

Abstract
Objective
To determine if magnetic resonance imaging/ultrasound (MRI/US) fusion biopsy facilitates longitudinal re-sampling of the same clonal focus of prostate cancer (PCa) and to determine if high-grade cancers can evolve from low-grade clones.
Materials and Methods
All men on AS who underwent tracking MRI/US fusion biopsy of Gleason 6 PCa, on at least two distinct occasions, between 2012 and 2014 were enrolled. MRI/US fusion was used to track and re-sample specific cancer foci. Immunohistochemistry (IHC) for ERG and targeted RNA/DNA next generation sequencing (NGS) were performed on formalin-fixed paraffin-embedded (FFPE) prostate biopsy specimens to assess clonality.
Results
Thirty-one men with median age and PSA of 65 years and 4.6 ng/mL, respectively, were analyzed. The median sampling interval was 12 months (range 5 - 35). Of the 26 evaluable men, ERG IHC concordance was found between initial and repeat biopsies in 25 (96%), indicating re-sampling of the same clonal focus over time. Targeted NGS supported ERG IHC results and identified unique and shared driving mutations, such as IDH1 and SPOP, in paired specimens. Of the 9 (34.6%) men who were found to have Gleason ≥7 on repeat biopsy, all displayed temporal ERG concordance. Prioritized genetic alterations were detected in 50% (13/26) of paired samples. Oncogenic mutations were detected in 22% (2/9) of Gleason 6 cancers prior to progression and 44% (4/9) of Gleason ≥7 cancers when progression occurred.
Conclusions
Precise tracking of PCa foci via MRI/US fusion biopsy allowed subsequent re-sampling of the same clonal focus of cancer over time. Further research is needed to clarify the grade progression potential of Gleason 6 PCa.
Keywords: prostate cancer, low grade, clonality, ERG fusions, next generation sequencing
INTRODUCTION
Contemporary advances in our understanding of the biology and clinical trajectory of low risk prostate cancer (PCa) have led to the growing adoption of active surveillance (AS) strategies.(1-3) The main objective of AS is to reduce PCa overtreatment, while reserving curative therapy for when disease progression is detected.(4,5) Because of PCa multifocality, precise sampling of PCa foci to assess true disease status is paramount to optimizing AS strategies. Currently, this is typically done with either systematic, yet random sampling of the prostate or by cognitively directed prostate biopsy. Unfortunately, both of these techniques lack precision. Traditional transrectal ultrasound (TRUS)-guided biopsy platforms are confounded by PCa multifocality and sampling bias (i.e., only ~ 0.04% of the prostate is normally assessed).(6)
Recently, magnetic resonance imaging/ultrasound (MRI/US) fusion-guided prostate biopsy platforms have been introduced to facilitate targeted sampling of regions of interest (i.e., areas considered at risk of harboring high-grade PCa on imaging) as well as longitudinal assessment of specific sites.(7-10)Tracking biopsy sites within the prostate, based on needle tracks recorded by the Artemis MRI/US fusion device, we showed in prior work that the precision of MRI/US fusion biopsy for re-sampling areas is within 3 mm following initial biopsy.(9,11) However, the accuracy of tracking biopsy in longitudinal re-sampling of the same clonal focus of cancer is unknown. Furthermore, it is currently unknown if high-grade PCa may arise from low-grade cancers or rather are a byproduct of de novo outgrowth. (12-16) The former notion forms the clinical basis of current AS strategies.
Herein, using molecular techniques, we assessed the potential of repeat MRI/US fusion-guided prostate biopsy to sample the same clonal focus of cancer over time in a cohort of men undergoing AS. We hypothesized that MRI/US fusion guided prostate biopsy would allow precise re-assessment of the same clonal focus of PCa sampled at a later time. Further, we sought to shed light on the histopathological fate of Gleason 6 cancers, i.e., the ability of high-grade cancers to evolve from low-grade clones.
PATIENTS AND METHODS
Cohort description
Subjects were consecutive men with Gleason 6 PCa foci who underwent an initial diagnostic and a subsequent confirmatory biopsy between January 2012 and December 2014. All were enrolled in an IRB-approved AS registry at UCLA. Inclusion criteria for this analysis were that 2 biopsies were performed at least 4 months apart and that evaluable tissue was available at both time points. Exclusion criteria included any previous form of prostate ablative treatment, androgen deprivation therapy, or 5α-reductase inhibitor use. Characteristics of the group are shown in Table 1.
Table 1.
Demographic and Clinical Characteristics of the Study Cohort (N = 31)
| Variable | Value | |
|---|---|---|
| Age (years) | 65 [46 – 74] | |
| Race/Ethnicity | African American | 1 (3.2) |
| Asian | 3 (9.7) | |
| Caucasian | 26 (83.9) | |
| Hispanic | 1 (3.2) | |
| Family history of prostate cancer | 13 (41.9) | |
| Abnormal DRE | 5 (16.1) | |
| Serum PSA (ng/mL) | Initial biopsy | 4.56 [0.49 – 21.00] |
| Repeat biopsy | 4.60 [0.47 – 10.90] | |
| Prostate volume (cm3) | 42.0 [17.0 – 81.1] | |
| PSA density at initial biopsy (ng/mL/cm3) | 0.097 [0.023 – 0.328] | |
| Final Gleason Score* | 6 | 20 (64.5) |
| 3+4 | 8 (25.8) | |
| 4+3 | 2 (6.5) | |
| 8 - 10 | 1 (3.2) | |
| Cancer core length (mm) | Initial biopsy | 2.5 (0.5 – 7) |
| Repeat biopsy | 2.0 (0.5 – 13) | |
| Interval between biopsies (months) | 12 (5 – 35) | |
Median [range] and frequency (percentages) are presented for continuous and categorical variables respectively.
Abbreviations: DRE – digital rectal exam; PSA – Prostate specific antigen.
All patients had Gleason 6 disease at study entry.
Biopsy strategy
Biopsy methods and re-sampling technique using the Artemis device are shown in Figures 1, 2 and Supplementary Figure 1. Prior to the initial biopsy, multiparametric MRI (mpMRI) of the prostate was obtained using a 3T (Siemens Medical Solutions, Malvern, PA) magnet and a trans-abdominal coil, as previously described.(9,11) In 16 men, the PCa focus was within an MRI target (region of interest, ROI); in 15 men, the focus was not in a ROI. Segmentation and initial MRI/US fusion biopsy were performed as previously described, using the Artemis device to obtain samples from any ROI seen on mpMRI and systematically via the 12-point template incorporated into the device (Figure 1).(9,11) All biopsy sites were mapped, electronically tracked and saved to enable re-sampling of the same site. Follow-up biopsy was targeted at the previously identified PCa focus (obtaining 1 core every 3 mm along the longest axis of the lesion; 3 - 5 cores), which was recorded on the Artemis® device (Figure 2).(9,11) The ability of this system to re-sample the same site has been previously reported to be within 1.2±1.1 mm margin of error.(11,17) MRI was not repeated prior to undergoing re-sampling of tracked biopsy sites.
Figure 1. Targeted biopsy using MRI/US Fusion system.
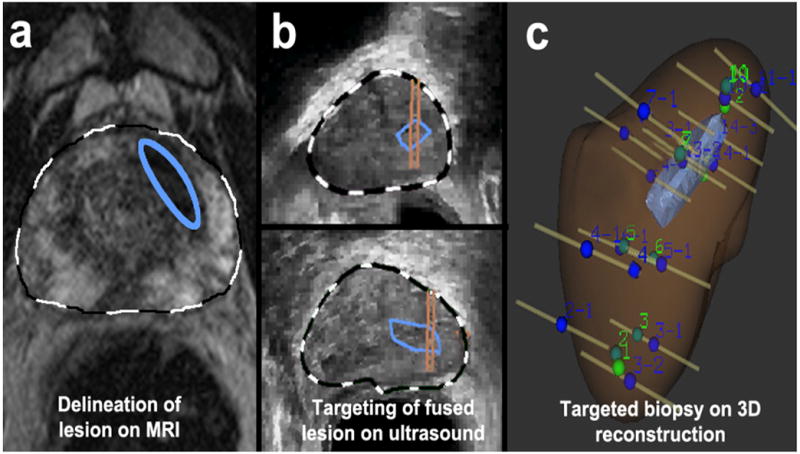
(a) A lesion was identified on MRI and delineated on T2WI by radiologist (blue ellipse). The MRI was fused with real-time ultrasound images. (b) Lesion was identified in sagittal and axial planes (blue enclosures), and biopsy targeting the lesion was established (parallel lines overlying blue enclosures). (c) Sites of targeted and 12-core biopsies were recorded in a 3D reconstruction, confirming that several targeted biopsies penetrated region of interest. (Reprinted with permission from Urol Oncol, 2011 vol. 29 (3 pp. 334-342)
Figure 2. Tracking technique for repeat sampling with Artemis device.
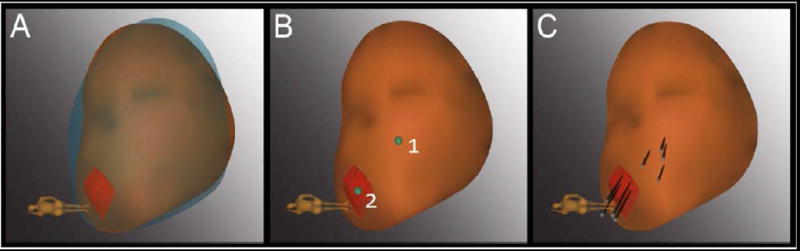
(A) The 3D model of the prostate from the 2nd biopsy (brown) was superimposed on the model from the 1st biopsy (blue), showing a close match in size and shape. The models were created in real-time at biopsy by the Artemis device. An MRI target (red) was displayed in the model. (B) The location of prior positive sites (1 & 2) was mapped by the device (green dots). Site 1 was a systematic site; Site 2 was from the MRI-targeted core. (C) Four cores (black cylinders) were taken from each site. (3D = 3-dimensional; Reprinted with permission from Urol Oncol, 2014 vol. 32 (7) pp.952-957)
Tissue preparation
Diagnostic formalin fixed and paraffin embedded (FFPE) prostate biopsy tissue obtained from the initial and repeat MRI/US fusion-guided prostate biopsy for each participant was procured. Pathology slides selected for next generation sequencing (NGS) were re-reviewed independently by two board certified anatomic pathologists with genitourinary pathology interest (J.H. and S.A.T) to confirm Gleason score, volume of cancer, and to identify areas for NGS (discrepancies were resolved by a third pathologist, L.P.K.). Immunohistochemistry (IHC) and NGS were performed with IRB approval on a single FFPE biopsy block with cancer (representing the highest Gleason score) per time point. 10-11 × 5 μm FFPE sections were cut from each block, with H&E staining performed on the first and last sections to confirm tumor. The penultimate slide was used for ERG IHC. The remaining slides were used for manual tumor dissection with a scalpel for DNA/RNA isolation.
ERG immunohistochemistry (IHC)
ERG rearrangement status is a clonal marker in PCa.(16,18-20). Thus, to determine clonality of cancer specimens, we assessed ERG status on cancerous tissues obtained from the same focus at both time points (Supplementary Figure 1). Immunohistochemistry (IHC) for ERG was performed using the Ventana Benchmark System and rabbit monoclonal anti ERG (clone 5B7, Ventana Medical Systems; Tucson, Arizona), as described.(21,22) ERG positivity was defined as diffuse, moderate to strong nuclear immunoreactivity.(21)
Targeted DNA/RNA next generation sequencing (NGS)
DNA and RNA were co-isolated from each specimen as previously described.(23) DNA and RNA libraries were generated per sample using the Ion Ampliseq Library kit (Life Technologies, Foster City, CA), as described.(23) We prepared templates for DNA and RNA libraries using the Ion PI Template OT2 200 Kit v3 on the Ion One Touch 2 and sequencing was performed on Ion Proton P1 chips using the Ion PI Sequencing 200 Kit v3 (200 base pair reads), essentially as described.(24,25) NGS data analysis was performed using Torrent Suite (4.2.0) and the Coverage Analysis Plug-ins (both v4.0-r73765), along with the Ion Reporter (4.2.0) Targeted NGS, fusion analysis workflow and in house validated pipelines as described in the Supplementary Methods.(24-27) A sample was classified as fusion positive if a fusion isoform was supported by ≥ 20 reads and ≥ 3.0% total mapped reads, otherwise is classified as fusion negative.
Data acquisition and analysis
Demographic, relevant clinical and pathologic data of the cohort were abstracted from medical records and entered into a secure electronic HIPAA-compliant database. Biopsies were performed at UCLA and genetic studies were performed at University of Michigan with IRB approval. For each tissue sample, genetic alterations were classified as present or absent, and compared between paired “initial” and “repeat” tissue samples to assess clonality and reclassification from Gleason 6 to Gleason >6 PCa. Statistical analyses were performed using R® (R Foundation for Statistical Computing, http://www.R-project.org). Two-tailed statistical tests were used for all comparisons and p-values <0.05 considered statistically significant.
RESULTS
Of the 275 men on AS for PCa during the study period, 31 met eligibility criteria of whom 26 (84%) had cancer present on initial and repeat biopsy sections in quantities sufficient for ERG staining. Concordant ERG status in initial and repeat biopsies was observed in 25 of 26 (96%) patients, with 10 (38%), 15 (58%) and 1 (4%) patient demonstrating concordant ERG+|+, concordant ERG−|− and discordant ERG-/ERG+ paired biopsies, respectively (Figure 3 and Supplementary Figure 1). Our observation of 96% concordance is highly significant compared to that expected by chance (expected: 50% [13/26] concordant (ERG+/+ and ERG−/−); observed: 96% [25/26] concordant (ERG+/+ and ERG−/−), p=0.0003). RNAseq using the Oncomine Comprehensive Panel (OCP), which targets recurrent cancer gene fusions (including all known 5’ and 3’ partners in PCa ETS gene fusions), was evaluable in paired initial and repeat specimens from 13 patients. Concordant ETS fusion status in initial and repeat samples was present in 12 of 13 (92%) patients, and RNAseq results were consistent with ERG IHC status in all samples with evaluable staining. Of interest, patient 29, who did not show grade progression on repeat biopsy, had ERG− and ERG+ cancer by IHC on initial and repeat biopsy, respectively. RNAseq identified a TMPRSS2:ETV1 fusion in the ERG− initial biopsy sample and a TMPRSS2:ERG fusion in the ERG+ repeat biopsy sample (Table 2), supporting the sampling of two distinct clonal foci in this case.
Figure 3. Examples of ERG immunohistochemistry (IHC) in paired initial and repeat prostate biopsies.

For patients (Pt) 17 and 25, H&E stain of biopsy cores and corresponding ERG IHC are shown for paired initial and repeat biopsy samples. Low power views of corresponding H&E and ERG IHC are shown on the left, with areas shown in higher power indicated by black (H&E, middle panels) and red (ERG IHC, right panels) dashed boxes. Areas of cancer are indicated by green arrows. For ERG IHC, staining of endogenous ERG in endothelial cells as an internal positive control is indicated by red arrowheads. Original magnification 2x (left panels) and 10x (middle and right panels). Overall in our cohort, 96% of patients showed concordant ERG IHC status between paired early and repeat biopsies, supporting frequent sampling of the same clonal focus.
Table 2.
Pathologic and Genetic Profile of the Biopsy Samples exhibiting Prioritized Genetic Alterations
| ID | ERG Status | Gleason Score | Prioritized Mutations | Variant Allele Freq. | ||||
|---|---|---|---|---|---|---|---|---|
| Initial | Repeat | Initial | Repeat | Gene | Mutation | Initial | Repeat | |
| 1 | + | + | 6 | 3+4 | SPEN | P2984L | 0.0% | 10.9% |
| 4 | - | N/A | 6 | 3+4 | SPOP | F133L | 0.4% | 12.9% |
| 7 | - | - | 6 | 6 | BRCA2 | K2524fs | 0.0% | 10.1% |
| 8 | + | + | 6 | 6 | ARID1B | Q1491R | 10.6% | 0.7% |
| NOTCH1 | P743S | 28.3% | 0.0% | |||||
| 9 | - | N/A | 6 | 6 | ZC3H13 | N55H | 31.3% | 8.6% |
| 18 | - | - | 6 | 3+4 | TP53 | R196P | 0.2% | 12.2% |
| PIK3CA | V344M | 0.1% | 11.4% | |||||
| 19 | + | + | 6 | 6 | APC | A1718V | 0.0% | 19.8% |
| 22 | - | - | 6 | 6 | SPOP | F125V | 5.7% | 7.6% |
| 23 | - | - | 6 | 3+4 | IDH1 | R132C | 32.0% | 19.0% |
| RNF213 | S411X | 50.9% | 46.9% | |||||
| 25 | + | + | 6 | 3+4 | GAS6 | P285L | 0.0% | 11.4% |
| NOTCH1 | P168S | 0.0% | 16.7% | |||||
| ATRX | M6I | 0.0% | 24.5% | |||||
| 27 | + | + | 6 | 4+4 | ARID1A | E1958K | 27.4% | 0.5% |
| KMT2B | R2092G | 36.6% | 32.5% | |||||
| 28 | + | + | 6 | 6 | KMT2B | A1964T | 16.9% | 0.0% |
| ARHGAP35 | V644I | 13.0% | 0.1% | |||||
| 29 | - | + | 6 | 6 | NCOR1 | S1750C | 15.4% | 0.0% |
Abbreviations: ID – Patient identification; N/A- Insufficient sample for analysis;
ERG status (by IHC) and Gleason score of profiled initial and repeat biopsies is indicated. The variant allele frequencies (in %) of high confidence somatic mutations identified in initial and/or repeat biopsies is given (bold indicates detected).
In addition to ETS gene fusions, SPOP and IDH1 mutations are early driving molecular alterations in PCa and define molecular subtypes (~10% and 1% of all PCa, respectively) that are mutually exclusive with ERG gene fusions.(24,28,29) Both alterations are targeted by the DNA component of the OCP; thus we assessed for SPOP and IDH1 mutation status as clonal markers in serial samples. In patient 4, who showed grade progression on repeat biopsy and was ERG− on initial (IHC−/RNAseq−) and repeat biopsy (IHCN/A/RNAseq−), we identified a high-confidence prioritized SPOP F133L mutation by targeted DNAseq exclusively in the repeat biopsy specimen (3/670 reads [0.4%] in the initial sample vs. 111/864 reads [13%] in the repeat sample), consistent with serial sampling of two clonally distinct foci. In contrast, patient 22, who did not show grade progression and was ERG− on initial and repeat biopsy (both IHC−/RNAseq−), harbored high-confidence prioritized SPOP F125V mutations by targeted DNAseq in both serial samples (7/124 reads [6%] in the initial sample vs. 20/264 reads [8%] in the repeat sample). Similarly, patient 23, who also progressed to Gleason score 3+4=7 on repeat biopsy and was ERG− on initial and repeat biopsy (both IHC−/RNAseq−), harbored high-confidence prioritized IDH1 R132C mutations by targeted DNAseq in both serial samples (128/400 reads [32%] in the initial sample vs. 85/448 reads [19%] in the repeat sample). IDH1 R132H mutations were confirmed in both samples by Sanger Sequencing (data not shown). Concordant IDH1 mutations in this case with grade progression supports a clonal relationship between serially sampled low and high-grade cancer components. Taken together, through IHC and targeted RNAseq/DNAseq, our data support MRI/US fusion as being able to sample the same PCa focus over time.
Overall, of the 26 evaluable cases, 9 (34.6%) progressed to high-grade disease. Repeat biopsies, however, showed only a small focus (≤ 10%) of a higher Gleason grade component in 3 of the 9 cases. All 9 (100%) cases that progressed to high-grade disease demonstrated ERG concordance between the initial and the repeat biopsies, strongly suggesting that these higher-grade cancers shared a clonal relationship with their lower-grade counterpart.
DISCUSSION
In this study we demonstrate, using electronic biopsy-site tracking and ERG IHC status as a clonal marker, that a clonal focus of PCa may be serially sampled over a median interval of 1-year. In men on active surveillance, a 96% ERG-status concordance between paired biopsies obtained from the same location over time was found using MRI/US fusion biopsy guidance. This finding strongly suggests that the tissues assayed at two different time points were of the same clonal origin.(18) Additionally, the results also provide support for the notion that high-grade PCa (i.e., ≥ Gleason 7) may arise clonally from Gleason 6 disease and further implies that some Gleason 6 PCa may not be indolent.
Effective AS rests on accurate patient selection and the ability to precisely detect changes in disease status over time. Results from our study, and others, suggest that MRI/US fusion-guided biopsy facilitates both.(14) It is not uncommon to obtain a negative trans-rectal ultrasound guided prostate needle biopsy during AS, even when biopsies are cognitively guided towards areas of presumed disease.(14) One explanation is the lack of precision of trans-rectal ultrasound in tracking and locating areas of previously diagnosed PCa. In prior work, the precision of MRI/US fusion biopsy for re-sampling areas to within 3mm was confirmed in phantom models and validated in a patient cohort. (9,11) Clinical validation in this study was assessed by targeting the same site as determined by repetitive 3D modeling during the same biopsy accession. Further, a recent report by Ukimura et al., employing a commercial cell cycle-based gene expression signature interrogating biopsy samples assayed 1 year apart, suggested a same site biopsy precision of 86% but could not determine clonality.(30) In the present study, we present molecular data to clearly show that MRI/US targeted biopsy can serially sample the same clonal focus of PCa.
At present, it is unclear whether high-grade PCa arises de novo, or if Gleason 6 cancers possess the biological potential for high-grade progression. In a recent epidemiological study, Penney et al. concluded that Gleason grade progression of PCa is uncommon.(31) Similar to other reports, the evidence presented stems from the decline in advanced stage disease in the PSA era compared to the pre-PSA era, without a corresponding decrease in the proportion of high-grade disease across the same time period.(31-33) Such analyses, however, are unable to evaluate the possibility of grade progression on an individual level. In a cross-sectional study evaluating the clonal origin of Gleason grades 3 and 4 cancer, Sowalsky et al. examined adjacent foci of disease.(16) The authors reported 100% concordance for the TMPRSS2:ERG gene fusion and identical TMPRSS2:ERG fusion breakpoints in selected cases, suggesting a common clonal origin between contiguous areas of cancer. In another analysis of multifocal PCa with metastasis, VanderWeele et al. concluded that: i) a single progenitor can give rise to both low- and high-grade disease; ii) early divergence occurs between low- and high-grade foci; and iii) late divergence occurs between high-grade foci and metastases. Although both studies suggest that high-grade disease may arise from low-grade lesions, neither study can ascertain the temporal progression of high-grade disease.(34)
The ability to longitudinally assess the same focus of PCa over time has only recently been made possible by MRI/US fusion biopsy platforms. In our cohort using MRI/US fusion biopsy with longitudinal sampling, we found that 100% of cases that progressed from Gleason 6 to ≥7 cancer demonstrated concordance for ERG status. These data obtained via molecular profiling, are the first to show that high-grade disease may arise clonally from Gleason 6 PCa over time. A notable corollary to this is the notion that some Gleason 6 cancers may not be indolent and should be followed carefully. A case report of a lethal clone arising from Gleason pattern 3 (although in the presence of additional distinct large high-grade tumors), is in line with this and suggests that heterogeneity may also exist within low-grade lesions vis-á-vis aggressive potential.(35) More work is needed to discern the molecular profile of Gleason 6 PCa destined to progress.
Our study has several limitations. First, repeat biopsy was performed after 5-35 months based on routine clinical practice, with the ideal time to track grade progression unknown. Second, as fusion biopsy technology continues to accumulate, the reproducibility of our findings with more generalized use of MRI/US fusion biopsy technology needs to be evaluated. Third, our cohort is relatively small and our findings should be tested in larger populations. And fourth, an element of sampling bias remains an important confounder of our study. We cannot say with certainty that high-grade lesions did not exist at the time of initial biopsy where low-grade lesions were observed. Since only a small focus of high-grade component was found in some repeat biopsies, initially non-sampled high-grade lesions that were present in close continuity with low-grade lesions might have given us the false impression of grade progression (Supplementary Figure 1). Future studies that aim to additionally sample areas around targeted biopsy sites, at high density, may address this issue.
The strength of this study lies in its longitudinal nature and the performance of contemporary molecular techniques on minute FFPE biopsy samples. The present findings provide molecular data to support the clinical use of MRI/US fusion biopsy platforms in the management of men on AS. In addition, our results add to our understanding of the biology of low-grade PCa and suggest that some Gleason 6 cancers may not be indolent. If these findings are confirmed, MRI/US fusion biopsy may become a new standard for monitoring the growing number of men on AS. Additional work is needed to confirm our findings and to develop genomic predictors of Gleason 6 PCa progression.
CONCLUSION
In this study, we demonstrate that serial MRI/US targeted prostate biopsy allows accurate assessment of the same clonal focus of cancer over time, even in the absence of an MRI target. Molecular profiling of tissue obtained in a longitudinal fashion suggests that Gl eason ≥7 PCa may arise clonally from Gleason 6 disease. These findings may have significant impact on the clinical management of the growing number of men with low-grade PCa being managed with AS. Larger studies are needed to validate our findings and to definitively determine the risk of low-grade PCa progression.
Supplementary Material
Statement of translational relevance.
Use of active surveillance to manage ‘low-risk’ prostate cancer (PCa) is increasing rapidly. Follow-up biopsy is the main method to determine if continued surveillance or active intervention is most appropriate. However, follow-up biopsy has until recently been a blind procedure where the detection of progressive cancer, not suitable for surveillance, may be missed as a result of sampling error at the outset. Herein we report the use of biopsy site tracking via magnetic resonance imaging/ultrasound (MRI/US) fusion to sample a specific locus of cancer cells serially over a median interval of 1 year. Through detailed molecular study of temporally paired cancer tissues, we determined that serial sampling of a specific prostate cancer clone over time is possible. We also observed molecular evidence that some low grade PCa harbor deleterious genetic alterations and may progress to higher grade disease during AS. Our findings provide rationale for employing MRI/US fusion biopsy strategies to monitor patients on AS and suggest that some Gleason 6 cancers may not be indolent.
Acknowledgments
Financial Support: Supported in part by award number R01CA158627 (L.S.M.) and 5 P50 CA186786-05 (G.S.P) from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Additional support was provided by the Beckman Coulter Fo`undation, the Jean Perkins Foundation, and the Steven C. Gordon Family Foundation. Supported in part by the Department of Defense PC130652 (S.A.T.), W81XWH-14-1-0287 (T.M.M.). S.S.S. is supported by the Urology Care Foundation. S.A.T. and T.M.M. are supported by the A. Alfred Taubman Medical Research Institute and the Prostate Cancer Foundation. Research reported in this publication was supported by the Pilot Training Grant in Translational Research to K.R.V. provided by the Department of Pathology, University of Michigan.
S.A.T has received travel support from, and had a sponsored research agreement with Compendia Bioscience/Life Technologies/ThermoFisher that provided access to the sequencing panel used herein. No other aspect of this study was supported by Compendia Bioscience/Life Technologies/ThermoFisher. The University of Michigan has been issued a patent on ETS gene fusions in prostate cancer on which S.A.T. is a co-inventor. The diagnostic field of use has been licensed to Hologic/Gen-Probe, Inc., which has sublicensed rights to Roche/Ventana Medical Systems. S.A.T. has served as a consultant for and received hornoraria from Roche/Ventana Medical Systems, Jansenn, AbbVie and Astellas/Medivation. S.A.T. has a sponsored research agreement with Astellas. S.A.T. is a co-founder and consultant for Strata Oncology.
Footnotes
Presented in a podium session at the 2016 AUA Annual Meeting in San Diego, CA
Disclosures: The other authors have no competing interests to declare.
References
- 1.Carter HB, Sauvageot J, Walsh PC, Epstein JI. Prospective evaluation of men with stage T1C adenocarcinoma of the prostate. J Urol NIH Public Access. 1997;157:2206–9. [PMC free article] [PubMed] [Google Scholar]
- 2.Klotz L, Zhang L, Lam A, Nam R, Mamedov A, Loblaw A. Clinical results of long-term follow-up of a large, active surveillance cohort with localized prostate cancer. J Clin Oncol American Society of Clinical Oncology. 2010;28:126–31. doi: 10.1200/JCO.2009.24.2180. [DOI] [PubMed] [Google Scholar]
- 3.Tosoian JJ, Mamawala M, Epstein JI, Landis P, Wolf S, Trock BJ, et al. Intermediate and Longer-Term Outcomes From a Prospective Active-Surveillance Program for Favorable-Risk Prostate Cancer. J Clin Oncol American Society of Clinical Oncology. 2015;62:5764. doi: 10.1200/JCO.2015.62.5764. JCO.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson I, Thrasher JB, Aus G, Burnett AL, Canby-Hagino ED, Cookson MS, et al. Guideline for the management of clinically localized prostate cancer: 2007 update. J Urol. 2007:2106–31. doi: 10.1016/j.juro.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Mohler JL, Kantoff PW, Armstrong AJ, Bahnson RR, Cohen M, D’Amico AV, et al. Prostate cancer, version 2.2014. J Natl Compr Canc Netw. 2014:686–718. doi: 10.6004/jnccn.2014.0072. [DOI] [PubMed] [Google Scholar]
- 6.Fleshner NE, O’Sullivan M, Fair WR. Prevalence and predictors of a positive repeat transrectal ultrasound guided needle biopsy of the prostate. J Urol. 1997;158:505–8. discussion508–9. [PubMed] [Google Scholar]
- 7.Pinto PA, Chung PH, Rastinehad AR, Baccala AA, Kruecker J, Benjamin CJ, et al. Magnetic resonance imaging/ultrasound fusion guided prostate biopsy improves cancer detection following transrectal ultrasound biopsy and correlates with multiparametric magnetic resonance imaging. J Urol. 2011;186:1281–5. doi: 10.1016/j.juro.2011.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rastinehad AR, Turkbey B, Salami SS, Yaskiv O, George AK, Fakhoury M, et al. Improving detection of clinically significant prostate cancer: magnetic resonance imaging/transrectal ultrasound fusion guided prostate biopsy. J Urol Elsevier. 2014;191:1749–54. doi: 10.1016/j.juro.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonn GA, Filson CP, Chang E, Natarajan S, Margolis DJ, Macairan M, et al. Initial experience with electronic tracking of specific tumor sites in men undergoing active surveillance of prostate cancer. Urol Oncol. 2014;32:952–7. doi: 10.1016/j.urolonc.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filson CP, Natarajan S, Margolis DJA, Huang J, Lieu P, Dorey FJ, et al. Prostate cancer detection with magnetic resonance-ultrasound fusion biopsy: The role of systematic and targeted biopsies. Cancer. 2016;122:884–92. doi: 10.1002/cncr.29874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natarajan S, Marks LS, Margolis DJA, Huang J, Macairan ML, Lieu P, et al. Clinical application of a 3D ultrasound-guided prostate biopsy system. Urol Oncol. 2011;29:334–42. doi: 10.1016/j.urolonc.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Epstein JI, Walsh PC, Carter HB. Dedifferentiation of prostate cancer grade with time in men followed expectantly for stage T1c disease. J Urol. 2001;166:1688–91. [PubMed] [Google Scholar]
- 13.Sheridan TB, Carter HB, Wang W, Landis PB, Epstein JI. Change in prostate cancer grade over time in men followed expectantly for stage T1c disease. J Urol. 2008;179:901–4. doi: 10.1016/j.juro.2007.10.062. discussion904–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porten SP, Whitson JM, Cowan JE, Cooperberg MR, Shinohara K, Perez N, et al. Changes in prostate cancer grade on serial biopsy in men undergoing active surveillance. J Clin Oncol American Society of Clinical Oncology. 2011;29:2795–800. doi: 10.1200/JCO.2010.33.0134. [DOI] [PubMed] [Google Scholar]
- 15.Lavery HJ, Droller MJ. Do Gleason patterns 3 and 4 prostate cancer represent separate disease states? J Urol. 2012;188:1667–75. doi: 10.1016/j.juro.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 16.Sowalsky AG, Ye H, Bubley GJ, Balk SP. Clonal progression of prostate cancers from Gleason grade 3 to grade 4. Cancer Res American Association for Cancer Research. 2013;73:1050–5. doi: 10.1158/0008-5472.CAN-12-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bax J, Cool D, Gardi L, Knight K, Smith D, Montreuil J, et al. Mechanically assisted 3D ultrasound guided prostate biopsy system. Med Phys. 2008;35:5397–410. doi: 10.1118/1.3002415. [DOI] [PubMed] [Google Scholar]
- 18.Furusato B, Tan S-H, Young D, Dobi A, Sun C, Mohamed AA, et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis Nature Publishing Group. 2010;13:228–37. doi: 10.1038/pcan.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young A, Palanisamy N, Siddiqui J, Wood DP, Wei JT, Chinnaiyan AM, et al. Correlation of urine TMPRSS2:ERG and PCA3 to ERG+ and total prostate cancer burden. Am J Clin Pathol The Oxford University Press. 2012;138:685–96. doi: 10.1309/AJCPU7PPWUPYG8OH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haffner MC, Weier C, Xu MM, Vaghasia A, Gurel B, Gumuskaya B, et al. Molecular evidence that invasive adenocarcinoma can mimic prostatic intraepithelial neoplasia (PIN) and intraductal carcinoma through retrograde glandular colonization. J Pathol John Wiley & Sons, Ltd. 2016;238:31–41. doi: 10.1002/path.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomlins SA, Palanisamy N, Siddiqui J, Chinnaiyan AM, Kunju LP. Antibody-based detection of ERG rearrangements in prostate core biopsies, including diagnostically challenging cases: ERG staining in prostate core biopsies. Arch Pathol Lab Med College of American Pathologist 325 Waukegan Rd, Northfield, IL 60093. 2012;136:935–46. doi: 10.5858/arpa.2011-0424-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Udager AM, Shi Y, Tomlins SA, Alva A, Siddiqui J, Cao X, et al. Frequent discordance between ERG gene rearrangement and ERG protein expression in a rapid autopsy cohort of patients with lethal, metastatic, castration-resistant prostate cancer. Prostate. 2014;74:1199–208. doi: 10.1002/pros.22836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warrick JI, Hovelson DH, Amin A, Liu C-J, Cani AK, McDaniel AS, et al. Tumor evolution and progression in multifocal and paired non-invasive/invasive urothelial carcinoma. Virchows Arch Springer Berlin Heidelberg. 2015;466:297–311. doi: 10.1007/s00428-014-1699-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hovelson DH, McDaniel AS, Cani AK, Johnson B, Rhodes K, Williams PD, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015;17:385–99. doi: 10.1016/j.neo.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDaniel AS, Hovelson DH, Cani AK, Liu C-J, Zhai Y, Zhang Y, et al. Genomic Profiling of Penile Squamous Cell Carcinoma Reveals New Opportunities for Targeted Therapy. Cancer Res American Association for Cancer Research. 2015;75:5219–27. doi: 10.1158/0008-5472.CAN-15-1004. [DOI] [PubMed] [Google Scholar]
- 26.Cani AK, Hovelson DH, McDaniel AS, Sadis S, Haller MJ, Yadati V, et al. Next-Gen Sequencing Exposes Frequent MED12 Mutations and Actionable Therapeutic Targets in Phyllodes Tumors. Mol Cancer Res American Association for Cancer Research. 2015;13:613–9. doi: 10.1158/1541-7786.MCR-14-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDaniel AS, Stall JN, Hovelson DH, Cani AK, Liu C-J, Tomlins SA, et al. Next-Generation Sequencing of Tubal Intraepithelial Carcinomas. JAMA Oncol American Medical Association. 2015;1:1128–32. doi: 10.1001/jamaoncol.2015.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J-P, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ukimura O, Gross ME, de Castro Abreu AL, Azhar RA, Matsugasumi T, Ushijima S, et al. A novel technique using three-dimensionally documented biopsy mapping allows precise re-visiting of prostate cancer foci with serial surveillance of cell cycle progression gene panel. Prostate. 2015;75:863–71. doi: 10.1002/pros.22969. [DOI] [PubMed] [Google Scholar]
- 31.Penney KL, Stampfer MJ, Jahn JL, Sinnott JA, Flavin R, Rider JR, et al. Gleason grade progression is uncommon. Cancer Res American Association for Cancer Research. 2013;73:5163–8. doi: 10.1158/0008-5472.CAN-13-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crawford ED. Epidemiology of prostate cancer. Urology. 2003;62:3–12. doi: 10.1016/j.urology.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Falzarano SM, Magi-Galluzzi C. Prostate cancer staging and grading at radical prostatectomy over time. Adv Anat Pathol. 2011;18:159–64. doi: 10.1097/PAP.0b013e31820cb506. [DOI] [PubMed] [Google Scholar]
- 34.VanderWeele DJ, Brown CD, Taxy JB, Gillard M, Hatcher DM, Tom WR, et al. Low-grade prostate cancer diverges early from high grade and metastatic disease. Cancer Sci. 2014;105:1079–85. doi: 10.1111/cas.12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, et al. Tracking the clonal origin of lethal prostate cancer. J Clin Invest American Society for Clinical Investigation. 2013;123:4918–22. doi: 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.