds. J. Chem. Soc. 1, 3695–3705 (2000). [Google Scholar]
- 3.Yang Q., Chen Y. Z., Wang Z. U., Xu Q., Jiang H. L., One-pot tandem catalysis over Pd@MIL-101: Boosting the efficiency of nitro compound hydrogenation by coupling with ammonia borane dehydrogenation. Chem. Commun. 51, 10419–10422 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Downing R., Kunkeler P., van Bekkum H., Catalytic syntheses of aromatic amines. Catal. Today 37, 121–136 (1997). [Google Scholar]
- 5.Zinin N., Beschreibung einiger neuer organischer Basen, dargestellt durch die Einwirkung des Schwefelwasserstoffes auf Verbindungen der Kohlenwasserstoffe mit Untersalpetersäure. J. Prakt. Chem. 27, 140–153 (1842). [Google Scholar]
- 6.Sedghi R., Heravi M. M., Asadi S., Nazari N., Nabid M. R., Recently used nanocatalysts in reduction of nitroarenes. Curr. Org. Chem. 20, 696–734 (2016). [Google Scholar]
- 7.Kadam H. K., Tilve S. G., Advancement in methodologies for reduction of nitroarenes. RSC Adv. 5, 83391–83407 (2015). [Google Scholar]
- 8.Lara P., Philippot K., The hydrogenation of nitroarenes mediated by platinum nanoparticles: An overview. Catal. Sci. Technol. 4, 2445–2465 (2014). [Google Scholar]
- 9.Zhang S., Chang C. R., Huang Z. Q., Li J., Wu Z., Ma Y., Zhang Z., Wang Y., Qu Y., High catalytic activity and chemoselectivity of sub-nanometric Pd clusters on porous nanorods of CeO2 for hydrogenation of nitroarenes. J. Am. Chem. Soc. 138, 2629–2637 (2016). [DOI] [PubMed] [Google Scholar]
- 10.U. Siegrist, P. Baumeister, H. Blaser, M. Studer, Chemical Industries, F. Herkes, Ed. (Marcel Dekker, 1998), pp. 207–220. [Google Scholar]
- 11.Corma A., Serna P., Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 313, 332–334 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Mitsudome T., Mikami Y., Matoba M., Mizugaki T., Jitsukawa K., Kaneda K., Design of a silver–cerium dioxide core–shell nanocomposite catalyst for chemoselective reduction reactions. Angew. Chem. Int. Ed. 51, 136–139 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Wei H., Liu X., Wang A., Zhang L., Qiao B., Yang X., Huang Y., Miao S., Liu J., Zhang T., FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 5, 5634 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Shimizu K.-i., Heterogeneous catalysis for the direct synthesis of chemicals by borrowing hydrogen methodology. Catal. Sci. Technol. 5, 1412–1427 (2015). [Google Scholar]
- 15.Sarmah P. P., Dutta D. K., Chemoselective reduction of a nitro group through transfer hydrogenation catalysed by Ru0-nanoparticles stabilized on modified Montmorillonite clay. Green Chem. 14, 1086–1093 (2012). [Google Scholar]
- 16.Jia W.-G., Zhang H., Zhang T., Xie D., Ling S., Sheng E.-H., Half-sandwich ruthenium complexes with Schiff-base ligands: Syntheses, characterization, and catalytic activities for the reduction of nitroarenes. Organometallics 35, 503–512 (2016). [Google Scholar]
- 17.Westerhaus F. A., Jagadeesh R. V., Wienhöfer G., Pohl M.-M., Radnik J., Surkus A.-E., Rabeah J., Junge K., Junge H., Nielsen M., Brückner A., Beller M., Heterogenized cobalt oxide catalysts for nitroarene reduction by pyrolysis of molecularly defined complexes. Nat. Chem. 5, 537–543 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Jagadeesh R. V., Surkus A. E., Junge H., Pohl M. M., Radnik J., Rabeah J., Huan H., Schünemann V., Brückner A., Beller M., Nanoscale Fe2O3-based catalysts for selective hydrogenation of nitroarenes to anilines. Science 342, 1073–1076 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Zhang L. L., Wang A. Q., Wang W. T., Huang Y. Q., Liu X. Y., Miao S., Liu J. Y., Zhang T., Co-N-C catalyst for C-C coupling reactions: On the catalytic performance and active sites. ACS Catalysis 5, 6563–6572 (2015). [Google Scholar]
- 20.Aijaz A., Masa J., Rösler C., Xia W., Weide P., Botz A. J., Fischer R. A., Schuhmann W., Muhler M., Co@Co3O4 encapsulated in carbon nanotube-grafted nitrogen-doped carbon polyhedra as an advanced bifunctional oxygen electrode. Angew. Chem. Int. Ed. 55, 4087–4091 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Guo X., Shen D.-H., Li Y.-Y., Tian M., Liu Q., Guo C.-C., Liu Z.-G., Immobilization of metalloporphyrin on organosilicon microsphere mixed with ceria as a new catalyst for oxidation of cyclohexane. J. Mol. Catal. A Chem. 351, 174–178 (2011). [Google Scholar]
- 22.Lalande G., Côté R., Tamizhmani G., Guay D., Dodelet J., Dignard-Bailey L., Weng L., Bertrand P., Physical, chemical and electrochemical characterization of heat-treated tetracarboxylic cobalt phthalocyanine adsorbed on carbon black as electrocatalyst for oxygen reduction in polymer electrolyte fuel cells. Electrochim. Acta 40, 2635–2646 (1995). [Google Scholar]
- 23.Achar B. N., Lokesh K. S., Fohlen G. M., Kumar T. M., Characterization of cobalt phthalocyanine sheet polymer by gas chromatography mass spectrometry on its pyrolysis products. React. Funct. Polym. 63, 63–69 (2005). [Google Scholar]
- 24.Hou Y., Wen Z., Cui S., Ci S., Mao S., Chen J., An advanced nitrogen-doped graphene/cobalt-embedded porous carbon polyhedron hybrid for efficient catalysis of oxygen reduction and water splitting. Adv. Funct. Mater. 25, 872–882 (2015). [Google Scholar]
- 25.Wang Z.-L., Hao X.-F., Jiang Z., Sun X.-P., Xu D., Wang J., Zhong H.-X., Meng F.-L., Zhang X.-B., C and N hybrid coordination derived Co–C–N complex as a highly efficient electrocatalyst for hydrogen evolution reaction. J. Am. Chem. Soc. 137, 15070–15073 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Wang K., Wang R., Li H., Wang H., Mao X., Linkov V., Ji S., N-doped carbon encapsulated Co3O4 nanoparticles as a synergistic catalyst for oxygen reduction reaction in acidic media. Int. J. Hydrogen Energy 40, 3875–3882 (2015). [Google Scholar]
- 27.Yang Y., Jia L., Hou B., Li D., Wang J., Sun Y., The correlation of interfacial interaction and catalytic performance of N-doped mesoporous carbon supported cobalt nanoparticles for Fischer–Tropsch synthesis. J. Phys. Chem. C 118, 268–277 (2013). [Google Scholar]
- 28.Kudin K. N., Ozbas B., Schniepp H. C., Prud’Homme R. K., Aksay I. A., Car R., Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 8, 36–41 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Zheng B., Wang J., Wang F. B., Xia X.-H., Low-loading cobalt coupled with nitrogen-doped porous graphene as excellent electrocatalyst for oxygen reduction reaction. J. Mater. Chem. A 2, 9079–9084 (2014). [Google Scholar]
- 30.Morozan Adina, Jégou Pascale, Jousselme Bruno, Palacin Serge, Electrochemical performance of annealed cobalt–benzotriazole/CNTs catalysts towards the oxygen reduction reaction. Phys. Chem. Chem. Phys. 13, 21600–21607 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Arechederra R. L., Artyushkova K., Atanassov P., Minteer S. D., Growth of phthalocyanine doped and undoped nanotubes using mild synthesis conditions for development of novel oxygen reduction catalysts. ACS Appl. Mater. Interfaces 2, 3295–3302 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Xiao J., Chen C., Xi J., Xu Y., Xiao F., Wang S., Yang S., Core–shell Co@Co3O4 nanoparticle-embedded bamboo-like nitrogen-doped carbon nanotubes (BNCNTs) as a highly active electrocatalyst for the oxygen reduction reaction. Nanoscale 7, 7056–7064 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Feng J., Handa S., Gallou F., Lipshutz B. H., Safe and selective nitro group reductions catalyzed by sustainable and recyclable Fe/ppm Pd nanoparticles in water at room temperature. Angew. Chem. Int. Ed. 55, 8979–8983 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Mahata A., Rai R. K., Choudhuri I., Singh S. K., Pathak B., Direct vs. indirect pathway for nitrobenzene reduction reaction on a Ni catalyst surface: A density functional study. Phys. Chem. Chem. Phys. 16, 26365–26374 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Gómez S., Torres C., García Fierro J. L., Apesteguía C. R., Reyes P., Hydrogenation of nitrobenzene on Au/ZrO2 catalysts. J. Chil. Chem. Soc. 57, 1194–1198 (2012). [Google Scholar]
- 36.Ambrosi A., Denmark S. E., Harnessing the power of the water-gas shift reaction for organic synthesis. Angew. Chem. Int. Ed. 55, 12164–12189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He L., Wang L.-C., Sun H., Ni J., Cao Y., He H.-Y., Fan K.-N., Efficient and selective room-temperature gold-catalyzed reduction of nitro compounds with CO and H2O as the hydrogen source. Angew. Chem. Int. Ed. 48, 9538–9541 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Viganò M., Ragaini F., Buonomenna M. G., Lariccia R., Caselli A., Gallo E., Cenini S., Jansen J. C., Drioli E., Catalytic polymer membranes under forcing conditions: Reduction of nitrobenzene by CO/H2O catalyzed by ruthenium bis(arylimino)acenaphthene complexes. ChemCatChem 2, 1150–1164 (2010). [Google Scholar]
- 39.Mohapatra S. K., Sonavane S. U., Jayaram R. V., Selvam P., Heterogeneous catalytic transfer hydrogenation of aromatic nitro and carbonyl compounds over cobalt(II) substituted hexagonal mesoporous aluminophosphate molecular sieves. Tetrahedron Lett. 43, 8527–8529 (2002). [Google Scholar]
- 40.Hohloch S., Suntrup L., Sarkar B., Arene–ruthenium (II) and –iridium(III) complexes with “click”-based pyridyl-triazoles, bis-triazoles, and chelating abnormal carbenes: Applications in catalytic transfer hydrogenation of nitrobenzene. Organometallics 32, 7376–7385 (2013). [Google Scholar]
- 41.Tamura M., Tomishige K., Redox properties of CeO2 at low temperature: The direct synthesis of imines from alcohol and amine. Angew. Chem. Int. Ed. 54, 864–867 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Chen B., Wang L., Gao S., Recent advances in aerobic oxidation of alcohols and amines to imines. ACS Catal. 5, 5851–5876 (2015). [Google Scholar]
- 43.Chen J., Huang S., Lin J., Su W., Recyclable palladium catalyst for facile synthesis of imines from benzyl alcohols and nitroarenes. Appl. Catal. A 470, 1–7 (2014). [Google Scholar]
- 44.Zanardi A., Mata J. A., Peris E., One-pot preparation of imines from nitroarenes by a tandem process with an Ir–Pd heterodimetallic catalyst. Chem. Eur. J. 16, 10502–10506 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Gilkey M. J., Xu B., Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 6, 1420–1436 (2016). [Google Scholar]
- 46.Sorribes I., Wienhöfer G., Vicent C., Junge K., Llusar R., Beller M., Chemoselective transfer hydrogenation to nitroarenes mediated by cubane-type Mo3S4 cluster catalysts. Angew. Chem. Int. Ed. 51, 7794–7798 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Gawande M. B., Rathi A. K., Tucek J., Safarova K., Bundaleski N., Teodoro O. M. N. D., Kvitek L., Varma R. S., Zboril R., Magnetic gold nanocatalyst (nanocat-Fe–Au): Catalytic applications for the oxidative esterification and hydrogen transfer reactions. Green Chem. 16, 4137–4143 (2014). [Google Scholar]
- 48.Wienhöfer G., Sorribes I., Boddien A., Westerhaus F., Junge K., Junge H., Beller M., General and selective iron-catalyzed transfer hydrogenation of nitroarenes without base. J. Am. Chem. Soc. 133, 12875–12879 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Wang L., Zhang B. S., Meng X. J., Su D. S., Xiao F. S., Hydrogenation of biofuels with formic acid over a palladium-based ternary catalyst with two types of active sites. ChemSusChem 7, 1537–1541 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/2/e1601945/DC1
fig. S1. Structure of cobalt phthalocyanine.
fig. S2. TGA of the cobalt phthalocyanine/silica composite under a N2 atmosphere.
fig. S3. Solid UV-Vis spectra of the samples after the pyrolysis of the cobalt phthalocyanine/silica composite at different temperatures.
fig. S4. TEM images of the Co/N-C-SiO2-X samples.
fig. S5. Particle size distribution of Co nanoparticles.
fig. S6. Higher-resolution TEM images of the Co/N-C-AT-X samples.
fig. S7. XRD patterns of the samples.
fig. S8. Higher-resolution Co 2p XPS spectra.
fig. S9. Higher-resolution C 1s XPS spectra.
fig. S10. Higher-resolution N 1s XPS spectra.
fig. S11. Reaction pathways of the reduction of nitrobenzene.
fig. S12. Time course of the molar percentage of each compound during the hydrogenation of the nitrobenzene process.
fig. S13. GC analysis of the hydrogenation of nitrobenzene over the Co/Nx-C-800-AT catalyst at low reaction temperatures.
fig. S14. Molar percentage of the samples at three different temperatures.
fig. S15. Plot of ln(Ct/C0) versus time for the reduction of nitrobenzene over the Co–Nx/C-800-AT catalyst at different temperatures.
fig. S16. Tandem reaction of nitrobenzene with primary amines to produce imines.
fig. S17. Reductive N-formylation of nitrobenzene to N-phenylformamide by formic acid.
fig. S18. Synthesis of benzimidazole with o-dinitrobenzene and formic acid.
table S1. The content of Co and N in the as-prepared catalysts.
table S2. Recycling results for the Co–Nx/C-800-AT catalyst.