

Abstract
To analyze next-generation sequencing data, multivariate functional linear models are developed for a meta-analysis of multiple studies to connect genetic variant data to multiple quantitative traits adjusting for covariates. The goal is to take the advantage of both meta-analysis and pleiotropic analysis in order to improve power and to carry out a unified association analysis of multiple studies and multiple traits of complex disorders. Three types of approximate F -distributions based on Pillai–Bartlett trace, Hotelling–Lawley trace, and Wilks's Lambda are introduced to test for association between multiple quantitative traits and multiple genetic variants. Simulation analysis is performed to evaluate false-positive rates and power of the proposed tests. The proposed methods are applied to analyze lipid traits in eight European cohorts. It is shown that it is more advantageous to perform multivariate analysis than univariate analysis in general, and it is more advantageous to perform meta-analysis of multiple studies instead of analyzing the individual studies separately. The proposed models require individual observations. The value of the current paper can be seen at least for two reasons: (a) the proposed methods can be applied to studies that have individual genotype data; (b) the proposed methods can be used as a criterion for future work that uses summary statistics to build test statistics to meta-analyze the data.
Introduction
Meta-analysis of multiple studies and pleiotropy analysis of multiple traits are two areas in association studies that recently have received extensive attention in the literature.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 To our knowledge, meta-analysis and pleiotropy analysis have been performed separately so far, and there are no gene-based meta-analysis methods for combining multiple studies together and for carrying out a unified pleiotropy analysis. Here, multivariate functional linear models (MFLM) are developed to connect genetic variant data to multiple quantitative traits adjusting for covariates in a meta-analysis context. The goal is to take the advantage of both meta-analysis and pleiotropy analysis in order to improve power and to carry out a unified analysis of multiple studies and multiple quantitative traits of complex disorders.
A noticeable feature of next-generation sequencing data is that dense panels of genetic variants are available via high-throughput sequencing technology, and so we face high-dimension genetic data.11, 12, 13, 14 The genetic data can consist of rare variants, or common variants, or a combination of the two, where the rare variants' minor allele frequencies (MAFs) are less than 0.01∼0.05. The high dimensionality of genetic data and the presence of dense rare variants raise huge challenges, and properly dealing with the high dimensionality and rare variants is one priority of statistical research in recent years.15
In our previous research as well as research from other groups, functional data techniques were used to reduce the dimensionality of genetic data and to build fixed effect functional regression models for association analysis of quantitative, dichotomous, and survival traits.10, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 In most cases, it was shown that the functional regression test statistics perform better than sequence kernel association test (SKAT), its optimal unified test (SKAT-O), and a combined sum test of rare and common variant effect (SKAT-C) of mixed models.4, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 30, 31, 32, 33 Specifically, mixed model-based SKAT/SKATO/SKAT-C performs well when (a) the number of causal variants is large and (b) each causal variant contributes a small amount to the traits, as the assumption of mixed models is satisfied under these circumstances.7, 21, 34 In most cases, however, fixed models perform better since the causal variants of complex disorders can be common or rare or a combination of the two and some causal variants may have relatively large effects.10, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 If the number of causal variants is large and each causal variant contributes a small amount to the traits, it would be hard to show association as the power of a test can be low.35 One may want to note that SKAT and SKAT-O were shown to have higher power than burden tests, which is another main method to analyze rare variants.4, 32, 36, 37, 38 Thus, fixed models can be useful in association studies of complex traits.
As functional regression models perform well in most cases, we are motivated to extend them to meta-analysis of pleiotropy traits. For individual studies, MFLM were built to perform pleiotropy analysis between multiple genetic variants and multiple quantitative traits adjusting for covariates in Wang et al.10 Similarly, functional linear models were developed to perform meta-analysis of a univariate quantitative trait in Fan et al.18 In this paper, we build MFLM to analyze multiple traits of multiple studies and introduce related approximate F-distributed test statistics to test for association based on multivariate analysis theory. The proposed methods are applied to analyze lipid traits in eight European cohorts. Simulation analysis is performed to evaluate the false-positive rates and power of the proposed tests.
Materials and methods
Consider a meta-analysis with L studies in a genomic region. For the l-th study, we assume that there are nl individuals who are sequenced in the region at ml variants. For each individual, we assume there are J quantitative trait phenotypes, J≥1. In this article, the research goal is to model association between the ml genetic variants and the J phenotypic traits by combining all the L studies as a whole. We assume that the ml variants are located with ordered physical positions 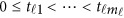 . To make the notation simpler, we normalized the region
. To make the notation simpler, we normalized the region 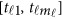 to be [0, 1]. For the i-th individual in the l-th study, let ylij denote her/his j-th quantitative trait (j=1,2,⋯,J),
to be [0, 1]. For the i-th individual in the l-th study, let ylij denote her/his j-th quantitative trait (j=1,2,⋯,J), 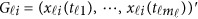 denote her/his genotypes of the ml variants, and
denote her/his genotypes of the ml variants, and 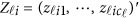 denote her/his
denote her/his 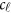 covariates. Hereafter, ′ denotes the transpose of a vector or matrix. For the genotypes, we assume that
covariates. Hereafter, ′ denotes the transpose of a vector or matrix. For the genotypes, we assume that 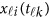 (=0,1,2) is the number of minor alleles of individual i at the k-th variant.
(=0,1,2) is the number of minor alleles of individual i at the k-th variant.
Multivariate functional linear models
We view the i-th individual's genotype data as a genetic variant function (GVF) 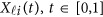 from the l-th study. To relate the GVF to the phenotypic traits adjusting for covariates, we consider the following MFLM for
from the l-th study. To relate the GVF to the phenotypic traits adjusting for covariates, we consider the following MFLM for 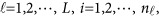
The notations used in the model (1) are defined below
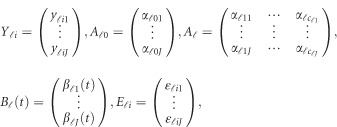 |
where 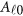 is a vector of overall means,
is a vector of overall means, 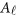 is a
is a 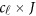 matrix of regression coefficients of covariates,
matrix of regression coefficients of covariates, 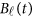 is a vector of genetic effect functions
is a vector of genetic effect functions 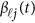 , and
, and 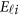 is a vector of error terms. For each pair of land i, the error vector
is a vector of error terms. For each pair of land i, the error vector 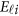 is normally distributed with a mean vector of zeros and a J × J variance–covariance matrix Σ. Moreover,
is normally distributed with a mean vector of zeros and a J × J variance–covariance matrix Σ. Moreover, 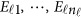 are assumed to be independent.
are assumed to be independent.
Expansion of Genetic Effect Function
The genetic effect functions 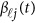 of
of 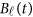 are assumed to be continuous/smooth functions of the position t. One may expand it by B-spline or Fourier basis functions. Formally, let us expand the genetic effect functions
are assumed to be continuous/smooth functions of the position t. One may expand it by B-spline or Fourier basis functions. Formally, let us expand the genetic effect functions 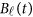 by a series of Kβ basis functions
by a series of Kβ basis functions 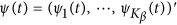 as
as
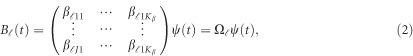 |
where 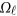 is a
is a 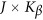 matrix of coefficients
matrix of coefficients 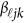 . We consider two types of basis functions: (1) the B-spline basis:
. We consider two types of basis functions: (1) the B-spline basis: 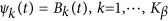 ; and (2) the Fourier basis:
; and (2) the Fourier basis: 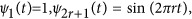 and
and 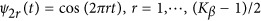 .39, 40, 41, 42
.39, 40, 41, 42
Estimation of GVF
To estimate the GVFs 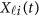 from the genotypes
from the genotypes 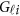 , we use an ordinary linear square smoother.16, 17, 18, 19, 20, 42, 43 Let φk(t), k=1, ⋯, K, be a series of K basis functions, such as the B-spline basis and Fourier basis functions. Denote φ(t)=(φ1(t), ⋯, φK(t))′. Let Φ denote the ml by K matrix containing the values
, we use an ordinary linear square smoother.16, 17, 18, 19, 20, 42, 43 Let φk(t), k=1, ⋯, K, be a series of K basis functions, such as the B-spline basis and Fourier basis functions. Denote φ(t)=(φ1(t), ⋯, φK(t))′. Let Φ denote the ml by K matrix containing the values 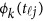 , where
, where 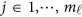 . Using the discrete realizations
. Using the discrete realizations 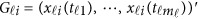 , we may estimate the GVF
, we may estimate the GVF 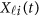 using an ordinary linear square smoother as follows:42
using an ordinary linear square smoother as follows:42
Revised MFLM
Replacing 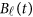 by the expansion (2) and
by the expansion (2) and 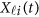 in the MFLM (1) by
in the MFLM (1) by 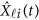 in (3), we have a revised multivariate linear regression model
in (3), we have a revised multivariate linear regression model
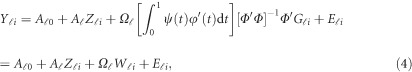 |
where 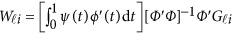 . In the above revised regression model, one needs to calculate
. In the above revised regression model, one needs to calculate 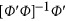 and
and 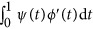 to get
to get 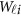 . In the statistical computing environment R, there are readily available R packages to calculate them.43
. In the statistical computing environment R, there are readily available R packages to calculate them.43
Dealing with missing genotype data
If some genotype data are missing, the estimation (3) can be modified to estimate GVF 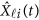 . For instance, there is no genotype information at the first variant for the i-th individual, ie, we only have
. For instance, there is no genotype information at the first variant for the i-th individual, ie, we only have 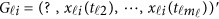 . Let Φ1 denote the
. Let Φ1 denote the 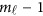 by K matrix containing the values
by K matrix containing the values 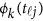 , where
, where 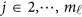 . Then, we may revise the estimation (3) as
. Then, we may revise the estimation (3) as
Note that the estimation (5) only depends on the available genotype data 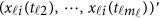 . Hence, each individual's GVF is estimated by his/her own data. This is one advantage of functional data analysis, which can be useful in practice. Using the estimation (5), one may revise the model (4) accordingly.
. Hence, each individual's GVF is estimated by his/her own data. This is one advantage of functional data analysis, which can be useful in practice. Using the estimation (5), one may revise the model (4) accordingly.
Beta-smooth-only MFLM
Model (1) is a theoretical MFLM.42 For analysis of dense genetic data, one may use a simplified MFLM as follows
where 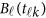 is a vector of the genetic effects at position
is a vector of the genetic effects at position 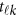 for the
for the  -th study, and the other terms are the same as those in the general MFLM (1).
-th study, and the other terms are the same as those in the general MFLM (1).
In model (6), 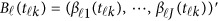 is a vector of the genetic effects at the position
is a vector of the genetic effects at the position 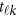 . We assume that
. We assume that 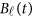 is a vector of genetic effect functions
is a vector of genetic effect functions 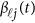 of the physical position t. Therefore,
of the physical position t. Therefore, 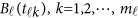 are the values of vector
are the values of vector 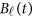 at the
at the 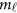 physical positions. The genetic effect functions
physical positions. The genetic effect functions 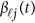 are assumed to be smooth. One may expand it by B-spline or Fourier basis functions. Replacing
are assumed to be smooth. One may expand it by B-spline or Fourier basis functions. Replacing 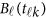 by expansion (2), model (6) can be revised as
by expansion (2), model (6) can be revised as
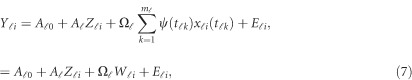 |
where 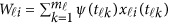 . In model (6) and its revised version (7), we use the raw genotype data
. In model (6) and its revised version (7), we use the raw genotype data 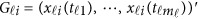 directly in the analysis. The genetic effect vector
directly in the analysis. The genetic effect vector 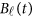 is assumed to be smooth or continuous. Hence, the models are called beta-smooth only.
is assumed to be smooth or continuous. Hence, the models are called beta-smooth only.
Dealing with Missing Genotype Data
If some genotype data are missing, eg, we only have 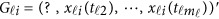 and
and 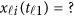 is missing, we may revise the MFLM (6) as
is missing, we may revise the MFLM (6) as
Again, the revised MFLM (8) only depends on the available genotype data 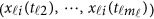 , and it can be revised accordingly to be a form of model (7) by expansion (2) as
, and it can be revised accordingly to be a form of model (7) by expansion (2) as
Traditional additive effect multivariate linear models
Traditionally, an additive effect model can be used to analyze the relation between the trait and the 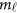 variants in the
variants in the  -study as Jung et al.44 and Anderson45
-study as Jung et al.44 and Anderson45
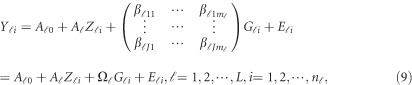 |
where 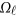 is a
is a 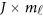 matrix of coefficients
matrix of coefficients 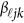 , which is the additive genetic effect of variant k for the j-th trait in the
, which is the additive genetic effect of variant k for the j-th trait in the  -th study, and the other terms are similar to those in the MFLM (1) and (6). There is only one difference between model (6) and model (9), ie, the genetic effect coefficients
-th study, and the other terms are similar to those in the MFLM (1) and (6). There is only one difference between model (6) and model (9), ie, the genetic effect coefficients 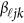 in model (9) do not depend on the physical position
in model (9) do not depend on the physical position 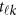 , whereas
, whereas 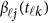 in model (6) depends on the physical position
in model (6) depends on the physical position 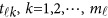 .
.
Approximate F-distributed test statistics
Consider the revised regression models (4), (7), and the multivariate linear model (9), which model the genetic effect of the J phenotypic traits simultaneously adjusting for covariates by combining the L studies together. First, assume that the genetic effects among the L studies are different/heterogeneous. In the test of association between the 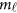 genetic variants and the J quantitative traits simultaneously, the null hypothesis is
genetic variants and the J quantitative traits simultaneously, the null hypothesis is 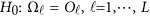 , where
, where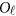 is a zero J × Kβ matrix OJ × Kβ for models (4) and (7) or a zero
is a zero J × Kβ matrix OJ × Kβ for models (4) and (7) or a zero 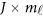 matrix
matrix 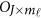 for model (9). We may test the null H0 by approximate F-distribution tests based on Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda using standard statistical approaches.45, 46 The approximate F-distributed test statistic is denoted as heterogeneous F-approximation test statistics (Het-F).
for model (9). We may test the null H0 by approximate F-distribution tests based on Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda using standard statistical approaches.45, 46 The approximate F-distributed test statistic is denoted as heterogeneous F-approximation test statistics (Het-F).
Consider the revised models (4) and (7). If the genetic effects are homogeneous, ie, 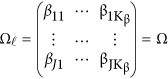 , we may test the association between the genetic variants and the J quantitative traits by testing a simplified null H0:Ω=OJ × Kβ. The null H0 can be tested by approximate F-distribution tests based on Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda using standard statistical approaches. The approximate F-distributed statistic is denoted as Hom-F.
, we may test the association between the genetic variants and the J quantitative traits by testing a simplified null H0:Ω=OJ × Kβ. The null H0 can be tested by approximate F-distribution tests based on Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda using standard statistical approaches. The approximate F-distributed statistic is denoted as Hom-F.
Assume that each individual of the L studies is sequenced at the same variants located at 0≤t1<⋯<tm and so 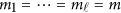 . In addition, assume that the genetic effects are homogenous. Let us denote
. In addition, assume that the genetic effects are homogenous. Let us denote 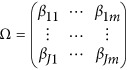 . Then, the model (9) is simplified as
. Then, the model (9) is simplified as
The null hypothesis of no association between the genetic variants and the quantitative traits is H0:Ω=OJ × m. The corresponding approximate F-distributed test statistic is denoted as Hom-F.
If there is only one study, ie, L=1, the approximate F-distribution tests are equivalent to those of Wang et al.10 and Het-F is the same as Hom-F. If we only have one quantitative trait, ie, J=1, the three approximate F-distribution tests based on Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda are equivalent to the F-test statistics of the standard multiple linear regression. The models proposed in this article and the related approximate F-distribution tests extend the models and the F-test statistics in Fan et al.18
In practice, we find that the results of the three approximate F-distribution tests based on Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda are similar to each other.10 In this article, we only report the results of approximate F-distribution tests based on Pillai-Bartlett trace.
Parameters of Functional Data Analysis
In the data analysis and simulations, we used two functions from the fda R package to create the basis:
Basis=create.bspline.basis(norder=order, nbasis=bbasis)
basis=create.fourier.basis(c(0,1), nbasis=fbasis)
The three parameters were taken as order=4, bbasis=15, fbasis=25 in all data analysis and simulations. To make sure that the results are valid and stable, we tried a wide range of parameters: (1) 10≤K=Kβ≤23 for the heterogeneous genetic effect model and (2) 10≤K=Kβ≤29 for the homogeneous genetic effect model. The results are similar to each other.
Results
A simulation study
To evaluate the performance of the proposed MFLM, we carried out simulation analyses for two cases: (1) the variants are all rare; (2) some variants are rare and some are common. Simulations were performed for three scenarios listed in Table 4 in Supplementary Materials.4, 18 For scenarios 1 and 2, we used the European-like (EUR) sequence data used in Lee et al.32 For scenario 3, we used both the EUR and African-American-like (AA) sequence data. Specifically, the EUR sequence data were generated using COSI's calibrated best-fit models, and the generated European haplotypes mimick CEPH Utah individuals with ancestry from northern and western Europe in terms of site frequency spectrum and linkage disequilibrium (LD) pattern (Figure 4 in Schaffner et al.47, 48). Similarly, the AA sequence data mimic individuals with 20:80 mixture of Europeans and Africans, together with parameters calibrated to model realistic demographic history (including bottleneck, population expansion, and migration events). The EUR sequence data included 10 000 chromosomes covering 1 Mb regions, and the AA sequence data included 45 000 chromosomes covering 0.1 Mb regions. Genetic regions of 3 kb length were randomly selected in the simulations for type I error and power calculations.
Type I error simulations
To evaluate the type I error rates of the proposed MFLM and related tests, we generated phenotype data sets by using the model
Three scenarios of covariates are given in Supplementary Table S1, in which three covariates are considered: z1 is a dichotomous covariate taking values 0 and 1 with a probability of 0.5, z2 and z3 are continuous covariates from a standard normal distribution N(0,1). The vector of error terms 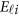 in model (11) follows a normal distribution with a mean vector of 0 and a 3 × 3 variance–covariance matrix
in model (11) follows a normal distribution with a mean vector of 0 and a 3 × 3 variance–covariance matrix
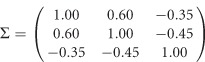 |
The 3 × 3 variance–covariance matrix Σ is taken from an empirical analysis of the three traits of the Trinity Students Study from Wang et al.10 For scenario 1 in Supplementary Table S1, the covariate regression coefficients are given by
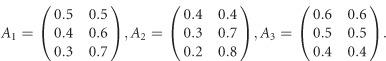 |
For scenarios 2 and 3 in Supplementary Table S1, the covariate regression coefficients are given by
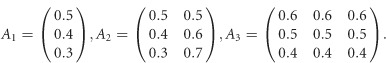 |
To obtain genotype data, 3 kb subregions were randomly selected in the 1 Mb region of EUR-like data and the 0.1 Mb region of AA-like data. The ordered genotypes were these SNPs in the 3 kb subregions. Note that the trait values are not related to the genotypes, and so the null hypothesis holds. The sample sizes were 1600 (study 1), 2200 (study 2), and 3200 (study 3). The simulation settings are summarized in Supplementary Table S1. For each sample size combination, 1.2 × 106 phenotype–genotype data sets were generated to fit the proposed models and to calculate the test statistics and related P-values. Then, an empirical type I error rate was calculated as the proportion of 1.2 × 106 P-values that were smaller than a given α level (ie, 0.05, 0.01, 0.001, and 0.0001, respectively).
Empirical power simulations
To evaluate the power of the proposed MFLM and related tests, we simulated data sets under the alternative hypothesis by randomly selecting 3 kb subregions to obtain causal variants for the phenotype values as follows. Once a 3 kb subregion was selected, a subset of 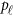 causal variants located in the 3 kb subregion for the
causal variants located in the 3 kb subregion for the  -th study was then randomly selected to obtain ordered genotypes
-th study was then randomly selected to obtain ordered genotypes 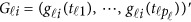 . Then, we generated the quantitative traits by
. Then, we generated the quantitative traits by
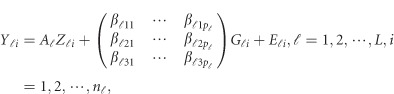 |
where 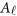 and
and 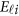 are the same as in the type I error model (11), and the βs are additive effect for the causal variants defined as follows. We used
are the same as in the type I error model (11), and the βs are additive effect for the causal variants defined as follows. We used 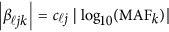 , where MAFk was the MAF of the k-th variant. Three genetic effect scenarios were used to perform power calculations: (1) all causal variants had positive effects; (2) 20%/80% causal variants had negative/positive effects; (3) 50%/50% causal variants had negative/positive effects. As in Fan et al.18 and Lee et al.,4 three different settings were considered: 5, 10, and 20% of variants in the 3 kb subregion are chosen as causal variants. When 5, 10, and 20% of the variants were causal, two parameter settings of genetic effects were considered for
, where MAFk was the MAF of the k-th variant. Three genetic effect scenarios were used to perform power calculations: (1) all causal variants had positive effects; (2) 20%/80% causal variants had negative/positive effects; (3) 50%/50% causal variants had negative/positive effects. As in Fan et al.18 and Lee et al.,4 three different settings were considered: 5, 10, and 20% of variants in the 3 kb subregion are chosen as causal variants. When 5, 10, and 20% of the variants were causal, two parameter settings of genetic effects were considered for 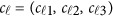 : (1) homogeneous and (2) heterogeneous (Supplementary Table S2). In the homogeneous case, the genetic effects are the same for the three studies, ie, c1=c2=c3. In the heterogeneous case, the genetic effects are different for the three studies, ie, c2=c1+(0.15,0.15,0.15),c3=c1−(0.15,0.15,0.15). For each setting, 1000 data sets were simulated to calculate empirical power as the proportion of P-values, which are smaller than an α=0.0001 level.
: (1) homogeneous and (2) heterogeneous (Supplementary Table S2). In the homogeneous case, the genetic effects are the same for the three studies, ie, c1=c2=c3. In the heterogeneous case, the genetic effects are different for the three studies, ie, c2=c1+(0.15,0.15,0.15),c3=c1−(0.15,0.15,0.15). For each setting, 1000 data sets were simulated to calculate empirical power as the proportion of P-values, which are smaller than an α=0.0001 level.
Type I error simulation results
The empirical type I error rates are reported in Supplementary Table S3 when the variants are only rare and in Supplementary Table S4 when some variants are rare and some are common. For each entry of empirical type I error rates, we generated 1.2 × 106 data sets. Results of four different α=0.05, 0.010.001, and 0.0001 levels were reported. For the proposed approximate F-distributed test statistics of MFLM (4) and (7) and additive model (9), all empirical type I error rates are around the nominal α levels for both B-spline basis and Fourier basis (columns 5–9 of Supplementary Tables S.3 and S.4). Therefore, the approximate F-distributed test statistics of MFLM controlled type I error rates correctly for all scenarios at all significance levels. The MFLM and related approximate F-distributed test statistics can be useful in both whole-genome and whole-exome association studies.
Power results
We compared the power of F-test of univariate and the approximate F-distributed tests of bivariate and trivariate traits based on the simulated COSI sequence data. The empirical power levels of the test statistics at α=0.0001 level were plotted in Figures 1 and 2. In the figures, 20%/80% causal variants had negative/positive effects for each trait. In the legend of all the Figures, ‘beta' means that the power level is from beta-smooth only model (7), and ‘add' means that the power level is from additive model (9). In Figure 1, the results of ‘Hom-F' were reported when the approximate F-distributed statistics were constructed using the homogeneous effect model (7) when the data were generated using the homogeneous models (Supplementary Table S2). Since the genotype data are different from study to study, there are no power levels for homogeneous additive model (10) in Figure 1. In Figure 2, the results of ‘Het-F' were reported that the approximate F-distributed statistics were constructed using heterogeneous effect models (7) and (9) when the data were generated using the heterogeneous models (Supplementary Table S2). Therefore, ‘correct models' were used to analyze simulated data in Figures 1 and 2.
Figure 1.

The empirical power of homogeneous approximate F-distributed Statistics (Hom-F) of the model (7) at α=0.0001, when the genetic effects were simulated as homogeneous. For each trait, 20%/80% causal variants had negative/positive effects.
Figure 2.

The empirical power of Het-F of the models (7) and (9) at α=0.0001, when the genetic effects were simulated as heterogeneous. For each trait, 20%/80% causal variants had negative/positive effects.
In general, the power levels of F-test of the univariate y1 trait are the lowest, the power levels of approximate F-distributed tests of the bivariate (y1,y2) trait are in the middle, and the power levels of approximate F-distributed tests of the trivariate (y1,y2,y3) trait are the highest for either beta-smooth only model (7) or additive model (9) in Figures 1 and 2. Therefore, it makes sense to perform multivariate analysis of pleiotropy traits.
Meta-analysis of lipid traits in eight European cohorts
Lipid traits from eight European cohorts were analyzed: five from Finland (FUSION Stage 2, D2d-2007, DPS, METSIM, and DRs EXTRA), two from Norway (HUNT and Tromso), and one from Germany (DIAGEN). The two Norwegian cohorts are combined into one study for these analyses. The genotype data were generated using the Metabochip, which was designed to fine map regions that have been associated with metabolic traits.49 For each cohort, 54 741 genetic variants were genotyped.
For our analysis, we utilized the existing literature as a reference for gene selection and found that 22 gene regions were fine mapped.5 We used Builder Mar. 2006 (NCBI36/hg18) to determine gene positions and 5 kb was used to extend the gene region on each side of a gene. The summary of 22 genes and the number of genetic variants in each region are given in Supplementary Table S5, Supplementary Materials. Four lipid traits were analyzed: high-density lipoprotein levels, low-density lipoprotein (LDL) levels, triglycerides (TG), and total cholesterol (CHOL). The sample sizes for each trait are provided in Supplementary Table S6, Supplementary Materials. For each trait, inverse normal rank transformation was performed to make sure that normality holds. For all studies except for METSIM, age, sex, and type 2 diabetes status were used as covariates. For METSIM, age and type 2 diabetes status were used as covariates since no females were included in the study. A significance threshold of P<3.1 × 10−6 was taken from Liu et al.5 (corresponding to 0.05/16 153 and allowing for the number of genes tested therein).
Using homogeneous F-approximation test statistics (Hom-F) based on Pillai-Bartlett trace, Table 1 reports results of three-trait and four-trait meta-analysis of lipid traits in European studies. For each combination of three to four traits, we observed association at five genes of APOB, APOE, LDLR, LPL, and PCSK9. For each of the five genes, we observed association for some of the traits in one-trait meta-analysis by homogeneous models (Table 1 of Fan et al.18 presented in Supplementary Table S7 in the Supplementary Materials). The results of two-trait meta-analysis of the lipid traits are presented in Supplementary Table S8, and association is observed for each of the five genes for some of the two-trait combinations.
Table 1. Three-trait and four-trait meta-analysis of lipid traits in European studies using Hom-F based on Pillai-Bartlett trace.
| P-values of the Hom-F | |||||
|---|---|---|---|---|---|
| Basis of Both GVF and βℓ(t) | Basis of beta-smooth only | ||||
| Traits | Gene | B-spline basis | Fourier basis | B-spline basis | Fourier basis |
| LDL, TG, CHOL | APOB | 1.29 × 10−10 | 1.73 × 10−5 | 9.16 × 10−10 | 2.21 × 10−6 |
| APOE | 1.82 × 10−88 | 5.22 × 10−90 | 9.03 × 10−89 | 1.31 × 10−90 | |
| LDLR | 3.14 × 10−11 | 2.25 × 10−9 | 3.51 × 10−9 | 8.49 × 10−8 | |
| LPL | 1.74 × 10−7 | 2.33 × 10−8 | 8.71 × 10−8 | 1.01 × 10−8 | |
| PCSK9 | 7.55 × 10−6 | 4.00 × 10−7 | 0.000196 | 2.16 × 10−6 | |
| HDL, LDL, TG | APOB | 6.47 × 10−10 | 6.32 × 10−6 | 4.89 × 10−10 | 1.15 × 10−6 |
| APOE | 6.22 × 10−95 | 3.11 × 10−97 | 4.77 × 10−95 | 2.03 × 10−96 | |
| LDLR | 1.03 × 10−11 | 2.16 × 10−10 | 1.34 × 10−10 | 4.51 × 10−9 | |
| LPL | 6.31 × 10−7 | 2.98 × 10−7 | 3.64 × 10−7 | 1.69 × 10−6 | |
| PCSK9 | 1.46 × 10−7 | 3.62 × 10−8 | 4.54 × 10−5 | 1.08 × 10−6 | |
| HDL, LDL, CHOL | APOB | 1.01 × 10−9 | 1.67 × 10−5 | 5.61 × 10−10 | 2.23 × 10−6 |
| APOE | 1.62 × 10−82 | 1.25 × 10−83 | 5.03 × 10−81 | 5.94 × 10−83 | |
| LDLR | 1.33 × 10−10 | 6.82 × 10−10 | 3.07 × 10−9 | 2.24 × 10−8 | |
| LPL | 2.32 × 10−7 | 8.76 × 10−8 | 2.24 × 10−7 | 1.33 × 10−7 | |
| PCSK9 | 1.18 × 10−6 | 3.25 × 10−8 | 7.88 × 10−5 | 1.57 × 10−7 | |
| HDL, TG, CHOL | APOB | 1.11 × 10−10 | 2.93 × 10−6 | 1.66 × 10−10 | 4.11 × 10−7 |
| APOE | 4.59 × 10−88 | 1.10 × 10−88 | 2.10 × 10−87 | 1.06 × 10−86 | |
| LDLR | 4.67 × 10−12 | 1.48 × 10−10 | 7.33 × 10−11 | 3.23 × 10−9 | |
| LPL | 1.86 × 10−9 | 6.25 × 10−11 | 1.47 × 10−9 | 2.33 × 10−10 | |
| PCSK9 | 7.62 × 10−8 | 1.47 × 10−8 | 1.13 × 10−5 | 1.35 × 10−7 | |
| HDL, LDL, TG, CHOL | APOB | 2.23 × 10−10 | 1.60 × 10−6 | 1.67 × 10−10 | 1.24 × 10−7 |
| APOE | 4.76 × 10−93 | 1.64 × 10−94 | 7.29 × 10−94 | 8.08 × 10−94 | |
| LDLR | 3.17 × 10−11 | 6.89 × 10−11 | 1.13 × 10−9 | 5.15 × 10−9 | |
| LPL | 7.41 × 10−8 | 8.79 × 10−9 | 7.14 × 10−8 | 2.20 × 10−9 | |
| PCSK9 | 1.41 × 10−7 | 1.51 × 10−7 | 8.07 × 10−5 | 4.49 × 10−7 | |
Abbreviations: GVF, genetic variant function; Hom-F, homogeneous F-approximation test statistics.
The associations that attain a threshold significance of P<3.1 × 10−6 are highlighted in bold.5 The results of ‘Basis of Both GVF and βℓ(t)' were based on smoothing both GVF and genetic effect functions βℓ(t) of model (4), and the results of ‘Basis of beta-smooth only' were based on smoothing βℓ(t) only approach of model (7).
Using Het-F based on Pillai-Bartlett trace, Tables 2 and 3 report results of three-trait meta-analysis of the lipid traits, and results of four-trait meta-analysis of Het-F are presented in Table 4. By Het-F of MFLM (4) and (7), we observe associations for some three-trait and four-trait combinations at APOB, APOE, CDC123, CDKAL1, CDKN2B, FTO, HMGA2, HNF1A, JAZF1, IDE, KCNQ1, KIF11, LDLR, LPL, OASL, PCSK9, and TSPAN8. The results of two-trait meta-analysis of lipid traits are presented in Supplementary Tables S9 and S10 and association is observed for some genes and some of the two-trait combinations. Three traits (LDL, TG, and CHOL) are associated with some genes in one-trait meta-analysis by heterogeneous models (Table 2 of Fan et al.18 presented in Supplementary Table S11 in the Supplementary Materials). The additive effect model (9) detects some association signals, but less than the MFLM (4) and (7).
Table 2. Three-trait meta-analysis of lipid traits in European studies using Het-F based on Pillai-Bartlett trace.
| P-values of the Het-F | ||||||
|---|---|---|---|---|---|---|
| Basis of Both GVF and βℓ(t) | Basis of beta-smooth only | |||||
| Traits | Gene | B-spline basis | Fourier basis | B-spline basis | Fourier basis | Additive model (9) |
| LDL, TG, CHOL | APOB | 1.20 × 10−10 | 2.01 × 10−8 | 3.92 × 10−6 | 1.39 × 10−6 | 2.06 × 10−7 |
| APOE | 3.80 × 10−69 | 8.84 × 10−68 | 1.07 × 10−63 | 2.62 × 10−65 | 2.79 × 10−64 | |
| CDKL1 | 3.77 × 10−6 | 2.97 × 10−7 | 5.90 × 10−6 | 3.03 × 10−7 | 0.001498 | |
| FTO | 1.15 × 10−6 | 0.000219 | 0.000242 | 0.001061 | 0.001042 | |
| HNF1A | 1.69 × 10−10 | 1.73 × 10−7 | 4.00 × 10−7 | 3.79 × 10−8 | 2.02 × 10−8 | |
| LPL | 5.98 × 10−7 | 2.86 × 10−6 | 1.11 × 10−6 | 2.48 × 10−8 | 0.000581 | |
| OSAL | 1.37 × 10−6 | 6.14 × 10−5 | 3.08 × 10−5 | 0.000824 | 0.000993 | |
| TSPAN8 | 3.31 × 10−8 | 5.38 × 10−9 | 2.30 × 10−8 | 2.80 × 10−9 | 3.06 × 10−8 | |
| PCSK9 | 2.28 × 10−8 | 8.68 × 10−10 | 2.49 × 10−10 | 1.63 × 10−10 | 5.87 × 10−11 | |
| HDL, LDL, TG | APOB | 7.90 × 10−13 | 2.40 × 10−10 | 4.29 × 10−8 | 7.68 × 10−9 | 5.52 × 10−5 |
| APOE | 5.25 × 10−77 | 1.01 × 10−75 | 1.97 × 10−71 | 1.16 × 10−73 | 2.83 × 10−72 | |
| CDC123 | 1.14 × 10−5 | 1.55 × 10−5 | 8.07 × 10−6 | 1.98 × 10−6 | 0.018755 | |
| CDKL1 | 1.37 × 10−8 | 4.18 × 10−9 | 4.90 × 10−9 | 3.34 × 10−10 | 2.72 × 10−5 | |
| CDKN2B | 6.12 × 10−7 | 1.95 × 10−6 | 1.64 × 10−6 | 1.51 × 10−6 | 3.72 × 10−6 | |
| FTO | 4.38 × 10−8 | 3.80 × 10−6 | 5.76 × 10−6 | 3.88 × 10−5 | 5.65 × 10−5 | |
| HNF1A | 1.48 × 10−10 | 1.04 × 10−8 | 4.48 × 10−9 | 1.55 × 10−9 | 1.16 × 10−9 | |
| JAZF1 | 1.58 × 10−6 | 2.31 × 10−6 | 2.80 × 10−6 | 9.99 × 10−6 | 0.003786 | |
| KIF11 | 1.74 × 10−6 | 0.000153 | 1.31 × 10−5 | 6.83 × 10−6 | 0.000198 | |
| LPL | 2.29 × 10−7 | 4.09 × 10−7 | 2.36 × 10−7 | 6.03 × 10−9 | 3.59 × 10−5 | |
| OSAL | 1.38 × 10−9 | 7.99 × 10−8 | 4.01 × 10−8 | 1.20 × 10−6 | 6.20 × 10−6 | |
| TSPAN8 | 1.95 × 10−11 | 1.28 × 10−12 | 9.49 × 10−12 | 6.69 × 10−13 | 1.43 × 10−11 | |
| PCSK9 | 4.08 × 10−10 | 3.75 × 10−12 | 6.47 × 10−11 | 6.24 × 10−11 | 2.50 × 10−11 | |
Abbreviations: GVF, genetic variant function; Het-F, heterogeneous F-approximation test statistics.
The associations that attain a threshold significance of P<3.1 × 10−6 are highlighted in bold.5 The results of ‘Basis of Both GVF and βℓ(t)' were based on smoothing both GVF and genetic effect functions βℓ(t) of model (4), the results of ‘Basis of beta-Smooth Only' were based on smoothing βℓ(t) only approach of model (7), and the results of ‘Additive Model (9)' were based on the additive effect model (9).
Table 3. Three-trait meta-analysis of lipid traits in European studies using Het-F based on Pillai-Bartlett trace.
| P-values of the Het-F | ||||||
|---|---|---|---|---|---|---|
| Basis of both GVF and βℓ(t) | Basis of beta-smooth only | |||||
| Traits | Gene | B-spline basis | Fourier basis | B-spline basis | Fourier basis | Additive model (9) |
| HDL, LDL, CHOL | APOB | 4.23 × 10−11 | 6.12 × 10−8 | 4.50 × 10−6 | 1.11 × 10−6 | 7.75 × 10−9 |
| APOE | 4.59 × 10−66 | 3.96 × 10−65 | 1.44 × 10−60 | 7.92 × 10−63 | 4.98 × 10−62 | |
| CDKL1 | 7.85 × 10−7 | 2.07 × 10−8 | 6.30 × 10−8 | 5.77 × 10−9 | 0.000183 | |
| FTO | 2.89 × 10−7 | 4.84 × 10−5 | 0.000331 | 0.000907 | 0.001266 | |
| HNF1A | 1.91 × 10−10 | 7.11 × 10−8 | 7.35 × 10−8 | 1.07 × 10−8 | 3.20 × 10−8 | |
| JAZF1 | 2.89 × 10−6 | 1.02 × 10−5 | 4.42 × 10−6 | 2.91 × 10−6 | 0.053595 | |
| LDLR | 5.87 × 10−7 | 2.07 × 10−7 | 8.56 × 10−8 | 2.87 × 10−8 | 6.70 × 10−8 | |
| LPL | 4.56 × 10−7 | 2.99 × 10−7 | 1.71 × 10−5 | 5.70 × 10−6 | 0.017553 | |
| OSAL | 2.81 × 10−9 | 6.62 × 10−7 | 8.13 × 10−7 | 3.67 × 10−5 | 0.000276 | |
| TSPAN8 | 5.79 × 10−12 | 1.20 × 10−13 | 8.15 × 10−13 | 6.81 × 10−14 | 1.36 × 10−12 | |
| PCSK9 | 5.23 × 10−10 | 1.64 × 10−11 | 4.65 × 10−11 | 8.36 × 10−11 | 3.58 × 10−10 | |
| HDL, TG, CHOL | APOB | 3.25 × 10−13 | 7.35 × 10−12 | 5.16 × 10−9 | 2.84 × 10−10 | 1.80 × 10−7 |
| APOE | 1.64 × 10−69 | 8.22 × 10−68 | 1.21 × 10−66 | 9.28 × 10−67 | 1.28 × 10−65 | |
| CDKL1 | 7.17 × 10−8 | 1.18 × 10−8 | 4.47 × 10−8 | 2.38 × 10−9 | 2.55 × 10−5 | |
| FTO | 2.86 × 10−8 | 6.38 × 10−7 | 2.41 × 10−6 | 2.03 × 10−5 | 2.91 × 10−5 | |
| HNF1A | 7.17 × 10−9 | 4.60 × 10−7 | 9.22 × 10−8 | 2.14 × 10−8 | 2.38 × 10−8 | |
| KIF11 | 2.68 × 10−6 | 9.95 × 10−5 | 2.74 × 10−5 | 1.35 × 10−5 | 0.001113 | |
| LPL | 5.18 × 10−8 | 9.15 × 10−8 | 2.73 × 10−7 | 1.13 × 10−9 | 1.50 × 10−5 | |
| OSAL | 9.59 × 10−8 | 4.81 × 10−7 | 1.17 × 10−7 | 1.20 × 10−6 | 4.65 × 10−6 | |
| TSPAN8 | 3.34 × 10−9 | 1.15 × 10−10 | 4.38 × 10−10 | 4.82 × 10−11 | 7.09 × 10−10 | |
| PCSK9 | 8.29 × 10−11 | 2.89 × 10−11 | 2.95 × 10−10 | 4.21 × 10−10 | 1.59 × 10−11 | |
Abbreviations: GVF, genetic variant function; Het-F, heterogeneous F-approximation test statistics.
The associations that attain a threshold significance of P<3.1 × 10−6 are highlighted in bold.5 The results of ‘basis of both GVF and βℓ(t)' were based on smoothing both GVF and genetic effect functions βℓ(t) of model (4), the results of ‘basis of beta-smooth only' were based on smoothing βℓ(t) only approach of model (7), and the results of ‘additive model (9)' were based on the additive effect model (9).
Table 4. Four-trait meta-analysis of lipid traits in European studies using Het-F based on Pillai-Bartlett trace.
| P-values of the Het-F | ||||||
|---|---|---|---|---|---|---|
| Basis of both GVF and βℓ(t) | Basis of beta-smooth only | |||||
| Traits | Gene | B-spline basis | Fourier basis | B-spline basis | Fourier basis | Additive model (9) |
| HDL, LDL, TG, CHOL | APOB | 7.23 × 10−13 | 1.66 × 10−11 | 7.62 × 10−8 | 3.28 × 10−9 | 5.26 × 10−15 |
| APOE | 7.45 × 10−74 | 3.26 × 10−72 | 4.64 × 10−67 | 1.61 × 10−69 | 3.18 × 10−68 | |
| CDC123 | 4.12 × 10−5 | 0.000519 | 5.53 × 10−7 | 3.87 × 10−7 | 0.016618 | |
| CDKAL1 | 2.37 × 10−8 | 2.66 × 10−9 | 5.03 × 10−9 | 8.06 × 10−10 | 0.000182 | |
| FTO | 1.73 × 10−9 | 2.29 × 10−6 | 2.77 × 10−6 | 2.88 × 10−5 | 3.83 × 10−6 | |
| HMGA2 | 1.92 × 10−5 | 4.07 × 10−5 | 6.97 × 10−6 | 1.34 × 10−6 | 2.99 × 10−8 | |
| HNF1A | 4.40 × 10−14 | 9.32 × 10−10 | 2.99 × 10−10 | 5.91 × 10−12 | 2.05 × 10−10 | |
| IDE | 7.57 × 10−6 | 1.92 × 10−6 | 5.52 × 10−7 | 2.29 × 10−6 | 0.057381 | |
| KCNQ1 | 8.64 × 10−7 | 2.16 × 10−7 | 0.000121 | 4.23 × 10−5 | 1.85 × 10−5 | |
| KIF11 | 4.54 × 10−7 | 3.00 × 10−5 | 4.33 × 10−6 | 2.50 × 10−6 | 0.000326 | |
| LDLR | 6.34 × 10−7 | 7.06 × 10−7 | 3.60 × 10−7 | 2.02 × 10−7 | 3.96 × 10−7 | |
| LPL | 3.62 × 10−9 | 3.71 × 10−8 | 9.08 × 10−9 | 2.48 × 10−11 | 1.11 × 10−5 | |
| OASL | 1.11 × 10−10 | 1.64 × 10−8 | 3.87 × 10−9 | 1.88 × 10−7 | 6.23 × 10−7 | |
| PCSK9 | 7.64 × 10−10 | 3.82 × 10−11 | 2.57 × 10−11 | 1.37 × 10−10 | 5.53 × 10−10 | |
| TSPAN8 | 8.05 × 10−13 | 9.58 × 10−15 | 2.03 × 10−13 | 6.01 × 10−15 | 3.48 × 10−13 | |
Abbreviations: GVF, genetic variant function; Het-F, heterogeneous F-approximation test statistics.
The associations that attain a threshold significance of P<3.1 × 10−6 are highlighted in bold.5 The results of ‘basis of Both GVF and βℓ(t)' were based on smoothing both GVF and genetic effect functions βℓ(t) of model (4), the results of ‘basis of beta-smooth only' were based on smoothing βℓ(t) only approach of model (7), and the results of ‘additive model (9)' were based on the additive effect model (9).
In study-based pleiotropy analysis of Wang et al.,10 which analyzes each data set separately, association was observed at only two genes, APOE and LDLR, in some studies (Supplementary Table S12 in the Supplementary Materials from Table 1 of Wang et al.10). Thus, it is more advantageous to perform meta-analysis of multiple studies.
Discussion
Here we develop MFLM for meta-analysis of multiple quantitative traits adjusting for covariates. On the basis of the MFLM, approximate F-distributed statistics of Pillai-Bartlett trace, Hotelling-Lawley trace, and Wilks's Lambda are introduced to test for association between multiple quantitative traits and multiple genetic variants. Simulation analysis is performed to show that the approximate F-distributed tests control the false-positive rates accurately. By evaluating power performance, it is shown that it can be advantageous to perform the proposed pleiotropy analysis instead of individual trait analysis.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 27, 44 Among other merits, the MFLM can handle missing genotype data naturally.
The proposed methods were used to analyze four lipid traits in eight European cohorts. When we use the homogeneous MFLM to analyze three traits and four traits together, association is observed at five genes of APOB, APOE, LDLR, LPL, and PCSK9. For each of the five genes, we only observed association for some traits in one-trait meta-analysis and two-trait meta-analyses (Table 1 of Fan et al.18 presented in Supplementary Table S7 and Supplementary Table S8 in the Supplementary Materials). Similarly, the proposed heterogeneous MFLM detected more and stronger association signals by three-trait or four-trait analysis than one-trait or two-trait analysis.
One special feature of MFLM is that functional data analysis techniques are used to reduce the dimensionality of the next-generation sequencing data.39, 40, 41, 42, 43 The key idea is that multiple genetic variants of an individual is treated as a realization of an underlying stochastic process.50 Therefore, the genome of an individual is viewed as a continuous stochastic function that contains both genetic position and LD information of the genetic markers. In real data analysis, one may test whether the genetic effects are heterogeneous or homogeneous, ie, to test H0: Ω1=· · ·=ΩL=Ω. If the H0 is rejected, the genetic effects are heterogeneous; otherwise, they are homogeneous.
In linkage analysis, it is well known that the genetic data are treated as functions of the recombination fraction51, 52 to order genes along a chromosome.53 Thus, it is reasonable and esirable to treat genetic data as functions. In linkage analysis, one needs to estimate the recombination fractions based on pedigree data. In next-generation sequencing data, the physical positions in terms of base pairs are available in almost all studies and one does not need to estimate them. However, in association studies, the genetic data are usually treated as discrete and the physical positions are simply ignored in most literature except in recent functional regression models.10, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 Our functional regression models provide a way to properly utilize the physical positions in gene-based association studies.
In genetic meta-analysis, summary statistics from different studies are usually used to meta-analyze the data as individual data are not always available.5, 54 In our case, the European cohorts individual genetic data are available for analysis. Therefore, we build our MFLM using the individual-level data. If only summary statistics of functional regression models are available from different studies, it is still an open question if those statistics can be used to meta-analyze the data. It is known that meta-analysis using individual data are advantageous over meta-analysis of summary statistics in non-genetics studies.55, 56, 57 It would be interesting to evaluate the pros and cons of two approaches in genetic association analysis in the future studies. Note that the functional regressions are simply ordinary regressions after revising the theoretical functional models by functional data analysis techniques, and so the strategy of usual meta-analysis would be useful.54 It should be possible to use results from functional regression models for a meta-analysis across cohorts. However, the details are still waiting for further work.
Acknowledgments
Two anonymous reviewers and Editor-in-Chief, Professor Dr Gertjan van Ommen, provided very good and insightful comments for us to improve the manuscript. We greatly thank the European cohort investigators for letting us analyze the data and use them as examples. Dr Heather M Stringham and Dr Tanya M Teslovich kindly sent us the data of the European cohorts and patiently answered many questions about the cohorts, and we greatly appreciated them. This study was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Maryland (Ruzong Fan and Chi-yang Chiu), by Wei Chen's NIH grants R01EY024226 and R01HG007358 and the University of Pittsburgh (Ruzong Fan is an unpaid collaborator on the grant R01EY024226). This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Computer program
The methods proposed in this paper are implemented by using procedures of the R functional data analysis (fda) package. The R codes for data analysis and simulations are available from the web site: http://www.nichd.nih.gov/about/org/diphr/bbb/software/fan/Pages/default.aspx.
The authors declare no conflict of interest.
Supplementary Material
References
- Gianola D, de los Campos G, Toro MA, Naya H, Schön CC, Sorensen D: Do molecular markers inform about pleiotropy? Genetics 2015; 201: 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Liu Z, Wang X, Zhang H: Genetic association test for multiple traits at gene level. Genet Epidemiol 2013; 37: 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Jannink JL: Multiple trait genomic selection methods increase genetic value prediction accuracy. Genetics 2012; 192: 1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Teslovich TM, Boehnke M, Lin X: General framework for meta-analysis of rare variants in sequencing association studies. Am J Hum Genet 2013; 93: 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DJ, Peloso GM, Zhan X et al: Meta-analysis of gene-level tests for rare variant association. Nat Genet 2014; 46: 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity A, Sullivan PF, Tzeng JY: Multivariate phenotype association analysis by marker set kernel machine regression. Genet Epidemiol 2012; 36: 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadaway KA, Cutler DJ, Duncan R et al: A statistical approach for testing cross-phenotype effects of rare variants. Am J Hum Genet 2016; 98: 525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier R, Moser G, Chen GB et al: Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet 2015; 96: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Sluis S, Dolan CV, Li J et al: MGAS: a powerful tool for multivariate gene-based genome-wide association analysis. Bioinformatics 2015; 31: 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Liu AY, Mills JL et al: Pleiotropy analysis of quantitative traits at gene level by multivariate functional linear models. Genet Epidemiol 2015; 39: 259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER: Next-generation DNA sequencing methods. Annu Rev Genom Hum Genet 2008; 9: 387–402. [DOI] [PubMed] [Google Scholar]
- Metzker ML: Sequencing technologies the next generation. Nat Rev Genet 2010; 11: 31–34. [DOI] [PubMed] [Google Scholar]
- Rusk N, Kiermer V: Primer: sequencingthe next generation. Nat Methods 2008; 5: 15. [DOI] [PubMed] [Google Scholar]
- Shendure J, Ji H: Next-generation DNA sequencing. Nat Biotechnol 2008; 26: 1135–1145. [DOI] [PubMed] [Google Scholar]
- Bansal V, Libiger O, Torkamani A, Schork NJ: Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet 2010; 11: 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan RZ, Wang YF, Mills JL, Wilson AF, Bailey-Wilson JE, Xiong MM: Functional linear models for association analysis of quantitative traits. Genet Epidemiol 2013; 37: 726–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan RZ, Wang YF, Mills JL et al: Generalized functional linear models for case-control association studies. Genet Epidemiol 2014; 38: 622–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan RZ, Wang YF, Boehnke M et al: Gene level meta-analysis of quantitative traits by functional linear models. Genetics 2015; 200: 1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan RZ, Wang YF, Chiu CY et al: Meta-analysis of complex diseases at gene level by generalized functional linear models. Genetics 2016; 202: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan RZ, Wang YF, Qi Y et al: Gene-based association analysis for censored traits via functional regressions. Genet Epidemiol 2016; 40: 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan RZ, Chiu CY, Jung JS et al: A comparison study of fixed and mixed effect models for gene level association studies of complex traits. Genet Epidemiol 40: 702–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Boerwinkle E, Xiong MM: Association studies for next-generation sequencing. Genome Res 2011; 21: 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Zhu Y, Xiong MM: Quantitative trait locus analysis for next-generation sequencing with the functional linear models. J Med Genet 2012; 49: 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Zhu Y, Xiong MM: Smoothed functional principal component analysis for testing associa- tion of the entire allelic spectrum of genetic variation. Eur J Hum Genet 2013; 21: 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svishcheva GR, Belonogova NM, Axenovich TI: Region-based association test for familial data under functional linear models. PLoS ONE 2015; 10: e0128999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vsevolozhskaya OA, Zaykin DV, Greenwood MC, Wei C, Lu Q: Functional analysis of variance for association studies. PLoS ONE 2014; 9: e105074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vsevolozhskaya OA, Zaykin DV, Barondess DA, Tong X, Jadhav S, Lu Q: Uncovering local trends in genetic effects of multiple phenotypes via functional linear models. Genet Epidemiol 2016; 40: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Boerwinkle E, Xiong MM: Epistasis analysis for quantitative traits by functional regres- sion models. Genome Res 2014; 24: 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JY, Zhu Y, Xiong MM: Genome-wide gene-gene interaction analysis for next-generation sequencing. Eur J Hum Genet 2016; 24: 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lumley T, Brody J et al: Sequence kernel association test for survival traits. Genet Epidemiol 2014; 38: 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X: Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet 2013; 92: 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Emond MJ, Bamshad MJ et al: Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012; 91: 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X: Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011; 89: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA: The correlation between relatives on the supposition of Mendelian inheritance. Philos Trans R Soc Ed 1918; 52: 399–433. [Google Scholar]
- Zuk O, Schaffner SF, Samocha K et al: Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci USA 2014; 111: E455E464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Leal SM: Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 2008; 83: 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen BE, Browning SR: A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet 2009; 5: e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Zeggini E: An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol 2010; 34: 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boor C: A Practical Guide to Splines, Revised Version. New York, NY, USA: Springer, 2001. [Google Scholar]
- Ferraty F, Romain Y: The Oxford Handbook of Functional Data Analysis. New York, NY, USA: Oxford University Press, 2010. [Google Scholar]
- Horváth L, Kokoszka P: Inference for Functional Data With Applications. New York, NY, USA: Springer, 2012. [Google Scholar]
- Ramsay JO, Silverman BW: Functional Data Analysis, 2nd edn. New York, NY, USA: Springer, 2005. [Google Scholar]
- Ramsay JO, Hooker G, Graves S: Functional Data Analysis With R and Matlab. New York, NY, USA: Springer, 2009. [Google Scholar]
- Jung JS, Zhong M, Liu L, Fan RZ: Bi-variate combined linkage and association mapping of quantitative trait loci. Genet Epidemiol 2008; 32: 396–412. [DOI] [PubMed] [Google Scholar]
- Anderson TW: An Introduction to Multivariate Statistical Analysis, 2nd edn. New York, NY, USA: John Wiley & Sons, 1984. [Google Scholar]
- Rao CR: Linear Statistical Inference and its Applications, 2nd edn. New York, NY, USA: John Wiley & Sons, 1973. [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D: Calibrating a coalescent simulation of human genome sequence variation. Genome Res 2005; 15: 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium: A map of human genome variation from population scale sequencing. Nature 2010; 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SM: Stochastic Processes, 2nd edn. New York, NY, USA: John Wiley & Sons, 1996. [Google Scholar]
- Lange K: Mathematical and Statistical Methods for Genetic Analysis, 2nd edn. New York, NY, USA: Springer, 2002. [Google Scholar]
- Ott J: Analysis of Human Genetic Linkage, 3rd edn. Baltimore and London: Johns Hopkins University Press, 1999. [Google Scholar]
- Sturtevant AH: The linear arrangement of six sex-linked factors in Drosophila, as shown by their mode of association. J Exp Zool 1913; 14: 43–59. [Google Scholar]
- Lin DY, Zeng D: Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genet Epidemiol 2010; 34: 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debray TP, Moons KG, Abo-Zaid GM, Koffijberg H, Riley RD: Individual participant data metaanalysis for a binary outcome: one-stage or two-stage? PLoS One 2012; 8: e60650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JP, Whitehead A, Turner RM, Omar RZ, Thompson SG: Meta-analysis of continuous outcome data from individual patients. Stat Med 2001; 20: 2219–2241. [DOI] [PubMed] [Google Scholar]
- Mathew T, Nordström K: Comparison of one-step and two-step meta-analysis models using indi- vidual patient data. Biometric J 2010; 52: 271–287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.