

Abstract
Nucleotide excision repair (NER) eliminates a broad variety of helix-distorting DNA lesions that can otherwise cause genomic instability. NER comprises two distinct sub-pathways: global genomic NER (GG-NER) operating throughout the genome, and transcription-coupled NER (TC-NER) preferentially removing DNA lesions from transcribing DNA strands of transcriptionally active genes. Several NER factors undergo post-translational modifications, including ubiquitination, occurring swiftly and reversibly at DNA lesion sites. Accumulating evidence indicates that ubiquitination not only orchestrates the spatio-temporal recruitment of key protein factors to DNA lesion sites but also the productive assembly of NER preincision complex. This review will be restricted to the latest conceptual understanding of ubiquitin-mediated regulation of initial damage sensors of NER, i.e., DDB, XPC, RNAPII and CSB. We project hypothetical NER models in which ubiquitin-specific segregase, valosin-containing protein (VCP)/p97, plays an essential role in timely extraction of the congregated DNA damage sensors to functionally facilitate the DNA lesion elimination from the genome.
Graphical Abstract
INTRODUCTION
In the beginning there was ‘light repair’ (1), also known as photolyase- and light-driven host cell reactivation (2). Immediately afterwards, the so called ‘dark repair’ now known as excision repair, discovered by Richard Setlow (3), heralded a new expansive era in DNA repair research with many unique offshoots and intense areas of enquiry that culminated in the ultimate recognition and award of 2015 Nobel Prize in chemistry (4). In between the elapsed span of time, many unique discoveries have continued to shed bright light on intricate nuances of DNA repair. While much of the basic mechanistic understanding has already been delineated, ever-increasing knowledge, from peeling of the ever-complex regulatory layers of the DNA repair phenomenon and its wide-ranging implications to human health, has continued to infuse enthusiasm in new entrants as well as kept propelling the field forward. Alongside many stalwarts and preeminent laboratories around the world, our research program has also toiled in the DNA repair arena with a sharper focus on nucleotide excision repair, a sub-area championed by the Nobelist Aziz Sancar.
Genome is the most precious resource to sustain organismal life. Yet, genome is constantly challenged by exposures to ubiquitously occurring endogenous and exogenous DNA damaging agents. Repair of DNA lesions enables cells to overcome genotoxicity, maintain genome integrity and retain normal cellular functions. Genomic instability resulting from a cell’s inability to effectively repair its DNA damage has clear association with aging and disease pathogenesis, ranging from inherent susceptibility to induction of frank neoplasia. The significance and causative relationship between the genotoxic source, (e.g., solar ultraviolet (UV) radiation), the target (DNA), the culprit (a genomic DNA lesion), the mechanism (faulty repair causing sequence alterations in an essential gene), the regulatory breakdown (e.g., inability to exercise epigenetic control) and the deleterious repercussions (forms of carcinoma), is irrefutable and experimentally demonstrable in almost any DNA repair research laboratory around the globe. Much has already been unraveled and many clear concepts have evolved regarding the basic components and function of DNA repair machinery needed to ensure genomic stability (5–7). Latest DNA repair research has embraced newer complexities and delves into new interaction ‘pathway boxes’ to discern and integrate the effects exerted by other structural and functional features of the cell (8–10). For example, our research program has lately strived to dissect the dynamics of DNA damage recognition and its elaborate processing against the regulatory backdrop of specific ubiquitination and deubiquitination processes. Identification of these and other critical factors that influence the overall genomic stability, so fundamental to normal cellular existence, has obvious significance in understanding the cause and treating human diseases. In this regard, the DNA repair-related ubiquitin story has launched one of the most exciting new areas in molecular and cellular biology with significant biomedical implications (11).
NUCLEOTIDE EXCISION REPAIR
Among several DNA repair pathways, NER is the most versatile DNA repair system. NER eliminates a broad variety of helix-distorting DNA lesions, including UV-induced cyclobutane pyrimidine dimers (CPDs), 6-4 photoproducts (6-4PPs), and various chemically induced bulky adducts. NER consists of two distinct sub-pathways: global genomic NER (GG-NER), which operates throughout genome, and transcription-coupled NER (TC-NER), which removes DNA lesions from transcribed DNA strands of transcriptionally active genes (12,13). Impaired NER activity is associated with several rare autosomal recessive genetic disorders, illustrating its biological significance. For example, Xeroderma pigmentosum (XP) syndrome, characterized by extreme sun/UV sensitivity and predisposition of skin cancer, is caused by mutations in genes encoding proteins (e.g., XPA, XPB, XPC, XPD, XPE, XPF and XPG) involved in GG-NER (14). Whereas, TC-NER defects in Cockayne syndrome (CS) or UV-sensitive syndrome can result from mutations in CSA and CSB as well as UV-stimulated scaffold protein A (UVSSA) genes (15–17). The Cockayne Syndrome (CS) has distinctive features of growth retardation, UV sensitivity and premature ageing.
GG-NER
GG-NER can be reconstituted in vitro in a “cut and paste” reaction with only a few core components which include XPA, XPC-hHR23B, TFIIH, ERCC1-XPF, XPG, RPA, PCNA, DNA polymerase and DNA ligase I. The entire process constitutes damage recognition, dual incision, and gap-filling DNA synthesis steps (18–20).
The main damage recognition complex in GG-NER is XPC-hHR23B-Centrin-2 (heretofore referred as XPC) (21,22). XPC complex uses a multistep damage recognition strategy for ensuring the high level of damage discrimination (22–24). The complex, however, does not directly contact the DNA strand containing the lesion but the opposite DNA strand, while dislocates damage-containing base pairs inducing a flipped-out conformation (25). This binding allows XPC complex to serve as a platform for recruiting TFIIH (26,27), to verify DNA lesions with the help of XPA. The damage verification is carried out by a so-called tripartite DNA lesion selection system at levels of base-pairing, strand-treading and altered nucleotide binding (28–30). First, XPC detects DNA bubbles with base-pairing disruptions. Second, the TFIIH complex is loaded 5′ to the lesion site and uses its XPB and XPD components to translocate along both DNA strands and unwind DNA towards the lesion (31). When TFIIH encounters a bulky lesion on the DNA strand being scanned by XPD, both XPB and XPD motors are stalled and the XPA enhances the stalling (29). Third, after CAK complex disassociates from TFIIH (32), the core TFIIH and XPA together demarcate a lesion-containing DNA bubble structure with the RPA binding to the undamaged DNA strand (33). Subsequently, the XPF-ERCC1 and XPG nucleases are recruited for making the dual incision to allow the removal of ~24–32 nt oligo nucleotide containing the DNA lesion (34–37). The incision on 5′ side is carried out by the heterodimer XPF-ERCC1, followed by the incision on 3′ side by XPG (36). The lesion-containing oligonucleotide remains associated with TFIIH until eventually released and further processed by nuclease(s) (38). Subsequent gap-filling DNA synthesis is performed by the concerted action of pol δ, ε or κ that is aided by the cofactors PCNA, RFC and RPA (39) and the nicks are finally sealed by specific DNA ligases to complete the repair (19,40).
Besides XPC, UV damaged DNA binding protein (UV-DDB or DDB), a heterodimer of DDB1 and DDB2 proteins, also recognizes DNA lesions (41–43). GG-NER of CPDs in vivo requires both DDB and XPC proteins and is carried out at a much slower rate than that of 6-4PPs. Perhaps, CPDs cause minor helix distortion than 6-4PPs, thus the recognition of CPDs by XPC requires additional support from DDB, especially for the repair in the constrained chromatin environment (44–46). The crystal structure of DDB1-DDB2 complex showed that DDB2 interacts with two (BPA and BPC) of three seven-bladed beta-propellers (BPA, BPB and BPC) of DDB1. The lesion is held exclusively by the WD40 domain of DDB2 (47), where a DDB2 hairpin inserts into the minor groove, extrudes the photodimer into a binding pocket. The tightly localized probing of the photolesions, combined with proofreading in the photodimer pocket, enables DDB2 to detect lesions refractory to detection by other damage surveillance proteins (47).
TC-NER
While GG-NER employs DDB heterodimer and XPC complex to initiate the repair process, TC-NER utilizes elongating RNA polymerase II (RNAPII) and CSB as damage sensors (48,49). When elongating RNAPII stalls at the transcription-blocking lesions in transcribed strand, the stalled RNAPII stabilizes its interaction with CSB (50,51). Subsequently, the stalled RNAPII and CSB together recruit CSA [in CSA-DDB1-Cullin 4A (Cul4A) complex associated with COP9 signalosome], TFIIH and core NER factors as well as histone acetyltransferase p300 (52). Factors such as TFIIS, HMGN1 and XAB2 come to stalled RNAPII in a CSA-dependent manner. Yet, the exact roles of CSB, CSA and other non-NER factors are not clear (53–56). Nonetheless, once lesions are verified by TFIIH, the stalled RNAPII with other components of elongating machinery is resolved or backtracks, and the transcription elongation process funneled into assembly of preincision complex of NER.
UBIQUITINATION IN GG-NER
DDB2
DDB2 is the first GG-NER factor known to be regulated at posttranslational level by the ubiquitin-proteasome system (UPS) (57–59). In eukaryotic cells, UPS mediates a selective degradation of many cellular proteins (60). In UPS, the target proteins are modified by multiple moieties of ubiquitin, a highly conserved protein of 76 amino acids, via a cascade reaction of three enzymes, namely, E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme and E3 ubiquitin ligase. The first ubiquitin is attached to lysine residue of target protein, followed by chain extension at lysine (K) 6, 11, 27, 29, 33, 48 or 63 on ubiquitin. The ubiquitin chains with different linkages have distinct structure and properties (61,62). The K48-linked polyubiquitin chains are well established as the canonical signal for proteasomal degradation (63). Whereas, K63 linkages nonproteolytically regulate the protein-interacting properties of ubiquitinated proteins (64). For example, K63-linked ubiquitin chains stabilize the snRNP complex. The similar chains on histone H2A/X serve as transient mediators of protein interactions, recruiting subsequent E3 ubiquitin ligases to initiate a cascade of ubiquitin signaling events in response to DNA damage.
It has been known that UV-DDB heterodimer is part of E3 ubiquitin ligase complex that contains Cul4A (and to a lesser extent, Cul4B) and RING subunit Rbx1 (also known as Roc1) (65). It is the best characterized member of the DDB1-Cul4-Rbx1 (CRL4) ubiquitin ligase family (66). CRL4 complex utilizes DDB1- and Cul4A-associated factors (DCAFs) as substrate receptors and therefore is termed as CRL4DCAF ubiquitin ligase (e.g., CRL4DDB2, CRL4CSA and CRL4Cdt2). All DCAFs harbor a WD40 repeat that mediates substrate association. The WD40 repeats of DDB2, however, have additional capacity of DNA lesion binding (47). The ligase activity of CRL4DDB2 is tightly regulated by its association with COP9 signalosome (CSN) (65). Studies of CRL4DDB2 architecture, targeting and activation have revealed that DDB1 contains three seven-bladed beta-propellers (BPA, BPB and BPC) and a helical C-terminal domain (CTD). While the BPA and BPC domains interact DDB2, the BPB domain interacts with Cul4A (67). The DDB2 lesion-binding interface is specifically tailored toward CPD recognition. In vivo, CRL4DDB2 aids in accessing and detecting CPD lesions embedded within nucleosomes.
CRL4DDB2 mediates auto-ubiquitination of DDB2, mainly at K5, K11, K35 and K40 of its unstructured N terminus, which leads to proteolysis of DDB2 following UV exposure (67). Without DNA damage, CRL4DDB2 is held in an inactive state in complex with CSN. In particular, CSN inhibition of CRL4DDB2 is non-enzymatic, independent of CSN5-mediated cleavage of ubiquitin-like protein Nedd8 (67), whose conjugation to Cullins activates Cullin-based ubiquitin ligase (68). The CSN inhibition of CRL4DDB2 is relieved upon damage binding to the DDB2 module within CSN-CRL4DDB2, which results in steric displacement of CSN by DDB2 substrate binding (67). Importantly, the DDB2 binding to CPD itself does not trigger conformational changes that could serve as a signal for CRL4DDB2 activation, but is rather triggered by substrate binding. Thus, the ligase activity is activated towards its target substrates XPC, histones H2A, H3, and H4 (69–72). These substrates may be within the zone of rotational mobility of CRL4DDB2 ligase.
What could be the functional relevance of DDB2 ubiquitination? Intuitively, DDB2 ubiquitination could affect the lesion handover from DDB to XPC, or from DDB to TFIIH through XPC, since XPC recruits TFIIH for lesion verification (29) and the excised lesion-containing oligo is held by TFIIH (38). Evidently, DDB2 ubiquitination leads to two consequential events- rapid DDB2 degradation (73) and loss of DNA damage binding (71,74). Inhibition of DDB2 degradation by proteasome inhibition delays the departure of DDB2 from DNA damage sites (72) and decreases GG-NER (75). Similarly, mutation of majority of lysine residues within and outside N terminus (K146, K151, K187, K233 and K278) renders DDB2 resistant to self-ubiquitination and UV-induced degradation, but decreases GG-NER of CPDs and survival of mutant DDB2 expressing cells (74). The mutant DDB2 can bind UV-damaged chromatin and the CRL4mutant DDB2 can perform normal XPC ubiquitination. However, as it cannot leave the site and resides for a longer duration on damage chromatin, it negatively affects the TFIIH and XPA recruitment to damage sites. Thus, DDB2 ubiquitination impacts GG-NER via dissociation of DDB2/CRL4DDB2 from damaged chromatin. Recent discovery of involvement of valosin-containing protein (VCP)/p97 in extracting ubiquitinated DDB2 and XPC from damaged chromatin (discussed below) sheds additional light on how DDB2 ubiquitination regulates GG-NER (76,77).
XPC
XPC is a well characterized substrate of CRL4DDB2 (71,78). XPC ubiquitination occurs as early as 15 min following UV-induced damage triggering the rapid accumulation of DDB2, Cul4A and XPC at damage sites (69,72). The levels of XPC-ubiquitin conjugates begin to decline through reconversion to native XPC state at approximately 4 to 8 h after UV exposure. It is not known whether CRL4DDB2 forms K48- or K63-linked ubiquitin chain on XPC. However, CRL4DDB2 forms K48-linked ubiquitin chain on DDB2, which drives pronounced proteolysis of DDB2. Interestingly, UV irradiation does induce demonstrable XPC proteolysis when XPC is not protected by cellular ubiquitin-specific protease 7 (USP7) (77). Thus, it is highly likely that CRL4DDB2 forms K48-linked ubiquitin chain on XPC.
CRL4DDB2-mediated XPC ubiquitination, however, does not appear to drive XPC degradation in normal cells (71,77). XPC ubiquitination increases DNA and damaged DNA binding of XPC without altering its specificity (71). Surprisingly, the ubiquitination inhibits cell-free dual incision in vitro. Thus, tighter association of ubiquitinated XPC to damaged DNA may prevent efficient assembly of preincision complex. In accord, CRL4DDB2-mediated ubiquitination around DNA damage was demonstrated to contribute to XPA recruitment (79). To reconcile these observations, we speculate that the ubiquitinated DDB2 and XPC are required for their extraction from damaged chromatin to enable productive assembly of preincision complex.
XPC is also characterized as a substrate of RNF111 which is identified as a new SUMO (small ubiquitin-like modifier)-targeted ubiquitin ligase (STUbL) (80,81). RNF111 recognizes SUMOylated substrates through three adjacent SUMO-interacting motifs (SIMs) and selectively promotes UV-induced ubiquitination on SUMOylated XPC. The RNF111-mediated ubiquitination can be reconstituted in vitro in two-step SUMOylation and ubiquitination systems, using SUMO-2 and UBC9 for SUMOylation followed by using Ubc13/Mms2 as E2 conjugating enzyme and RNF111 as E3 ligase for ubiquitination (80). Thus, the ubiquitin chain formed on XPC by RNF111 is presumably the non-proteolytic K63-linked ubiquitin.
GG-NER function is regulated by UV-induced SUMOylation and RNF111-mediated ubiquitination (80). RNF111 knockdown results in increased accumulation of XPC to localized DNA damage sites, but decreases UV-induced DNA repair synthesis. Other studies have indicated that RNF111-mediated ubiquitination promotes the release of XPC from damaged DNA and is needed for stable incorporation of XPG and XPF into preincision complex, thereby contributing to efficient GG-NER (81).
Further characterization of XPC SUMOylation also appears to support a functional link between XPC SUMOylation and GG-NER via DDB2 (82). XPC is SUMOylated at multiple sites, which reside within the SUMO-1 consensus sequence ψKxE (ψ= hydrophobic amino acid, K=lysine, x=any amino acid and E=Glutamic acid) (82,83). XPC SUMOylation affects neither its DNA and damaged DNA binding ability nor the dual incision in vitro (82). However, mutant XPC lacking major SUMOylation sites shows moderate GG-NER defect in vivo, which can be rescued by knocking out DDB2. The XPC SUMOylation mutation increases UV-induced DDB2-dependent XPC immobilization in vivo. The mutant XPC are normally recruited to damage sites but fail to support incorporation of TFIIH and XPA to damage sites. Thus, XPC SUMOylation regulate GG-NER in vivo via regulating productive assembly of preincision complex.
UBIQUITINATION IN TC-NER
CSB
CSB is a substrate of CRL4CSA E3 ligase (65,84), which uses CSA as a substrate receptor. The crystal structure of CRL4CSA has also been determined (67). It is no surprise that CRL4CSA and CRL4DDB2 exhibit high overall structural similarity. CSA contains a seven-bladed WD40 propeller and an N-terminal helix-loop-helix motif (HLH box), which interacts with propellers BPA and BPC of DDB1. Like CRL4DDB2, CRL4CSA is also regulated by CSN. It is not clear whether CSA auto-ubiquitination occurs in vivo. However, CSA, like DDB2, can be auto-ubiquitinated by CRL4CSA in vitro and the auto-ubiquitination is inhibited by the presence of CSN. The inhibition by CSN can be relieved by providing substrate CSB. Thus, displacement of CSN by CSB binding activates CRL4CSA ligase activity.
CSB ubiquitination mediated by CRL4CSA following induction of cellular DNA damage has been described (84–86). For instance, UV irradiation induces CSB ubiquitination in a CSA-dependent manner (85,86), and results in CSB degradation. How CRL4CSA-medicated CSB ubiquitination and subsequent degradation is related to the role of CSB and CSA in TC-NER is not yet clear.
In addition to CRL4CSA, the BRCA1-BARD E3 ubiquitin ligase complex is also suggested to ubiquitinate CSB and control its stability (87). BRCA1 was detected to localize to CPD sites via CSB and RNAPII-mediated transcription. UV induces BRCA1-dependent CSB ubiquitination in vivo and the ubiquitination can be reconstituted in vitro using purified BRCA1-BARD complex. The siRNA knockdown of BRCA1 decreases the removal of CPDs from transcribed strand. The existence of the two aforementioned independent pathways for ubiquitin-mediated CSB degradation may reflect the importance of ubiquitination and proteolysis of CSB in regulating TC-NER.
RNAPII
DNA damage-induced ubiquitination and subsequent proteasomal degradation of large subunit Rpb1 of RNAPII was first described by Bregman et al (54). Since RNAPII ubiquitination appears to be dependent on both CSA and CSB, it was reasonably suggested to have a role in TC-NER (54,88). This concept, however, has been challenged as more E3 ubiquitin ligases were found to mediate RNAPII ubiquitination (89), e.g., Elongin A ubiquitin ligase complex (90,91), BRCA1-BARD ligase (92,93) and Nedd4 ligase (53).
Elongin is a heterotrimeric complex composed of one large subunit Elongin A, and two small subunits B and C. Through Elongin B and C, the Elongin heterotrimer is linked to a heterodimeric module composed of Cul2 or Cul5 and RING finger proteins Rbx1 or Rbx2 to form a multisubunit complex which can function as E3 ubiquitin ligase (94,95). The Elongin A ubiquitin ligase complex uses Von Hippel-Lindau (VHL) tumor suppressor protein as substrate receptor for Rpb1 (91). The involvement of VHL in TC-NER was suggested by the examination of TCR-NER-dependent toxicity of antitumor compound Et743 (96). It is known that Et743 forms Et743-DNA adducts which cause toxicity via trapping TC-NER machinery while it attempts to excise the adducts. In particular, VHL is required for Et743-induced RNAPII degradation and Et743 toxicity. The yeast counterpart of Elongin A ubiquitin ligase contains Ela1 (Elongin A), Cul3, Elc1(Elongin C) and Rbx1. Yeast Elongin A ligase complex (97,98) works cooperatively with Rsp5 E3 ubiquitin ligase (99) to ubiquitinate Rpb1. Surprisingly, Rsp5 partner Def1 is not required for TC-NER (100,101). Also, Ela1, Cul3 or Elc1 appears to function via a separate pathway than Rad26 (CSB in yeast). Thus, Rpb1 degradation may not be an integral part of TC-NER, at least in yeast.
Despite the prevalent discrepancy, it can be argued that once DNA damage is sensed by elongating RNAPII, TC-NER can only proceed by the congregation of E3 ubiquitin ligase(s), and CSB and CSA as well as other factors such as TFIIS, HMGN1, XAB2 and VCP/p97. Of these factors, VCP/p97 plays a role in timely extraction of ubiquitinated proteins from damaged chromatin. Nevertheless, the involvement of multiple E3 ubiquitin ligases in Rpb1 ubiquitination clearly alludes to the importance of Rpb1 degradation in DNA damage response. Ditching a damage-arrested RNAPII via proteolytic clearance may allow RNAPII-blocking lesions to either repair by GG-NER and/or prevent catastrophic cellular events like cell death to occur.
PROCESSING OF UBIQUITINATED DNA DAMGAE SENSORS BY VCP/p97
VCP/p97, a facilitator of UPS
VCP/p97, known as CDC48 in yeast, belongs to AAA (ATPase-associated with various cellular activities) family (102). The Cdc48 allele was first identified three decades ago by a genetic screen of genes which affect cell growth at non-permissive temperature (103). Now, CDC48/p97 is recognized as the central player that integrates modification of proteins, their execution of function and proteasomal degradation mediated by ubiquitination, SUMOylation and neddylation. The structure, function and adaptors/cofactors of VCP/p97 are extensively reviewed (104–106). Substrates of CDC48/p97 include many nuclear factors involved in DNA replication, DNA repair, transcription, telomere maintenance, sister-chromatid segregation and SUMO-targeted ubiquitination (105).
VCP/p97 is a type II AAA+ ATPase with a large amino-terminal domain (N-domain), two ATPase domains D1 and D2, a N-D1 linker and a short D1-D2 linker. Unlike many bacterial AAA+ proteins, six VCP/p97 monomers structurally assemble into ring-like hexameric complex, with two centric N-D1 and D2 rings (106). It is generally believed that VCP/p97 undergoes dramatic conformational changes which generate the needed mechanical force for VCP/p97 to segregate ubiquitinated clients from their tightly bound partners or multimeric assemblies. Despite that many hexameric ring-like AAA+ proteins are protein unfoldases (by definition, unfoldase unfolds substrate into linear peptides), VCP/p97 may unfold some clients but not the others, which allows some of VCP/p97 clients to be rescued via removal of ubiquitin by deubiquitinating enzymes (DUBs)/USPs.
VCP/p97 cooperates with different sets of mutually exclusive cofactors/adaptors for different cellular functions. The cofactors/adaptors of VCP/p97 recognize ubiquitinated clients. Many cofactors/adaptors, including UFD1, NPL4, Ataxin-3 and UBX proteins, bind VCP/p97’s N-domain via ubiquitin regulatory X (UBX), or UBX-like (UBXL) domains or via VCP-interacting motif (VIM), VCP-binding motif (VBM) or SHP box (107). The UFD1-NPL4 is a known VCP/p97 substrate-recruiting cofactor, which is needed for extracting the misfolded proteins from endoplasmic reticulum (ER) membrane during ER-associated degradation (108–111).
Among many VCP/p97 cofactors, UBXD7 is one of five UBX proteins (p47, UBXD7, UBXD8, FAF1 and SAKS1) that also contain ubiquitin-associated (UBA) domain, and thus are classified as UBA-UBX proteins. UBXD7 is unique because it also binds to CRL ligases through its ubiquitin-interacting motif (UIM) (112–114). The UIM of UBXD7 can recognize ubiquitin-like modifier Nedd8. As mentioned above, CRL ligases are activated upon Nedd8 conjugation to Cullins. Although UBXD7 binds to activated CRLs, the functional significance of the UBXD7’s binding, however, is not fully understood. UBXD7 may play a role in coordinating substrate ubiquitination and VCP/p97-mediated factor extractions.
VCP/p97 mediates the timely extractions of DDB2 and XPC in GC-NER
Although CRLDDB2 assists damage recognition conducted by XPC via ubiquitination of XPC, neither CRLDDB2 nor XPC are the components of final preincision complex. Recent studies have revealed a role of VCP/p97 in extracting DDB2 and XPC from UV-induced damage sites (76,77), and thereby identified VCP/p97 as a new player operational in GG-NER.
Upon UV-induced DNA damage, VCP/p97 translocates to lesion sites within 15 min and forms distinct UV radiation-induced foci (UVRIF). The formation of VCP/p97 UVRIF is dependent on proteasomal function, CRLDDB2-mediated ubiquitination and partially on the presence of XPC (76). VCP/p97 physically binds to K48-polyubiquitinated DDB2, and knockdown of VCP/p97 prevents UV-induced DDB2 degradation and prolongs the retention of DDB2 and ubiquitinated XPC in chromatin. Functionally, VCP/p97 is required for efficient repair of both 6-4PPs and CPDs. VCP/p97 deficiency causes UV-induced chromosomal aberrations, which can be alleviated by a concomitant down regulation of DDB2 or XPC. Thus, timely extraction of both DDB2 and XPC by VCP/p97 leads to efficient GG-NER, whereas prolonged retention of DDB2 and XPC on damaged chromatin causes unexpected genotoxicity.
The VCP/p97-mediated XPC extraction was confirmed by independent studies in USP7-knockout HCT116 cells (77). Specifically, UV-induced XPC proteasomal degradation occurs without protection by USP7 which deubiquitinates XPC. In USP7-knockout cells, VCP/p97 inhibition prevents UV-induced XPC degradation and increases accumulation of ubiquitinated XPC in damaged chromatin. USP7 deletion results in severe repair defect in the removal of CPDs. Thus, VCP/p97 acts as a segregase rather than an unfoldase for XPC regardless of proteolysis, allowing ubiquitinated XPC to be rescued by USP7. The VCP/p97 cofactors UFD1-NPL4 and UBXD7 are involved in VCP/p97-mediated extraction of DDB2 and presumably XPC (76). However, the direct link between CRLDDB2 and UBXD7 remains to be investigated.
VCP/p97 participates in TCR-NER
VCP/p97 functions in ubiquitin-mediated degradation of RNAPII
Elongating RNAPII and CSB are damage sensors for TC-NER. Analogous to VCP/p97-mediated extraction of DDB2 and XPC in GG-NER, yeast CDC48/p97 was found to mediate UV-dependent turnover of RNAPII (115). The large RNAPII subunit Rpb1 was identified, by mass spectrometry of ubiquitin conjugates, to be associated with proteasome in cdc48 mutant cells. Further studies revealed that UV induces Rpb1 degradation in a CDC48-UFD1-NPL4-, Ubx4- and Ubx5-dependent manner. It was noted that Ubx5 and Cdc48 act downstream of Cul3 in UV-induced Rpb1 degradation. Cul3 not only physically binds Ubx5, but also is required for accumulation of Rpb1-ubiquitin conjugates in Ubx5 deleted cells. Thus, Cul3 appears to handoff ubiquitinated Rpb1 to Ubx5. It is noteworthy that Ubx5 is a yeast ortholog of UBXD7 (114). Interestingly, CDC48/p97-mediated Rpb1 degradation is prompted by the regulatory control exerted via ATP-dependent INO80 chromatin remodeler complex (116). INO80 physically and functionally interacts with CDC48/p97, in facilitating release of the ubiquitinated Rpb1 from its tight association with chromatin for degradation. Intriguingly, INO80 function in Rpb1 turnover is required for cell growth and survival during genotoxic stress. Thus, CDC48/p97 plays an obvious role in ubiquitin-mediated Rpb1 degradation in yeast.
In mammalian cells, our unpublished data suggest a similar VCP/p97 function. Surprisingly, the UV-induced Rpb1 ubiquitination and degradation occur in CSB-independent manner. The CSB presence transiently stabilizes Rpb1 and enhances Rpb1-VCP/p97 interaction. In-depth investigation is underway to obtain a clearer view of VCP/p97-mediated Rpb1 extraction in mammalian cells.
VCP/p97 functions in proteolytic processing of CSB
Our recent studies have uncovered a role of VCP/p97 in mediating proteolytic processing of CSB (86). Similar to their role in extracting DDB2 and XPC, VCP/p97, its cofactors UFD1-NPL4 and UBXD7 are involved in UV-induced CSB degradation. VCP/p97 physically interacts with both native and ubiquitin-conjugated forms of CSB. However, the manifestation of UVRIF of VCP/p97 is independent of CSB and UBXD7, suggesting that formation of VCP/p97 UVRIF is largely related to GG-NER. Inhibition of proteasome and VCP/p97 function allows the accumulation of both native and ubiquitinated CSB, and results in accumulation of UBXD7 and proteasome in chromatin. The investigation also revealed that CRL4CSA is associated with both VCP/p97 and UBXD7. Thus, VCP/p97 is involved in timely extraction of both ubiquitinated RNAPII and CSB during TC-NER.
UBIQUITNATED DNA DAMGAE SENSORS ARE DIFFERENTLY RESCUED BY USPS
The unique aspect of ubiquitin as a signal for monitoring DNA damage sensors and their function at DNA lesions during GG-NER and TC-NER is that the ubiquitination can also control the ultimate fate of the involved proteins. The editing of ubiquitin chains by DUBs/USPs provides an additional level of regulation by delaying or preventing premature ubiquitin-mediated degradation. Several USPs have been found to regulate the fate of damage sensors.
USP24 in GG-NER
USP24 has been identified as a DDB2-interacting partner by yeast two-hybrid screening (119). The interaction between USP24 and DDB2 was confirmed by co-immunoprecipitation. Knockdown of USP24 decreases steady-state level of DDB2. Thus, USP24 may be a candidate for DDB2 deubiquitination.
USP7 in GG-NER
USP7, also known as a herpesvirus-associated ubiquitin-specific protease, is one of the best characterized DUBs/USPs for tumor suppressor p53 and Mdm2 (120,121). Our recent work has identified USP7 as the deubiquitinating enzyme specific for XPC (77). USP7 binds and deubiquitinates XPC in vivo and in vitro. A unique characteristic of USP7-mediated XPC deubiquitination is that it protects XPC from VCP/p97-mediated proteasomal degradation, allowing XPC to recycle during GG-NER. Cells lacking USP7 rapidly degrades XPC and essentially fail to repair CPDs after UV irradiation. Thus, XPC is recycled to allow multiple rounds of cellular GG-NER. We envisage that by recycling XPC, cells could sustain GG-NER at an optimal level, by utilizing the lower available constitutive level of XPC which are essential to avoid any unwanted excision of XPC-binding DNA structures.
USP7 has three structurally recognizable domains: the N-terminal TRAF domain, the catalytic domain and the C-terminal tandem ubiquitin-like (UBL) domains. USP7 uses TRAF domain to recognize the P/AxxS motif in p53 and Mdm2 (122). Away from this substrate recognition mechanism, USP7 is found to bind XPC via UBL1 (77). It was recently noted that tandem UBL domains of USP7 recognize a consensus primary sequence containing KxxxK motif in substrates, e.g., DNMT1, UHRF1 and ICP0 (123). Such a KxxxK motif indeed exists in XPC within amino acid 328 to 440 (Figure 1). We suggest that USP7 recognizes XPC using a different mechanism than the one utilized for p53 and Mdm2.
Figure 1.

Putative KxxxK motif in XPC. The alignment of corresponding KxxxK sequences in DNMT1, UHRF1, ICP0, H3 and H2A in comparison with KxxxK sequence in XPC. Highly conserved residues are highlighted in dark green and conserved residues in light green. The putative conserved residues in XPC are in green letters.
Besides USP7, OTUD4 was recently identified as a XPC interaction partner (124). Intriguingly, OTUD4 was the DUB, previously characterized as an USP7 substrate-binding partner in deubiquinating DNA demethylases ALKBH2 and ALKBH3 (125). It is not known whether OTUD4 works independently or cooperatively with USP7 to deubiquitinate XPC.
USP7 and UVSSA in TC-NER
USP7 was also identified as a partner of UVSSA, whose gene mutations cause UV-sensitive syndrome (126–128). The UVSSA protein was shown to interact with RNAPII, CSA, CSB and TFIIH. However, whether UVSSA is recruited to DNA photolesions as an RNAPII interaction partner (126) or as a CSA interaction partner (85,127) remains to be established. UVSSA also interacts with TRAF domain of USP7 (123) and forms a UV-independent complex with USP7 (126). Through such interaction, USP7 can be recruited to TC-NER complex upon UV damage. Depletion of USP7 leads to a decrease in UV survival and in RNA synthesis recovery similar to UVSSA depletion, indicating a role of USP7-UVSSA in regulating TC-NER.
In the absence of UVSSA or USP7, UV-induced CSB degradation is accelerated. Therefore, it was suggested that UVSSA stabilizes CSB following UV by targeting USP7 to TC-NER complex to deubiquitinate UV-induced ubiquitin chain on CSB (126). In line with this, CSB recovery is also severely abolished in cells lacking USP7, at later times following UV exposures (86). We envisage that UVSSA acts as an interaction partner of USP7 to recruit it to trim the ubiquitin chain on CSB for maintaining its remodeler function and rescue it from VCP/p97-mediated proteolysis. Future in-depth dissection of the role of UVSSA and USP7 in CSB deubiquitination will help to fully understand the molecular mechanism of TC-NER.
HYPOTHETICAL MODELS
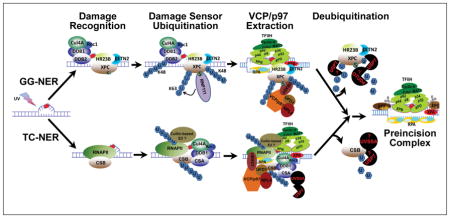
To summarize, NER can be conducted in vitro without any observable regulation by ubiquitination. However, in the native cellular environment, NER process is inevitably regulated by ubiquitination. With VCP/p97 in play, we feel comfortable in proposing overarching hypothetical models for the assembly of preincision complexes orchestrating the early events in GG-NER (Figure 2) and in TC-NER (Figure 3). In GG-NER, the ubiquitinated, lesion-holding DDB2 is extracted by VCP/p97 from CRLDDB2 and as a result, CRL is dispatched from chromatin following completion of its task of initial damage detection. The departure of CRLDDB2 facilitates the arrival of TFIIH and XPA. This hypothesis reconciles (i) structural characteristics of DDB and XPC proteins (25,47,67), (ii) the role of CRLDDB2 complex in recruiting TFIIH (60) and XPA (79), and (iii) the role of XPC and the effect of XPC SUMOylation in recruiting XPA and TFIIH (82). The CRLDDB2-mediated XPC ubiquitination enhances the inherent binding of XPC, which recruits TFIIH and positions both TFIIH and XPA for obligatory lesion verification (29,71). We speculate that both CRLDDB2-mediated XPC ubiquitination and RNF111-mediated SUMO-targeted XPC ubiquitination contributes to ubiquitin-dependent VCP/p97-mediated extraction of XPC (76,77,81). The departure of XPC in turn facilitates the arrival of XPG and XPF on the lesion-containing DNA strand and the RPA on the non-lesion strand, forming preincision complex without the presence of DDB and XPC.
Figure 2.

Schematic model depicting the role of VCP/p97 in the assembly of preincision complex during GG-NER. CRLDDB2 is recruited to DNA lesions situated in chromatin to recruit and support XPC. Arrival of XPC displaces CSN and activates CRLDDB2 to ubiquitinate DDB2 and XPC in a K48-linked ubiquitin chain. XPC ubiquitination enhances damage binding of XPC, which recruits TFIIH and XPA. The K48-polyubiquitinated DDB2 is extracted by VCP/p97, facilitating the arrival of TFIIH and XPA to the lesion. Meanwhile, RNF111 is recruited and mediates a UV-induced SUMO-targeted K63-ubiquitination of XPC. Subsequently, VCP/p97 mediates ubiquitin-dependent extraction of both K48- and K63-ubiquitinated XPC from lesion sites. The departure of XPC in turn facilitates the arrival of XPG and XPF as well as RPA, forming the final preincision complex. The ubiquitinated DDB2 undergoes proteolysis. Whereas, the ubiquitinated XPC is deubiquitinated by USP7, which restores the native state and constitutive levels of XPC.
Figure 3.

Schematic model depicting the role of VCP/p97 in the assembly of preincision complex during TC-NER. When elongating RNAPII encounters a transcription-blocking lesion, it is transiently installed by the lesion. The installed RNAPII stabilizes the interaction between RNAPII elongation machinery and preexisting CSB or recruits CSB de novo. The transiently stabilized RNAPII elongation machinery stimulates the formation of a functional Cullin-based Elongin A ubiquitin ligase complex or recruits a RNAPII E3 ubiquitin ligase. Simultaneously, CRL4CSA is recruited to the arrested machinery in a CSB-dependent manner. Consequently, RNAPII and CSB are independently ubiquitinated. The ubiquitin conjugates recruit VCP/p97 complex, which may dynamically interact with activated CRL4. Next, TFIIH and XPA arrive and replace RNAPII when RNAPII-CSB is co-extracted by VCP/p97 complex. Finally, the arrival of XPG and XPF as well as RPA completes the formation of preincision complex. The ubiquitinated CSB undergoes deubiquitination by USP7, whereas ubiquitinated RNAPII is degraded by proteasome.
As for the TC-NER (Figure 3), transcription-blocking DNA lesions are initially sensed by elongating RNAPII. The current model hypothesizes that the stalling of RNAPII by DNA lesions transiently pauses RNAPII elongation machinery, and the latter is stabilized by CSB, which dynamically interacts with this machinery during elongation. The stabilized RNAPII elongation machinery-CSB sets a stage for the action of Elongin A ubiquitin ligase complex, CRL4CSA and other core NER factors. Once again, the model emphasizes the regulation of TCR process by ubiquitination. Although RNAPII at lesions may not interfere with dual incision in vitro (117,118) and RNAPII-DNA interface may be remodeled by CSB in favor of TC-NER (48), but once ubiquitinated in vivo, RNAPII must be extracted and removed not only to process TC-NER but to ensure cell survival. In the model, CSB functions to stabilize lesion-arrested RNAPII elongation machinery, allowing CRL4CSA, TFIIH and XPA to come to the stage. RNAPII and CSB are independently ubiquitinated by their own ubiquitin ligases. However, in our hypothetical model we like to highlight the co-extraction of RNAPII and CSB by VCP/p97. We further speculate that the co-extraction ensures the timely removal of RNAPII and CSB for proper dispatching of the remaining components of elongation machinery and suitably positioning of TFIIH, XPA and RPA. In essence, VCP/p97-mediated extraction of RNAPII and CSB is a key process associated with TC-NER.
CONCLUDING REMARKS
In recent years, significant progress has been made to understand the highly coordinated molecular events in cellular processes like DNA replication and DNA damage response. It is becoming clear that both the recruitment and removal of proteins at replication forks or DNA damage sites/lesions are highly regulated to enable effective functions of the involved protein complexes. In this review, we have discussed the ubiquitination, ubiquitin-dependent extraction of DNA damage sensors and their functional consequences in sequential events of assembly of preincision complex in NER.
Multiple ubiquitination events occur in the process of NER. The ubiquitination of DNA damage sensors is important not only for regulating protein-DNA interaction but also for driving the transition through multiple steps of damage recognition and verification towards the assembly of preincision complex. Perhaps ubiquitination helps to set precise temporal sequence for clearing the factors from chromatin when their activity is no longer required or the occupied spaces are needed for downstream events or critical compositional changes are necessary for productive repair assembly. Another major benefit of ubiquitination as timing clock is in dictating the fate of the engaged protein(s)- to set them for a complete destruction or rescue them for additional rounds of DNA repair.
While many essential features of VCP/p97-mediated extraction of chromatin-associated DNA damage sensors have begun to form a clear conceptual foundation, many aspects of this complex phenomenon remain elusive. Various questions could be raised about the hypothesized models proposed in our compilation of molecular events. For example, does SUMOylation or RNF111-mediated non-proteolytic K63 ubiquitination of XPC contribute to its extraction by VCP/p97 complex? In the case of CRL4DDB2- and/or CRL4CSA-mediated ubiquitination, is the substrate ubiquitination coupled to substrate extraction by dynamic interaction between CRL4 and VCP/p97 complex? What does VCP/p97 cofactor UBXD7 do? The answers to such questions, and newer insights on compositional changes within the arrested RNAPII-containing elongation machinery, will undoubtedly shed more useful light on the role of VCP/p97 and related factors operational in NER.
Acknowledgments
Authors would like to thank Dr Qi-en Wang (Department of Radiology, The Ohio State University) for his insightful suggestions for the manuscripts. Authors also thank Jinshan He, Chunhua Han, Gulzar Wani, Alo Ray and all past alumni of our laboratory. This review would not be possible without their dedicated efforts and contribution. Our work, mentioned in this review, was supported by the National Institute of Health grants ES002388 and ES012991 (to AAW).
Abbreviations
- NER
nucleotide excision repair
- GG-NER
global genomic NER
- TC-NER
transcription-coupled NER
- DDB
damaged DNA binding protein
- XP
Xeroderma Pigmentosum
- XPC
Xeroderma Pigmentosum complementation group C
- RNAPII
RNA polymerase II
- CS
Cockayne syndrome
- CSB
Cockayne syndrome protein B
- CSA
Cockayne syndrome protein A
- VCP
valosin-containing protein
- UV
ultraviolet
- CPD
cyclobutane pyrimidine dimer
- 6-4PP
pyrimidine (6-4) pyrimidone photoproducts
- UVSSA
UV-stimulated scaffold protein A
- TFIIH
transcription factor II H
- CAK
CDK-activating kinase
- RPA
replication protein A
- PCNA
proliferating cell nuclear antigen
- ERCC1
excision repair cross-complementation group 1
- RFC
replication factor C
- RNAPII
RNA polymerase II
- Cul4A
Cullin 4A
- UPS
ubiquitin-proteasome system
- COP9
constitutive photomorphogenesis 9
- CSN
COP9 signalosome
- CRL4
DDB1-Cul4-Rbx1
- DCAF
DDB1- and Cul4A-associated factors
- SUMO
small ubiquitin-like modifier
- STUbL
SUMO-targeted ubiquitin ligase
- BRCA1
Breast cancer type 1 susceptibility protein
- BARD
BRCA1-associated RING domain protein
- USP7
ubiquitin-specific protease 7
- DUB
deubiquitinating enzyme
Biographies

Altaf Wani has been actively involved in delineating the intricate mechanisms of DNA repair. His research has utilized a variety of model systems and damaging agents to study several DNA repair pathways. Overarching goal of his research is to continue the molecular dissection of the processing of genomic lesions and unravel the cross-talk among closely related network of pathways with the ultimate aim of developing intervention strategies for elimination of diseases emanating from inherent instability of the human genome. Dr. Wani is Professor of Radiology at the Ohio State University (http://radiology.osu.edu/10724.cfm).

Qianzheng Zhu is interested in scientific research in the field of DNA damage repair, chromatin dynamics and ubiquitin proteasome system. His research topics are related to nucleotide excision repair, protein/histone ubiquitination and deubiquitination, VCP/p97 function in DNA damage response. He is a principal investigator (PI) and Assistant Professor at Department of Radiology, Wexner Medical Center at the Ohio State University (http://radiology.osu.edu/21787.cfm).
Footnotes
This article is part of the Special Issue highlighting Dr. Aziz Sancar’s outstanding contributions to various aspects of the repair of DNA photodamage in honor of his recent Nobel Prize in Chemistry.
REFFERENCES
- 1.Kelner A. Effect of visible light on the recovery of streptomyces griseus conidia from ultra-violet irradiation injury. Proc Natl Acad Sci U S A. 1949;35:73–79. doi: 10.1073/pnas.35.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Essen LO, Klar T. Light-driven DNA repair by photolyases. Cell Mol Life Sci. 2006;63:1266–1277. doi: 10.1007/s00018-005-5447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Setlow RB, Carrier WL. The disappearance of thymine dimers from DNA: An error-correcting mechanism. Proc Natl Acad Sci U S A. 1964;51:226–231. doi: 10.1073/pnas.51.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, Modrich P, Sancar A. The 2015 Nobel prize in chemistry the discovery of essential mechanisms that repair DNA damage. J Assoc Genet Technol. 2016;42:37–41. [PubMed] [Google Scholar]
- 5.Ljungman M. The DNA damage response-Repair or despair? Environ Mol Mutagen. 2010;51:879–889. doi: 10.1002/em.20597. [DOI] [PubMed] [Google Scholar]
- 6.Nouspikel T. DNA repair in mammalian cells : Nucleotide excision repair: variations on versatility. Cell Mol Life Sci. 2009;66:994–1009. doi: 10.1007/s00018-009-8737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugasawa K. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mut Res. 2010;685:29–37. doi: 10.1016/j.mrfmmm.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Lee J, Zhou P. Navigating the nucleotide excision repair threshold. J Cell Physiol. 2010;224:585–589. doi: 10.1002/jcp.22205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Q, Wani AA. Histone modifications: crucial elements for damage response and chromatin restoration. J Cell Physiol. 2010;223:283–288. doi: 10.1002/jcp.22060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi D, Grossman SR. Ubiquitin becomes ubiquitous in cancer: Emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol Ther. 2010;10 doi: 10.4161/cbt.10.8.13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 13.Ford JM, Hanawalt PC. Role of DNA excision repair gene defects in the etiology of cancer. Curr Top Microbiol Immunol. 1997;221:47–70. doi: 10.1007/978-3-642-60505-5_5. [DOI] [PubMed] [Google Scholar]
- 14.Petit C, Sancar A. Nucleotide excision repair: from E. coli to man. Biochimie. 1999;81:15–25. doi: 10.1016/s0300-9084(99)80034-0. [DOI] [PubMed] [Google Scholar]
- 15.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 16.Spivak G. UV-sensitive syndrome. Mutat Res. 2005;577:162–169. doi: 10.1016/j.mrfmmm.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Schwertman P, Vermeulen W, Marteijn JA. UVSSA and USP7, a new couple in transcription-coupled DNA repair. Chromosoma. 2013;122:275–284. doi: 10.1007/s00412-013-0420-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mu D, Park C-H, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- 19.Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 20.Aboussekhra A, Biggerstaff M, Shivji MKK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly J-M, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 21.Sugasawa K, Ng JMY, Masutani C, Iwai S, Van der Spek P, Eker A, Hanoaka F, Bootsma D, Hoeijmakers JHJ. Xeroderma pigmentosum group C complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 22.Araki M, Masutani C, Takemura M, Uchida A, Sugasawa K, Kondoh J, Ohkuma Y, Hanaoka F. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem. 2001;276:18665–18672. doi: 10.1074/jbc.M100855200. [DOI] [PubMed] [Google Scholar]
- 23.Sugasawa K, Shimizu Y, Iwai S, Hanaoka F. A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair (Amst) 2002;1:95–107. doi: 10.1016/s1568-7864(01)00008-8. [DOI] [PubMed] [Google Scholar]
- 24.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 26.Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F. The Xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem. 2000;275:9870–9875. doi: 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- 27.Volker M, Mone MJ, Karmakar P, Van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, Van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 28.Rademakers S, Volker M, Hoogstraten D, Nigg AL, Mone MJ, Van Zeeland AA, Hoeijmakers JH, Houtsmuller AB, Vermeulen W. Xeroderma pigmentosum group A protein loads as a separate factor onto DNA lesions. Mol Cell Biol. 2003;23:5755–5767. doi: 10.1128/MCB.23.16.5755-5767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li CL, Golebiowski FM, Onishi Y, Samara NL, Sugasawa K, Yang W. Tripartite DNA Lesion Recognition and Verification by XPC, TFIIH, and XPA in Nucleotide Excision Repair. Mol Cell. 2015;59:1025–1034. doi: 10.1016/j.molcel.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nocentini S, Coin F, Saijo M, Tanaka K, Egly JM. DNA damage recognition by XPA protein promotes efficient recruitment of transcription factor II H. Journal of Biological Chemistry. 1997;272:22991–22994. doi: 10.1074/jbc.272.37.22991. [DOI] [PubMed] [Google Scholar]
- 31.Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell. 2008;31:9–20. doi: 10.1016/j.molcel.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 33.Krasikova YS, Rechkunova NI, Maltseva EA, Petruseva IO, Lavrik OI. Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic Acids Res. 2010;38:8083–8094. doi: 10.1093/nar/gkq649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans E, Fellows J, Coffer A, Wood RD. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J. 1997;16:625–638. doi: 10.1093/emboj/16.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sijbers AM, De Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, De Jong MC, Rademakers S, De Rooij J, Jaspers NGJ, Hoeijmakers JHJ, Wood RD. Xeroderma pigmentosum group F caused by a defect in a structure- specific DNA repair endonuclease. Cell. 1996;86:811–822. doi: 10.1016/s0092-8674(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 36.Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsodikov OV, Ivanov D, Orelli B, Staresincic L, Shoshani I, Oberman R, Scharer OD, Wagner G, Ellenberger T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007;26:4768–4776. doi: 10.1038/sj.emboj.7601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemp MG, Reardon JT, Lindsey-Boltz LA, Sancar A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J Biol Chem. 2012;287:22889–22899. doi: 10.1074/jbc.M112.374447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LH, Yamashita S, Fousteri MI, Lehmann AR. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010;37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 40.Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH, Fousteri MI. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell. 2007;27:311–323. doi: 10.1016/j.molcel.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 41.Chu G, Chang E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science. 1988;242:564–567. doi: 10.1126/science.3175673. [DOI] [PubMed] [Google Scholar]
- 42.Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Molecular Cell. 2000;5:737–744. doi: 10.1016/s1097-2765(00)80252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nichols AF, Itoh T, Graham JA, Liu W, Yamaizumi M, Linn S. Human damage-specific DNA-binding protein p48 - Characterization of XPE mutations and regulation following UV irradiation. J Biol Chem. 2000;275:21422–21428. doi: 10.1074/jbc.M000960200. [DOI] [PubMed] [Google Scholar]
- 44.Fitch ME, Cross IV, Ford JM. p53 responsive nucleotide excision repair gene products p48 and XPC, but not p53, localize to sites of UV-irradiation-induced DNA damage, in vivo. Carcinogenesis. 2003;24:843–850. doi: 10.1093/carcin/bgg031. [DOI] [PubMed] [Google Scholar]
- 45.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 46.Wang QE, Zhu Q, Wani G, Chen J, Wani AA. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis. 2004;25:1033–1043. doi: 10.1093/carcin/bgh085. [DOI] [PubMed] [Google Scholar]
- 47.Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svejstrup JQ. Mechanisms of transcription-coupled DNA repair. Nat Rev Mol Cell Biol. 2002;3:21–29. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- 49.Vermeulen W, Fousteri M. Mammalian transcription-coupled excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012625. doi: 10.1101/cshperspect.a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Gool AJ, Citterio E, Rademakers S, van OR, Vermeulen W, Constantinou A, Egly JM, Bootsma D, Hoeijmakers JH. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997;16:5955–5965. doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van dB, Citterio VE, Hoogstraten D, Zotter A, Egly JM, van Cappellen WA, Hoeijmakers JH, Houtsmuller AB, Vermeulen W. DNA damage stabilizes interaction of CSB with the transcription elongation machinery. J Cell Biol. 2004;166:27–36. doi: 10.1083/jcb.200401056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fousteri M, Vermeulen W, Van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 53.Anindya R, Aygun O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28:386–397. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci U S A. 1996;93:11586–11590. doi: 10.1073/pnas.93.21.11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Citterio E, van dB, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cho I, Tsai PF, Lake RJ, Basheer A, Fan HY. ATP-dependent chromatin remodeling by Cockayne syndrome protein B and NAP1-like histone chaperones is required for efficient transcription-coupled DNA repair. PLoS Genet. 2013;9:e1003407. doi: 10.1371/journal.pgen.1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X, Zhang Y, Douglas L, Zhou P. UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J Biol Chem. 2001;276:48175–48182. doi: 10.1074/jbc.M106808200. [DOI] [PubMed] [Google Scholar]
- 58.Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol. 2001;21:6738–6747. doi: 10.1128/MCB.21.20.6738-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuda N, Azuma K, Saijo M, Iemura S, Hioki Y, Natsume T, Chiba T, Tanaka K, Tanaka K. DDB2, the xeroderma pigmentosum group E gene product, is directly ubiquitylated by Cullin 4A-based ubiquitin ligase complex. DNA Repair (Amst) 2005;4:537–545. doi: 10.1016/j.dnarep.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 60.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 61.Trempe JF. Reading the ubiquitin postal code. Curr Opin Struct Biol. 2011;21:792–801. doi: 10.1016/j.sbi.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 62.Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 63.Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73:3497–3506. doi: 10.1007/s00018-016-2255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 65.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 66.Scrima A, Fischer ES, Lingaraju GM, Bohm K, Cavadini S, Thoma NH. Detecting UV-lesions in the genome: The modular CRL4 ubiquitin ligase does it best! FEBS Lett. 2011;585:2818–2825. doi: 10.1016/j.febslet.2011.04.064. [DOI] [PubMed] [Google Scholar]
- 67.Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, Iwai S, Gut H, Sugasawa K, Thoma NH. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 68.Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12:220. doi: 10.1186/gb-2011-12-4-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci U S A. 2006;103:2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 71.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 72.El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M, Wani AA. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem. 2006;281:13404–13411. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- 73.Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 2002;30:2588–2598. doi: 10.1093/nar/30.11.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsumoto S, Fischer ES, Yasuda T, Dohmae N, Iwai S, Mori T, Nishi R, Yoshino K, Sakai W, Hanaoka F, Thoma NH, Sugasawa K. Functional regulation of the DNA damage-recognition factor DDB2 by ubiquitination and interaction with xeroderma pigmentosum group C protein. Nucleic Acids Res. 2015;43:1700–1713. doi: 10.1093/nar/gkv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang QE, Wani MA, Chen J, Zhu Q, Wani G, El-Mahdy MA, Wani AA. Cellular ubiquitination and proteasomal functions positively modulate mammalian nucleotide excision repair. Mol Carcinog. 2005;42:53–64. doi: 10.1002/mc.20065. [DOI] [PubMed] [Google Scholar]
- 76.Puumalainen MR, Lessel D, Ruthemann P, Kaczmarek N, Bachmann K, Ramadan K, Naegeli H. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat Commun. 2014;5:3695. doi: 10.1038/ncomms4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He J, Zhu Q, Wani G, Sharma N, Han C, Qian J, Pentz K, Wang QE, Wani AA. Ubiquitin-specific Protease 7 Regulates Nucleotide Excision Repair through Deubiquitinating XPC Protein and Preventing XPC Protein from Undergoing Ultraviolet Light-induced and VCP/p97 Protein-regulated Proteolysis. J Biol Chem. 2014;289:27278–27289. doi: 10.1074/jbc.M114.589812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang QE, Zhu Q, Wani G, El-Mahdy MA, Li J, Wani AA. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res. 2005;33:4023–4034. doi: 10.1093/nar/gki684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol. 2010;30:2708–2723. doi: 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Poulsen SL, Hansen RK, Wagner SA, van CL, van Belle GJ, Streicher W, Wikstrom M, Choudhary C, Houtsmuller AB, Marteijn JA, Bekker-Jensen S, Mailand N. RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J Cell Biol. 2013;201:797–807. doi: 10.1083/jcb.201212075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van CL, van Belle GJ, Turkyilmaz Y, Poulsen SL, Janssens RC, Theil AF, Sabatella M, Lans H, Mailand N, Houtsmuller AB, Vermeulen W, Marteijn JA. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat Commun. 2015;6:7499. doi: 10.1038/ncomms8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akita M, Tak YS, Shimura T, Matsumoto S, Okuda-Shimizu Y, Shimizu Y, Nishi R, Saitoh H, Iwai S, Mori T, Ikura T, Sakai W, Hanaoka F, Sugasawa K. SUMOylation of xeroderma pigmentosum group C protein regulates DNA damage recognition during nucleotide excision repair. Sci Rep. 2015;5:10984. doi: 10.1038/srep10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang QE, Praetorius-Ibba M, Zhu Q, El-Mahdy MA, Wani G, Zhao Q, Qin S, Patnaik S, Wani AA. Ubiquitylation-independent degradation of Xeroderma pigmentosum group C protein is required for efficient nucleotide excision repair. Nucleic Acids Res. 2007;35:5338–5350. doi: 10.1093/nar/gkm550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, Kisselev AF, Harel-Bellan A, Nakatani Y. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006;20:1429–1434. doi: 10.1101/gad.378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fei J, Chen J. KIAA1530 protein is recruited by Cockayne syndrome complementation group protein A (CSA) to participate in transcription-coupled repair (TCR) J Biol Chem. 2012;287:35118–35126. doi: 10.1074/jbc.M112.398131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He J, Zhu Q, Wani G, Sharma N, Wani AA. Valosin-containing Protein (VCP)/p97 Segregase Mediates Proteolytic Processing of Cockayne Syndrome Group B (CSB) in Damaged Chromatin. J Biol Chem. 2016;291:7396–7408. doi: 10.1074/jbc.M115.705350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wei L, Lan L, Yasui A, Tanaka K, Saijo M, Matsuzawa A, Kashiwagi R, Maseki E, Hu Y, Parvin JD, Ishioka C, Chiba N. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci. 2011;102:1840–1847. doi: 10.1111/j.1349-7006.2011.02037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ratner JN, Balasubramanian B, Corden J, Warren SL, Bregman DB. Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II -Implications for transcription-coupled DNA repair. J Biol Chem. 1998;273:5184–5189. doi: 10.1074/jbc.273.9.5184. [DOI] [PubMed] [Google Scholar]
- 89.Daulny A, Tansey WP. Damage control: DNA repair, transcription, and the ubiquitin-proteasome system. DNA Repair (Amst) 2009;8:444–448. doi: 10.1016/j.dnarep.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 90.Yasukawa T, Kamura T, Kitajima S, Conaway RC, Conaway JW, Aso T. Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J. 2008;27:3256–3266. doi: 10.1038/emboj.2008.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuznetsova AV, Meller J, Schnell PO, Nash JA, Ignacak ML, Sanchez Y, Conaway JW, Conaway RC, Czyzyk-Krzeska MF. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci U S A. 2003;100:2706–2711. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Starita LM, Horwitz AA, Keogh MC, Ishioka C, Parvin JD, Chiba N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J Biol Chem. 2005;280:24498–24505. doi: 10.1074/jbc.M414020200. [DOI] [PubMed] [Google Scholar]
- 93.Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL. BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005;19:1227–1237. doi: 10.1101/gad.1309505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kamura T, Burian D, Yan Q, Schmidt SL, Lane WS, Querido E, Branton PE, Shilatifard A, Conaway RC, Conaway JW. Muf1, a novel Elongin BC-interacting leucine-rich repeat protein that can assemble with Cul5 and Rbx1 to reconstitute a ubiquitin ligase. J Biol Chem. 2001;276:29748–29753. doi: 10.1074/jbc.M103093200. [DOI] [PubMed] [Google Scholar]
- 95.Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Jr, Elledge SJ, Conaway RC, Harper JW, Conaway JW. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 96.Aune GJ, Takagi K, Sordet O, Guirouilh-Barbat J, Antony S, Bohr VA, Pommier Y. Von Hippel-Lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of RNA polymerase II in response to trabectedin. Clin Cancer Res. 2008;14:6449–6455. doi: 10.1158/1078-0432.CCR-08-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ribar B, Prakash L, Prakash S. ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells. Mol Cell Biol. 2007;27:3211–3216. doi: 10.1128/MCB.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ribar B, Prakash L, Prakash S. Requirement of ELC1 for RNA polymerase II polyubiquitylation and degradation in response to DNA damage in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:3999–4005. doi: 10.1128/MCB.00293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beaudenon SL, Huacani MR, Wang G, McDonnell DP, Huibregtse JM. Rsp5 ubiquitin-protein ligase mediates DNA damage-induced degradation of the large subunit of RNA polymerase II in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:6972–6979. doi: 10.1128/mcb.19.10.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reid J, Svejstrup JQ. DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro. J Biol Chem. 2004;279:29875–29878. doi: 10.1074/jbc.C400185200. [DOI] [PubMed] [Google Scholar]
- 101.Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 2002;415:929–933. doi: 10.1038/415929a. [DOI] [PubMed] [Google Scholar]
- 102.Stolz A, Hilt W, Buchberger A, Wolf DH. Cdc48: a power machine in protein degradation. Trends Biochem Sci. 2011;36:515–523. doi: 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 103.Moir D, Stewart SE, Osmond BC, Botstein D. Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties, and pseudoreversion studies. Genetics. 1982;100:547–563. doi: 10.1093/genetics/100.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 105.Franz A, Ackermann L, Hoppe T. Ring of Change: CDC48/p97 Drives Protein Dynamics at Chromatin. Front Genet. 2016;7:73. doi: 10.3389/fgene.2016.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xia D, Tang WK, Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene. 2016;583:64–77. doi: 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Buchberger A, Schindelin H, Hanzelmann P. Control of p97 function by cofactor binding. FEBS Lett. 2015;589:2578–2589. doi: 10.1016/j.febslet.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 108.Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S. Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J. 2002;21:615–621. doi: 10.1093/emboj/21.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- 111.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 112.Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804–816. doi: 10.1016/j.cell.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bandau S, Knebel A, Gage ZO, Wood NT, Alexandru G. UBXN7 docks on neddylated cullin complexes using its UIM motif and causes HIF1alpha accumulation. BMC Biol. 2012;10:36. doi: 10.1186/1741-7007-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.den BW, Verma R, Kleiger G, Oania RS, Deshaies RJ. NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat Struct Mol Biol. 2012;19:511–6. S1. doi: 10.1038/nsmb.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Verma R, Oania R, Fang R, Smith GT, Deshaies RJ. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol Cell. 2011;41:82–92. doi: 10.1016/j.molcel.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lafon A, Taranum S, Pietrocola F, Dingli F, Loew D, Brahma S, Bartholomew B, Papamichos-Chronakis M. INO80 Chromatin Remodeler Facilitates Release of RNA Polymerase II from Chromatin for Ubiquitin-Mediated Proteasomal Degradation. Mol Cell. 2015;60:784–796. doi: 10.1016/j.molcel.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tremeau-Bravard A, Riedl T, Egly JM, Dahmus ME. Fate of RNA polymerase II stalled at a cisplatin lesion. J Biol Chem. 2004;279:7751–7759. doi: 10.1074/jbc.M309853200. [DOI] [PubMed] [Google Scholar]
- 118.Selby CP, Drapkin R, Reinberg D, Sancar A. RNA polymerase II stalled at a thymine dimer: Footprint and effect on excision repair. Nucleic Acids Res. 1997;25:787–793. doi: 10.1093/nar/25.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang L, Lubin A, Chen H, Sun Z, Gong F. The deubiquitinating protein USP24 interacts with DDB2 and regulates DDB2 stability. Cell Cycle. 2012;11:4378–4384. doi: 10.4161/cc.22688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell. 2005;18:565–576. doi: 10.1016/j.molcel.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 121.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 122.Sheng Y, Saridakis V, Sarkari F, Duan S, Wu T, Arrowsmith CH, Frappier L. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat Struct Mol Biol. 2006;13:285–291. doi: 10.1038/nsmb1067. [DOI] [PubMed] [Google Scholar]
- 123.Cheng J, Li Z, Gong R, Fang J, Yang Y, Sun C, Yang H, Xu Y. Molecular mechanism for the substrate recognition of USP7. Protein Cell. 2015;6:849–852. doi: 10.1007/s13238-015-0192-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lubin A, Zhang L, Chen H, White VM, Gong F. A human XPC protein interactome--a resource. Int J Mol Sci. 2014;15:141–158. doi: 10.3390/ijms15010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao Y, Majid MC, Soll JM, Brickner JR, Dango S, Mosammaparast N. Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. EMBO J. 2015;34:1687–1703. doi: 10.15252/embj.201490497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek AC, Laffeber C, Hoeijmakers JH, Demmers JA, Fousteri M, Vermeulen W, Marteijn JA. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat Genet. 2012;44:598–602. doi: 10.1038/ng.2230. [DOI] [PubMed] [Google Scholar]
- 127.Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S, Tahara H, Neilan EG, Honma M, Nohmi T, Yasui A, Tanaka K. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat Genet. 2012;44:593–597. doi: 10.1038/ng.2228. [DOI] [PubMed] [Google Scholar]
- 128.Nakazawa Y, Sasaki K, Mitsutake N, Matsuse M, Shimada M, Nardo T, Takahashi Y, Ohyama K, Ito K, Mishima H, Nomura M, Kinoshita A, Ono S, Takenaka K, Masuyama R, Kudo T, Slor H, Utani A, Tateishi S, Yamashita S, Stefanini M, Lehmann AR, Yoshiura K, Ogi T. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat Genet. 2012;44:586–592. doi: 10.1038/ng.2229. [DOI] [PubMed] [Google Scholar]