

Abstract
The p53 inhibitor Mdm4 is present at high levels in multiple human cancers. Overexpression of Mdm4 in mice drives spontaneous development of mostly lymphomas and sarcomas. In this study, we explored the ability of Mdm4 to cooperate with other lesions in tumour development. The Mdm4 transgene contributed to mammary tumour development in a BALB/cJ background. High levels of Mdm4 enhanced tumour development in a mutant p53R172H heterozygous background and reduced the need to lose the wild type p53 allele as compared to the mice heterozygous only for the p53R172H mutation. Additionally, high levels of Mdm4 cooperated with an oncogenic K-ras mutation to drive lung tumorigenesis in vivo. Lastly, we examined p53-independent functions of Mdm4 by studying the contribution of Mdm4 to tumour development in the absence of p53. While the overall survival of p53-null mice with and without the Mdm4 transgene was similar, male mice with both alterations showed a significantly shorter survival and exhibits differences in tumour spectrum as compared to p53-null male mice, demonstrating a p53-independent function of Mdm4 in tumorigenesis. Furthermore, p53-null mice with the highest level of Mdm4 tended to have multiple tumours. Thus, a detailed analysis of Mdm4 transgenic mice in various genetic backgrounds shows synergy in tumour development in vivo. Mdm4 may thus serve as a therapeutic target in cancers.
Keywords: Mammary tumor genesis, p53 LOH, Lung adenocarcinoma, p53 independent function
Introduction
Mdm2 and Mdm4 are critical inhibitors of the p53 tumour suppressor and are often overexpressed in human cancers [1]. Loss of either of these genes in mice leads to embryo-lethal phenotypes that are completely rescued by p53 deletion [2–6]. While Mdm2 is an E3 ligase that targets p53 for proteasomal degradation, the homolog Mdm4 inhibits p53 activity by binding to p53 amino terminal transactivation domain [7,8]. Although Mdm4 itself does not have E3 ubiquitin ligase function, it modulates p53 stability by interacting with Mdm2 through its RING domain [9]. Disruption of this interaction leads to p53-dependent embryo lethal phenotypes [10,11]. Thus, Mdm4 is a bona fide p53 negative regulator in development.
Conversely, a variety of human cancers have MDM4 amplification and overexpression resulting in high levels of MDM4 protein [1,12–16]. In most cases, human tumours with high levels of MDM4 do not have alterations in TP53 further emphasizing the mutually exclusive relationship between MDM4 and TP53 alterations [1]. However, some tumours do have both high MDM4 and TP53 mutations, prompting us to examine the cooperativity of the two events in tumour development in mouse models. For example, while 14% of invasive breast cancers have amplified MDM4, 3% also have mutations in TP53 (www.Cbioportal.com). The relationship between Mdm4 and mutant p53 is complex. Mdm4 binds the amino terminus of wild type p53 and likely binds mutant p53 as the latter retains the amino terminus. Since Mdm4 binding interferes with Mdm2 and Mdm4 lacks E3 ubiquitin ligase function, this interaction likely contributes to mutant p53 stability. Increased stability of mutant p53 proteins leads to gain-of-function activities [17], these data suggest increased Mdm4 levels may cooperate with mutant p53 in tumorigenesis. Additional in vivo experiments are needed to determine the effects of increased Mdm4 levels and p53 mutations on tumour development.
In addition, we examined the cooperativity of high levels of Mdm4 with other alterations in tumour development. A large proportion (36%) of lung adenocarcinoma patients have KRAS mutations. Of these, 3.5% have both KRAS mutations and MDM4 amplification in the TCGA dataset (www.Cbioportal.com). Since it is known that p53 loss cooperates with oncogenic K-ras in lung tumour models [18], we also examined whether Mdm4 overexpression could cooperate with oncogenic K-ras in lung adenocarcinoma tumorigenesis.
Moreover, the fact that some tumours have both high MDM4 levels and TP53 mutations suggests possible p53-independent functions of MDM4. Besides binding to p53 and Mdm2, Mdm4 also interacts with other proteins such as p21, 14-3-3gamma, ARF, HAUSP and Nbs1 [19–25], suggesting Mdm4 has p53-independent functions. Specifically, Mdm4 promotes genomic instability and increases cell transformation independent of p53 and Mdm2 by interacting with Nbs1 [25], further suggesting Mdm4 has a p53-independent function. This relationship has not been examined in tumour models in vivo.
We have previously generated Mdm4-overexpressing transgenic mice with varying levels of Mdm4, ranging from lowest to highest in Mdm4Tg1, Mdm4Tg6 and Mdm4Tg15. Mdm4 overexpression in mice in a mixed C57BL/6J and129/SvJ background leads to development of lymphomas, sarcomas, and a few carcinomas[26], demonstrating that Mdm4 acts as an oncogene when overexpressed. Since the BALB/cJ background is more permissive to development of mammary tumours [27–29], we crossed Mdm4 transgenic mice into BALB/cJ background to assess the role of Mdm4 in mammary tumour. These mice developed mammary tumours. Mdm4 overexpression in a p53R172H/+ background decreased survival and increased tumour incidence. Mdm4 overexpression cooperated with oncogenic K-ras in lung adenocarcinoma genesis. Additionally, p53-independent functions of Mdm4 were observed in male mice in a p53-null background. In summary, our results support that Mdm4 is a bona fide oncogene as it cooperates with other genetic lesions to drive tumour development.
Materials and methods
Mice and tumour analyses
Mouse experiments were performed in compliance with MD Anderson Cancer Center’s Institutional Animal Care and Use Committee. Mdm4Tg1, Mdm4Tg6, Mdm4Tg15 mice [26] were back-crossed to BALB/cJ wild type mice (Jackson Laboratory, Bar Harbor, Maine) at least six generations. p53R172H/+ mice in a BALB/cJ background were generated previously [30]. K-rasLA1/+ mice [18] were purchased from Jackson Laboratory and crossed to Mdm4Tg6 or Mdm4Tg15 mice in a mixed C57BL/6J and 129/SvJ background. Mouse cohorts were monitored daily for tumorigenesis. Moribund mice were sacrificed and tissues prepared for pathological analyses.
Immunohistochemistry
Mdm4 immunohistochemistry (IHC) was performed as described previously using AB112 antibody [13]. FL-393 (Santa Cruz Biotechnology) was used for p53 IHC (1:50 dilution). Vector DAB Substrate Kit (Burlingame, CA) was used for chromogenic detection.
p53 loss of heterozygosity (LOH) assay
PCR amplification was performed using tumour DNA samples and the products were sequenced. p53 LOH was determined as previously described [31].
Western blot analysis and reverse transcription-quantitative PCR (RT-qPCR)
Mouse primary tumour cell lines from Mdm4Tg15 (No.43) and p53−/− mice (No.32 and 46) were generated and cultured in DMEM with 10% fetal bovine serum. MTT assays were performed using 5,000 cells in 24-well plates. Mdm4 shRNAs were obtained from M.D. Anderson Cancer Center ShRNA and ORFeome Core Facility. Mouse Mdm4 cDNAs were cloned into the vector pBabe-puro and transfected into Phoenix cells to generate Mdm4 overexpressing cell lines. Antibodies used for western blots were: Mdm4 antibody (MX82 at 1:500) and ϒ tubulin (at 1:1000) (Sigma, MO). Total RNAs were prepared from cells using Trizol reagent (Invitrogen, CA), and then treated with DNase I (Roche, NJ). Complementary DNAs were made using a first strand reverse transcriptase kit (GE Healthcare, UK). The primer sequences of the p53 downstream target genes p21, Mdm2, BBC3 (Puma), and Noxa were used as reported previously [32]. The primers for other mutant p53 target genes are listed below: Hmgcr (GTGCTGAGCAGCGACATCAT and TGTACAGGATGGCGATGCA), Fdrs (GGAGAGGTGGCTTGGTTTCC and CAGGACTGGACACCCATATGC), Mvk (TCTGCTTGCCTTTCTCTACCTGTA and CTCGGGAGTGTCCTCTGCTT), Mvd (TGGTGAGCGCCGACAAG and TCTCCACGCTGGTCTGCAT), Sqle (CGACACTTCTTTTCCGTTGCA and CCCACGGCTCTGATTTGAA), Dhcr7 (CAACGCTCCCAAAGTCAAGAG and GGCCCCATTGTCCTTGAGAT), Lss (GGAACGTCCTTCACAAAAAAGG and CCAGAACTTCCCCCAGGAA), Mll1 (CGGGCTCATCAACGATAAGC and CAGGCCCAGATGTCAGGTG), Mll2 (GGATCTATGACAGGGCTTTCCC and ACCATGTGACATCATTCCTTGC), Moz (GTGCTGCTACACCGATGGTG and ACCATGTGACATCATTCCTTGC), Sharp1 (TGAATGCATTGCTCAGCTGAA and TGCCCCAGTGTTGTCAATTTC), Ccng2 (CCAGGCTGGCGGAAGAA and GACTGATGCGGATCACATCGT), Pdgfrb (CTGTGAATGCCGTGCAGACT and AATGCACCGGATGGTGATG).
Statistical analysis
Student’s t-tests and Kaplan-Meier survival analyses were performed using Prism 5 software (GraphPad Software). P-values <0.05 were considered statistically significant.
Results
Overexpression of Mdm4 drives mammary tumour development in a BALB/cJ background
Mdm4Tg6 and Mdm4Tg15 mice were generated previously in a C57BL/6J and 129/SvJ mixed background using a promoter that expresses high levels of Mdm4 in multiple tissues [26]. To investigate whether overexpression of Mdm4 plays a causative role in mammary tumorigenesis, Mdm4Tg6 and Mdm4Tg15 mice were back crossed to BALB/cJ mice which are predisposed to spontaneous breast tumour development [27–29]. All the mice were monitored for tumorigenesis within the same time frame. Mdm4Tg6 and Mdm4Tg15 mice developed different types of tumours with a median overall survival of 538 and 489 days, respectively (Figure 1a). Mdm4Tg6 mice developed lymphomas (56%), haemangiosarcomas (26%) and carcinomas (7%). Similarly, Mdm4Tg15 mice developed lymphomas (34%), sarcomas (25%) and carcinomas (31%) (Figure 1b and Table 1). Moreover, 25% of Mdm4Tg6 and 26% of Mdm4Tg15 mice had two or more tumours (Figure 1C) demonstrating that Mdm4 is a potent oncogene that drives tumour development in vivo. Interestingly, although with low penetrance, both Mdm4Tg6 (1/20) and Mdm4Tg15 (2/23) transgenic mice developed mammary adenocarcinoma (Figure 2A and Table 1), indicating that overexpression of Mdm4 indeed can induce mammary tumorigenesis in vivo. The mammary tumours ranged from moderately differentiated to focally poorly-differentiated with increased mitosis, angiogenesis, and fibrosis. Also, Mdm4Tg6 and Mdm4Tg15 mice both developed testicular interstitial cell (Leydig) adenoma, and tumours in Mdm4Tg15 mice even progressed to Leydig cell carcinoma (one metastasized to lung) (Figure 2C and 2D). Notably Leydig cell carcinomas are not observed in Mdm4Tg15 mice in a C57BL/6J -129/SvJ mixed background [26], indicating that BALB/cJ background may contribute to Leydig cell tumorigenesis in these mice. Furthermore, all the mammary adenocarcinomas (n=3) and Leydig cell tumours (n=4) from the transgenic mice were stained positively for Mdm4 by immunohistochemistry (IHC) (Figure 2B, 2E), indicating overexpression of Mdm4 contributed to tumorigenesis. Consistent with previous studies [26], 70% of tumours (7 of 10) from Mdm4Tg6 and 67% of tumours (6 of 9) from Mdm4Tg15 mice showed p53 IHC staining. More than half of the tumours (10 of 19) had positive staining for both Mdm4 and p53 (Fig 2F and 2G), which was consistent with our previous data [26]. The lymphomas were stained for T-cell (CD3) and B-cell specific markers (B220) to determine the cell of origin; lymphomas from the Mdm4Tg6 mice were all of B cell origin (5/5), and those from Mdm4Tg15 mice were of both B and T cell origin (Figure 2H–J and data not shown). To determine whether Mdm4 is suppressing p53 activity and propelling tumour growth, Mdm4 knockdown experiments were performed in a primary sarcoma tumour cell line from an Mdm4Tg15 mouse (Figure 2K). Mdm4 knockdown significantly increased the transcript levels of p53 downstream target genes (Figure 2L), and slowed proliferation (Figure 2M). Thus, decreasing Mdm4 levels allowed reactivation of functional p53 in these cells.
Figure 1.
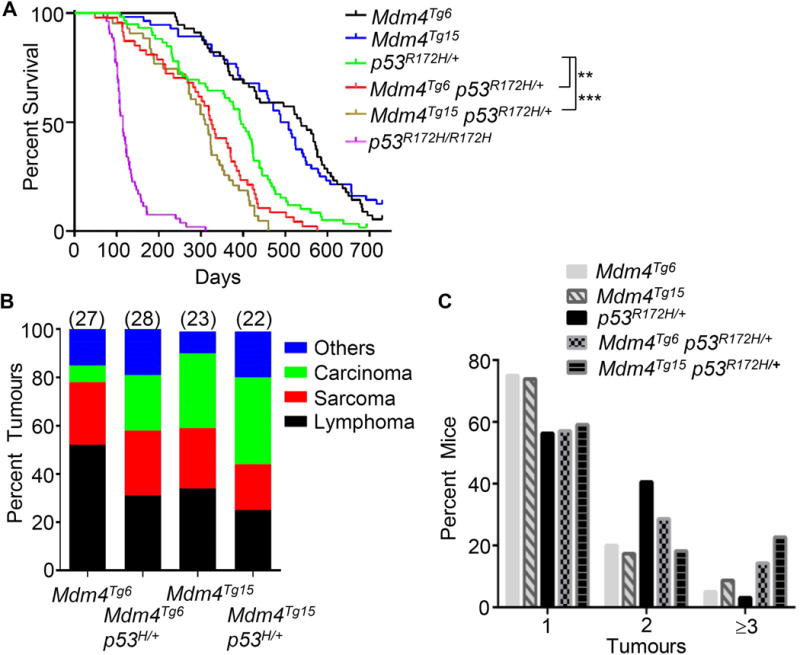
Mdm4 overexpression accelerates tumorigenesis in p53R172H/+mice. Kaplan-Meier survival curves of Mdm4Tg6 (n=53), Mdm4Tg15 (n=49), p53R172H/+ (n=58), Mdm4Tg6 p53R172H/+ (n=47), Mdm4Tg15 p53R172H/+ (n=43), and p53R172H/R172H (n=53) mice with BALB/cJ background. (B) The tumour spectrum of Mdm4Tg6, Mdm4Tg15, Mdm4Tg6, p53R172H/+ and Mdm4Tg15 p53R172H/+ mice presented as the percentage of each tumour type out of the total number of tumours for each genotype. The total number of tumours examined by pathologists for each genotype is indicated in parentheses above each bar. (C) Overexpression of Mdm4 increased multiple tumour incidences in the double mutant mice. ** indicates p<0.01 and *** indicates p < 0.001.
Table 1.
Tumour spectrum in Mdm4 transgenic mice in the BALB/cJ background
| Mdm4Tg6 (n=20) | Mdm4Tg15 (n=23) | ||
|---|---|---|---|
|
| |||
| Lymphoma | 56% | Lymphoma | 34% |
| Sarcoma | 26% | Sarcoma | 25% |
| Haemangiosarcoma | 7(26%) | Haemangiosarcoma | 4(13%) |
| Fibrosarcoma | 1(3%) | ||
| Osteosarcoma | 3(9%) | ||
|
| |||
| Carcinoma | 7% | Carcinoma | 31% |
| Mammary Adenocarcinoma | 1(4%) | Mammary Adenocarcinoma | 2(6%) |
| Alveolar-Bronchiolar | 1(4%) | Alveolar-Bronchiolar | 4(13%) |
| Leydig cell* | 4(13%) | ||
|
| |||
| Other tumour | 11% | Other tumour | 9% |
| Testicular interstitial cell adenoma | 2(8%) | Testicular interstitial cell adenoma | 3(9%) |
| Alveolar-bronchiolar Adenoma | 1(4%) | ||
|
| |||
| Tumour totals | 27 | 32 | |
One Leydig cell carcinoma metastasized to the lung
Figure 2.
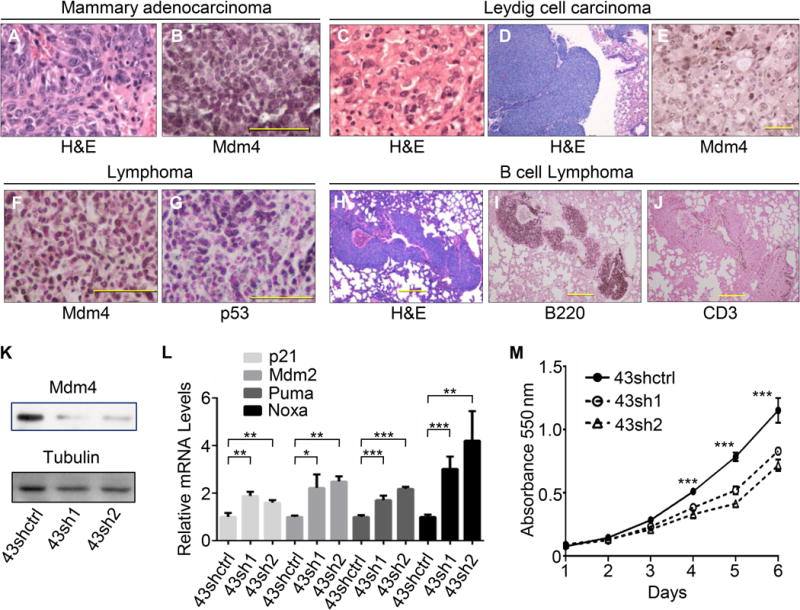
Mdm4 overexpression induces different tumour types and stabilizes p53 in the tumors. Representative H&E staining of a mammary adenocarcinoma (A) and a Leydig cell carcinoma (C) in Mdm4Tg15 mice (40× objective magnification). H&E staining of a Leydig cell carcinoma metastasized to lung (10× objective magnification) (D). IHC staining for Mdm4 in a mammary adenocarcinoma (B), and in a Leydig cell carcinoma (E). Double positive IHC staining for Mdm4 and p53 in a Mdm4Tg6 lymphoma (G, H). Mdm4Tg6 transgenic mice developed B cell lymphoma (H–J) (10× objective magnification). Knockdown of Mdm4 in Mdm4Tg15 tumour cell line 43 (K) upregulated p53 downstream targets (L) and decreased tumour cell proliferation (M). Scale bar = 200 μm in (B) (D) (E) (F) (G), and 500 μm in (H). *, ** and *** indicate p <0.05, p <0.01 and p < 0.001, respectively.
Overexpression of Mdm4 cooperates with p53R172H/+ in tumorigenesis with significant reduction of p53 LOH
The p53 pathway is dampened or altered by multiple mechanisms and a combination of molecular events may cooperate to undermine p53 function [1,33]. To investigate the effects of Mdm4 overexpression in p53R172H/+ mice, we crossed Mdm4Tg6 and Mdm4Tg15 transgenic mice to p53R172H/+ mice (all mice were in a BALB/cJ background). The median survival of Mdm4Tg6 p53R172H/+ (325 days) and Mdm4Tg15 p53R172H/+ (310 days) was significantly shorter than p53R172H/+ mice (395 days) (Figure 1A), demonstrating that high levels of Mdm4 cooperate with mutant p53R172H in tumorigenesis. Noticeably, Mdm4Tg6 p53R172H/+ and Mdm4Tg15 p53R172H/+ mice had more Leydig cell carcinomas than p53R172H/+ mice, suggesting again that overexpression of Mdm4 contributes to Leydig cell tumorigenesis (Table 2). RT-qPCR was used to examine the level of expression of Mdm4 in 20 tumour samples from double-mutant mice. All showed significantly higher Mdm4 mRNA levels in double mutant tumours than those in p53R172H/+ tumours (Figure 3A). Consistently, 82% (9 of 11) of tumours from Mdm4Tg6 p53R172H/+ and 89% (8 of 9) of tumours from Mdm4Tg15 p53R172H/+ mice had positive IHC staining for Mmd4 (Figure 3B and data not shown), strongly supporting that high levels of Mdm4 contributed to the worse tumour phenotypes in the double mutant mice. Additionally, only one p53R172H/+ mouse (3% of the cohort) had three tumours, but four of Mdm4Tg6 p53R172H/+ mice had three or more tumours (14% of the cohort) and five of Mdm4Tg15 p53R172H/+ mice had three tumours (23% of the cohort); the average tumour incidence per mouse for p53R172H/+, Mdm4Tg6 p53R172H/+ and Mdm4Tg15 p53R172H/+ were 1.41, 1.71 and 1.64 (Figure 1C), respectively, indicating that overexpression of Mdm4 contributed to a worse tumour phenotype in the presence of a p53 missense mutation.
Table 2.
Tumour spectrum in Mdm4 transgene and p53R172H/+ double mutant mice in the BALB/cJ background
|
p53R172H/+ (n=32) |
Mdm4Tg6 p53R172H/+ (n=28) |
Mdm4Tg15 p53R172H/+ (n=22) |
p53H/H (n=24) |
||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Lymphomas | 36% | Lymphomas | 40% | Lymphomas | 33% | Lymphomas | 67% |
| Sarcomas | 22% | Sarcomas | 27% | Sarcomas | 19% | Sarcomas | 20% |
| Haemangiosarcoma | 2(4%) | Haemangiosarcoma | 9(19%) | Haemangiosarcoma | 4(11%) | Haemangiosarcoma | 5(17%) |
| Osteosarcoma | 6(13%) | Osteosarcoma | 1(2%) | Leiomyosarcoma | 2(6%) | Undifferentiated sarcoma | 1(3%) |
| Anaplastic | 2(4%) | Salivary | 1(2%) | Rhabdomyosarcoma | 1(3%) | ||
| Spindle cell | 1(2%) | ||||||
| Rhabdomyosarcoma | 1(2%) | ||||||
|
| |||||||
| Carcinomas | 36% | Carcinomas | 23% | Carcinomas | 36% | Carcinomas | 7% |
| Adenocarcinoma | Adenocarcinoma | Adenocarcinoma | Alveolar-Bronchiolar | 1(3%) | |||
| Mammary | 12(27%) | Mammary | 4(8%) | Mammary | 7(19%) | Leydig cell | 1(3%) |
| Bladder | 1(2%) | Alveolar-Bronchiolar | 3(6%) | Alveolar-Bronchiolar | 1(3%) | ||
| Squamous cell | 1(2%) | Leydig cell | 4(8%) | Leydig cell | 5(14%) | ||
| Leydig cell | 1(2%) | ||||||
| Carcinoma NOS | 1(2%) | ||||||
|
| |||||||
| Other tumours | 7% | Other tumours | 10% | Other tumours | 11% | Other Tumours | 7% |
| Adenoma | Spindle cell tumour | 1(2%) | Pheochromocytoma | 1(3%) | Pleomorphic tumour | 1(3%) | |
| Bronchioloalveolar | 1(2%) | Anaplastic tumour | 1(2%) | Adenoma | Adenoma | 1(3%) | |
| Hepatoma | 1(2%) | Adenoma | Bronchioloalveolar | 1(3%) | |||
| Haematoma | 1(2%) | Bronchioloalveolar | 3(6%) | Salivary gland | 1(3%) | ||
| Papillary | 1(3%) | ||||||
|
| |||||||
| Tumour totals | 45 | 48 | 36 | 30 | |||
Figure 3.
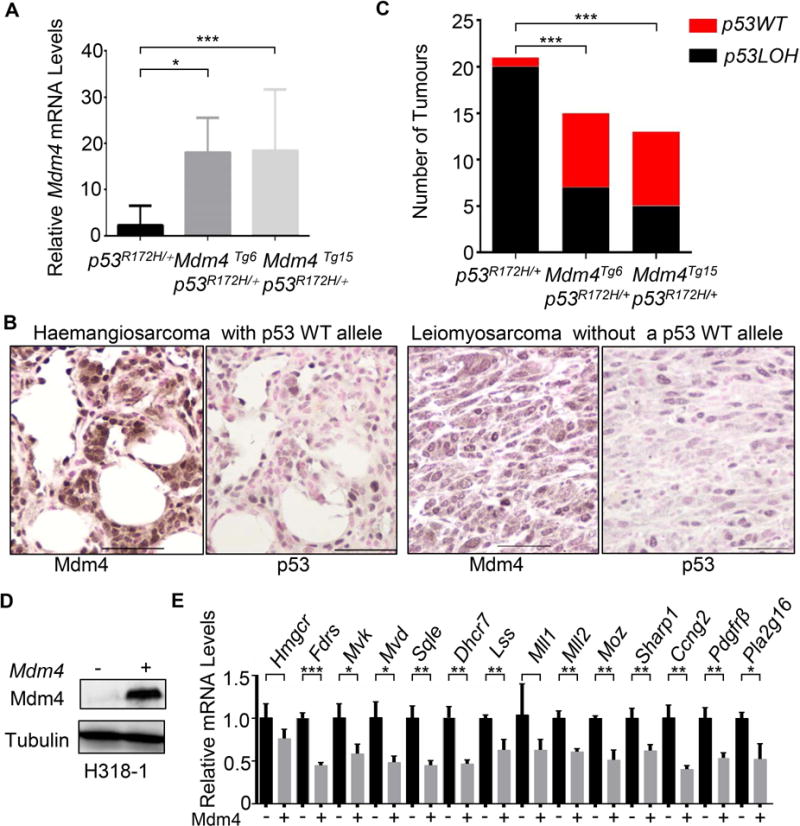
Mdm4 overexpression decreases p53 Loss of Heterozygosity (LOH) in Mdm4Tg6 p53R172H/+ and Mdm4Tg15 p53R172H/+ tumours. (A) The expression levels of Mdm4 were higher in the double mutant tumours as determined by RT-qPCR. (B) Representative IHC staining for Mdm4 and p53 in Mdm4Tg6 p53R172H/+ tumours with or without p53 LOH. (C) Decreased frequency of p53 LOH in double mutant tumours. (D) Overexpression of Mdm4 in H318-1 osteosarcoma cell line repressed the expression of mutant p53 target genes (E). *, ** and *** indicate p <0.05, p <0.01 and p < 0.001, respectively.
We next investigated the status of the p53 wild type allele in p53R172H/+, Mdm4Tg6 p53R172H/+ and Mdm4Tg15 p53R172H/+ tumours. Twenty of 21 (95%) tumours examined from p53R172H/+ mice had lost the wild type p53 allele (Figure 3C), similar to the previous reports from p53+/− tumours in a BALB/cJ background [28,34]. These data indicated that the presence of p53R172H allele does not mitigate the selective pressure for p53 LOH in the p53R172H/+ tumours. Strikingly, 47% (7/15) of the tumours from Mdm4Tg6 p53R172H/+ and 38% (5/13) from Mdm4Tg15 p53R172H/+ mice retained the wild type p53 allele, demonstrating that overexpression of Mdm4 reduced the selective pressure to lose the wild type p53 in these tumours during tumour evolution (Figure 3C). Consistently, IHC performed on 20 tumours from Mdm4Tg6 p53R172H/+ and Mdm4Tg15 p53R172H/+ showed that only tumours with Mdm4 overexpression had stabilized p53, including 10 tumours with p53 LOH (tumours only having mutant p53 protein) and 6 tumours without p53 LOH (tumours having both mutant and wild type p53 proteins) (Figure 3B). These data indicated that overexpression of Mdm4 stabilizes p53 irrespective of its mutation status in tumours.
Mutant p53 shows gain-of-function activities through multiple downstream regulators and pathways including activation of the mevalonate pathway [35], chromatin methyltransferases and acetyltransferases [36], TGFβ/p63 [37], PDGFRβ [38] and Pla2g16 [30]. To investigate whether increased Mdm4 levels could also affect mutant p53 gain of function, Mdm4 was over expressed in H318-1 osteosarcoma cells (Figure 3D), a mouse primary tumour cell line generated from p53R172H/+ osteosarcomas that had lost the wild-type p53 allele [39]. Analysis of mRNA levels of the reported mutant p53 target genes showed decreased expression of these genes except for Hmgcr and Mll1 (Figure 3E). Thus, increased Mdm4 levels stabilized mutant p53 and decreased its gain-of-function potential by masking its transcriptional activation domain.
Overexpression of Mdm4 cooperates with oncogenic K-ras in tumorigenesis
p53 loss accelerates lung adenocarcinoma development in K-rasLA1/+ mice [18]. Since approximately 6% of lung adenocarcinoma patients with K-ras mutations have increased copy number of MDM4 (www.Cbioportal.com), we asked whether Mdm4 overexpression could cooperate with the K-RasG12D mutation in lung tumorigenesis in vivo. We therefore crossed both Mdm4Tg6 and Mdm4Tg15 mice to K-rasLA1/+ mice in a C57BL/6J and 129/SvJ mixed background. Overexpression of Mdm4 significantly accelerated tumorigenesis in both Mdm4Tg6 K-rasLA1/+ (n=20) and Mdm4Tg15 K-rasLA1/+ (n=14) mice compared to K-rasLA1/+ littermates (Figure 4A). Compared with the median survival of K-rasLA1/+ mice (n=20) at 341 days, both double mutant mice had significantly shorter median survival at 267 and 255 days, respectively. Additionally, Mdm4 IHC staining was positive in 60% of lung adenocarcinomas from the double mutant mice (Figure 4B), suggesting that overexpression of Mdm4 contributes to lung tumorigenesis.
Figure 4.
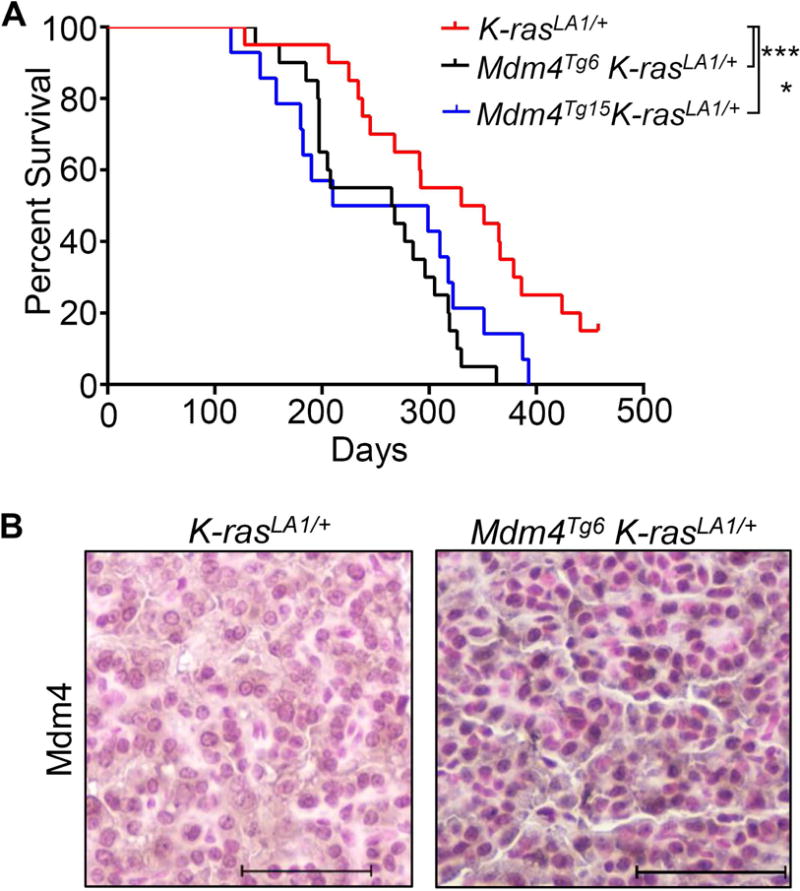
Mdm4 overexpression accelerates tumorigenesis in K-rasLA1mice. (A) Kaplan-Meier survival curves of K-rasLA1 (n=17), Mdm4Tg6 K-rasLA1 (n=20), and Mdm4Tg15 K-rasLA1 (n=14). (B) Positive IHC staining for Mdm4 in a representative lung adenocarcinoma. * and *** indicate p <0.05 and p <0.001, respectively.
Mdm4 has p53-independent function in vivo
MDM4 interacts with a variety of proteins in different human cell lines, such as p21, 14-3-3gamma, ASPP, ARF, p300, Smads, and RB [19,21–23,40–42], suggesting it has p53-independent functions. Also, MDM4 promotes genomic instability by associating with Nbs1 to promote cell transformation [25]. In order to investigate whether different levels of Mdm4 overexpression contribute to its p53-independent functions, we chose Mdm4Tg1 mice with a relatively low expression level and Mdm4Tg15 mice with a higher expression level to generate the cohorts of Mdm4Tg1 p53−/− (n=39) and Mdm4Tg15 p53−/− (n=27). The survival curves of these two cohorts were indistinguishable from p53−/− (n=53) mice (Figure 5A). The median survival of p53−/−, Mdm4Tg1 p53−/−, and Mdm4Tg15 p53−/− was at 118, 114.5 and 115.5 days, respectively. However, when the survival data were segregated in a gender-specific manner, the median survival of Mdm4Tg15, p53−/− (n=15) male mice was significantly shorter than p53−/− male mice (n=25) (98 vs 130 days, p=0.0042), but the median survival of p53−/− female mice was similar to Mdm4Tg15, p53−/− female mice (data not shown), indicating Mdm4 overexpression accelerated tumorigenesis in a p53-null background through p53-independent functions in these male mice (Figure 5B). In mice with or without the Mdm4Tg1 transgene, p53 loss did not alter the median survival in either male or female cohorts suggesting that the p53-independent function of Mdm4 may depend on the expression levels of Mdm4 (data not shown). Another interesting observation is that six different types of sarcomas arose in p53−/− mice, while both Mdm4Tg1 p53−/− and Mdm4Tg15 p53−/− mice developed a more limited sarcoma type (Table 3). While 34% of p53−/− mice (10/29) and 32% of Mdm4Tg1 p53−/− mice (6/19) had two or more tumours, 55% of Mdm4Tg15 p53−/− mice (6/11) had two or more tumours (Figure 5C), implying overexpression of Mdm4 at a higher level led to a worse tumorigenic phenotype even in the absence of p53. In addition, we also observed Mdm4 overexpression in 57% of Mdm4Tg15 p53−/− tumours (4 out of 7 tumours), but not in any of the tumours from p53−/− (8 tumours) or Mdm4Tg1 p53−/− (7 tumours) mice (Figure 5D). Overexpression of Mdm4 in two primary tumour cell lines from p53−/− mice (Figure 5E) showed increased cell proliferation (Figure 5F) suggesting Mdm4 contributed to cell proliferation independent of p53. Taken together, these data indicate that overexpression of Mdm4 in the absence of p53 contributed to tumour development.
Figure 5.
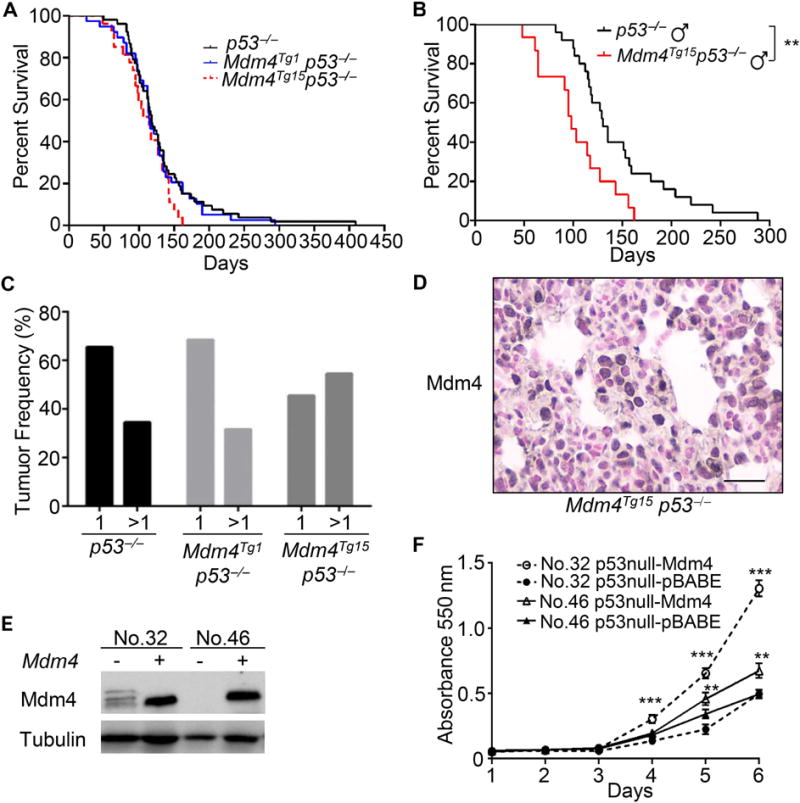
Mdm4 overexpression reveals p53 independent function. (A) Similar Kaplan-Meier survival curves of p53−/− (n=51), Mdm4Tg1 p53−/− (n=36) and Mdm4Tg15 p53−/− (n=28). (B) Mdm4Tg15 p53−/− male mice (n=25) had significant shorter survival than p53−/− male mice (n=15). (C) Mdm4Tg15 p53−/− mice had higher incidence of multiple tumours. (D) Positive IHC staining for Mdm4 in a representative Mdm4Tg15 p53−/− haemangiosarcoma. (E) Overexpression of Mdm4 in two p53−/− primary tumour cell lines (No. 32 and No. 46) accelerated cell proliferation (F). ** and *** indicate p <0.01 and p <0.001, respectively.
Table 3.
Tumour spectrum in Mdm4 transgene and p53−/− double mutant mice in the BALB/cJ background
|
p53−/− (n=29) |
Mdm4Tg1 p53−/− (n=19) |
Mdm4Tg15 p53−/− (n=11) |
|||
|---|---|---|---|---|---|
|
| |||||
| Lymphoma | 52% | Lymphoma | 72% | Lymphoma | 47% |
| Sarcoma | 43% | Sarcoma | 20% | Sarcoma | 53% |
| Haemangiosarcoma | 12(40%) | Haemangiosarcoma | 4(16%) | Haemangiosarcoma | 8(47%) |
| Rhabdomyosarcoma | 1(2%) | Osteosarcoma | 1(4%) | Anaplastic sarcoma | 1(6%) |
| Anaplastic sarcoma | 1(2%) | ||||
| Osteosarcoma | 1(2%) | ||||
| Pleomorphic | 2(5%) | ||||
| Poor differentiated | 1(2%) | ||||
|
| |||||
| Carcinoma | 2% | Carcinoma | 0% | Carcinoma | 0% |
| Adenocarcinoma | 1(2%) | ||||
|
| |||||
| Other tumour | 2% | Other tumour | 8% | Other tumour | 0% |
| Testicular tumour | 1(2%) | chondroma | 1(4%) | ||
| phenochromocytoma | 1(4%) | ||||
|
| |||||
| Tumour totals | 42Δ | 25# | 17@ | ||
9 mice had two tumours and two mice had three tumours.
Six mice had two tumours.
Six mice had two tumours and one mouse had three tumours.
Discussion
High levels of MDM4 were first observed in approximately 19% of breast cancer patients [12]. Subsequently, 14% of invasive lobular breast cancers were also found to have MDM4 amplification [43], and TCGA data demonstrated that MDM4 amplification is a common event in breast cancer [14]. In this study, using two Mdm4 transgenic mouse lines in a BALB/cJ background which is sensitized to breast tumour development, we demonstrated that Mdm4 overexpression directly contributes to mammary tumorigenesis. However, the low incidence of mammary tumours in Mdm4 overexpression transgenic mice suggests that additional genetic changes are required. The other possibility is that high Mdm4 levels cooperated more readily in lymphomagenesis in Mdm4 overexpressing mice.
Mdm4 overexpression in a p53R172H/+ background also accelerated tumorigenesis and reduced the selective pressure to lose wild type p53 in tumours demonstrating that high Mdm4 levels indeed contribute to tumorigenesis in the presence of a hot spot p53R172H mutation. Thus tumours with both high Mdm4 and p53 mutations are likely to retain the wild type p53 allele suggesting that it will be beneficial to use Mdm4 inhibitors to treat these cancers. In the above examples, high levels of Mdm4 stabilized mutant p53 in many but not all tumours, yet no obvious gain-of-function phenotypes of mutant p53 were observed, suggesting Mdm4 binding to mutant p53 may hinder its association with other proteins to inhibit gain of function activities.
Finally, overexpression of Mdm4 changed the tumour spectrum in p53−/− mice. Specifically, a higher percentage of Mdm4Tg15 p53−/− mice developed multiple tumours compared to p53−/− mice. Mdm4Tg15 p53−/− male mice had a significant shorter median survival compared to p53−/− male mice, demonstrating that Mdm4 overexpression had a p53-independent function. It is interesting to note that overexpression of Mdm4 accelerated tumorigenesis only in male mice. Mdm4 binds numerous other proteins besides p53 such as Smad, Nbs1 and UXT; perhaps one or more of these proteins are present at different levels in males versus females [25,41,44]. Additionally, high levels of Mdm4 also contribute to worse tumour phenotypes in Mdm4Tg6 p53R172H/+ and Mdm4Tg15 p53R172H/+ mice, which could be at least partly due to p53 independent function of Mdm4. But the mechanism of these p53-independent functions of Mdm4 in tumorigenesis will need further investigation. Since MDM4 overexpression or amplification is a common event in a variety of human cancers, and Mdm4 overexpression reduces the need to lose the wild type p53 allele, Mdm4 inhibitors may activate p53. Moreover, Mdm4 inhibition induces less toxicity in adult mice compared to Mdm2 in vivo [32,45–47], thus making Mdm4 a more attractive therapeutic target to activate p53 in the tumours with wild type p53.
Acknowledgments
We thank Ana Elizondo-Fraire for help with mouse maintenance and dissections. Drs. Laura Pageon, Mark McArthur, and Elizabeth Whitley provided some pathology support. This work was supported by NIH grant CA47296 to G. Lozano.
Footnotes
Conflict of interest statement:
The authors declare that there is no conflict of interest.
Author contributions
SX was responsible for the experimental design, undertaking the experiments, data analysis and interpretation and manuscript drafting; VP, NA and YZ carried out some of the immunohistochemical staining, RT-qPCR assays and data analyses; MJY and DK performed pathological analyses of H&E-stained tissue sections; and GL conceived the experiments, interpreted data and revised and finalized the manuscript.
References
- 1.Wasylishen AR, Lozano G. Attenuating the p53 pathway in human cancers: many means to the same end. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026211. pii: a026211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 3.Jones SN, Roe AE, Donehower LA, et al. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 4.Parant J, Chavez-Reyes A, Little NA, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 5.Migliorini D, Denchi EL, Danovi D, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–5538. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finch RA, Donoviel DB, Potter D, et al. mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002;62:3221–3225. [PubMed] [Google Scholar]
- 7.Shvarts A, Steegenga WT, Riteco N, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. Embo J. 1996;15:5349–5357. [PMC free article] [PubMed] [Google Scholar]
- 8.Pant V, Xiong S, Jackson JG, et al. The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 2013;27:1857–1867. doi: 10.1101/gad.227249.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh M, Huang K, Berberich SJ. Overexpression of Mdm2 and MdmX fusion proteins alters p53 mediated transactivation, ubiquitination, and degradation. Biochemistry. 2003;42:2291–2299. doi: 10.1021/bi0271291. [DOI] [PubMed] [Google Scholar]
- 10.Pant V, Xiong S, Iwakuma T, et al. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci U S A. 2011;108:11995–12000. doi: 10.1073/pnas.1102241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang L, Yan Z, Liao X, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci U S A. 2011;108:12001–12006. doi: 10.1073/pnas.1102309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danovi D, Meulmeester E, Pasini D, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835–5843. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valentin-Vega YA, Barboza JA, Chau GP, et al. High levels of the p53 inhibitor MDM4 in head and neck squamous carcinomas. Hum Pathol. 2007;38:1553–1562. doi: 10.1016/j.humpath.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gembarska A, Luciani F, Fedele C, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18:1239–1247. doi: 10.1038/nm.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 17.Terzian T, Suh YA, Iwakuma T, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 19.Jin Y, Zeng SX, Sun XX, et al. MDMX promotes proteasomal turnover of p21 at G1 and early S phases independently of, but in cooperation with, MDM2. Mol Cell Biol. 2008;28:1218–1229. doi: 10.1128/MCB.01198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeBron C, Chen L, Gilkes DM, et al. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. EMBO J. 2006;25:1196–1206. doi: 10.1038/sj.emboj.7601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin Y, Dai MS, Lu SZ, et al. 14-3-3gamma binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. Embo J. 2006;25:1207–1218. doi: 10.1038/sj.emboj.7601010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JH, Lu H. 14-3-3Gamma inhibition of MDMX-mediated p21 turnover independent of p53. J Biol Chem. 2011;286:5136–5142. doi: 10.1074/jbc.M110.190470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson MW, Lindstrom MS, Berberich SJ. MdmX binding to ARF affects Mdm2 protein stability and p53 transactivation. J Biol Chem. 2001;276:25336–25341. doi: 10.1074/jbc.M010685200. [DOI] [PubMed] [Google Scholar]
- 24.Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci. 2007;120:371–378. doi: 10.1242/jcs.03362. [DOI] [PubMed] [Google Scholar]
- 25.Carrillo AM, Bouska A, Arrate MP, et al. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene. 2015;34:846–856. doi: 10.1038/onc.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong S, Pant V, Suh YA, et al. Spontaneous tumorigenesis in mice overexpressing the p53-negative regulator Mdm4. Cancer Res. 2010;70:7148–7154. doi: 10.1158/0008-5472.CAN-10-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ullrich RL, Bowles ND, Satterfield LC, et al. Strain-dependent susceptibility to radiation-induced mammary cancer is a result of differences in epithelial cell sensitivity to transformation. Radiation Res. 1996;146:353–355. [PubMed] [Google Scholar]
- 28.Kuperwasser C, Hurlbut GD, Kittrell FS, et al. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. a model for Li-Fraumeni syndrome. Am J Pathol. 2000;157:2151–2159. doi: 10.1016/S0002-9440(10)64853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch JG, Gu X, Han Y, et al. Mammary tumor modifiers in BALB/cJ mice heterozygous for p53. Mamm Genome. 2007;18:300–309. doi: 10.1007/s00335-007-9028-2. [DOI] [PubMed] [Google Scholar]
- 30.Xiong S, Tu H, Kollareddy M, et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc Natl Acad Sci U S A. 2014;111:11145–11150. doi: 10.1073/pnas.1404139111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Post SM, Quintas-Cardama A, Pant V, et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell. 2010;18:220–230. doi: 10.1016/j.ccr.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong S, Van Pelt CS, Elizondo-Fraire AC, et al. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci U S A. 2006;103:3226–3231. doi: 10.1073/pnas.0508500103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eischen CM, Lozano G. The Mdm network and its regulation of p53 activities: a rheostat of cancer risk. Hum Mutat. 2014;35:728–737. doi: 10.1002/humu.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umesako S, Fujisawa K, Iiga S, et al. Atm heterozygous deficiency enhances development of mammary carcinomas in p53 heterozygous knockout mice. Breast Cancer Res. 2005;7:R164–170. doi: 10.1186/bcr968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freed-Pastor WA, Mizuno H, Zhao X, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Sammons MA, Donahue G, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–211. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adorno M, Cordenonsi M, Montagner M, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 38.Weissmueller S, Manchado E, Saborowski M, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–394. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lang GA, Iwakuma T, Suh YA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Bergamaschi D, Samuels Y, Zhong S, et al. Mdm2 and mdmX prevent ASPP1 and ASPP2 from stimulating p53 without targeting p53 for degradation. Oncogene. 2005;24:3836–3841. doi: 10.1038/sj.onc.1208535. [DOI] [PubMed] [Google Scholar]
- 41.Kadakia M, Brown TL, McGorry MM, et al. MdmX inhibits Smad transactivation. Oncogene. 2002;21:8776–8785. doi: 10.1038/sj.onc.1205993. [DOI] [PubMed] [Google Scholar]
- 42.Zhang H, Hu L, Qiu W, et al. MDMX exerts its oncogenic activity via suppression of retinoblastoma protein. Oncogene. 2015;34:5560–5569. doi: 10.1038/onc.2015.11. [DOI] [PubMed] [Google Scholar]
- 43.Ciriello G, Gatza ML, Beck AH, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi M, Ganapathy S, Zeng W, et al. UXT, a novel MDMX-binding protein, promotes glycolysis by mitigating p53-mediated restriction of NF-kappaB activity. Oncotarget. 2015;6:17584–17593. doi: 10.18632/oncotarget.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ringshausen I, O’Shea CC, Finch AJ, et al. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10:501–514. doi: 10.1016/j.ccr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Grier JD, Xiong S, Elizondo-Fraire AC, et al. Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol Cell Biol. 2006;26:192–198. doi: 10.1128/MCB.26.1.192-198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia D, Warr MR, Martins CP, et al. Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 2011;25:1746–1757. doi: 10.1101/gad.16722111. [DOI] [PMC free article] [PubMed] [Google Scholar]