

Abstract
The fields of telomere biology and DNA repair have enjoyed a great deal of cross-fertilization and convergence in recent years. Telomeres function at chromosome ends to prevent them from being falsely recognized as chromosome breaks by the DNA damage response and repair machineries. Conversely, both canonical and non-conical functions of numerous DNA repair proteins have been found to be critical for preserving telomere structure and function. In 2009 Elizabeth Blackburn, Carol Greider, and Jack Szostak were awarded the Nobel prize in Physiology or Medicine for the discovery of telomeres and telomerase. Four years later pioneers in the field of DNA repair, Aziz Sancar, Tomas Lindahl and Paul Modrich, were recognized for their seminal contributions by being awarded the Nobel Prize in Chemistry. This review is part of a special issue meant to celebrate this amazing achievement, and will focus in particular on the convergence of nucleotide excision repair and telomere biology, and will discuss the profound implications for human health.
Graphical abstract
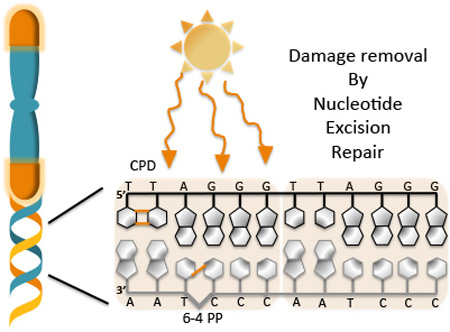
Telomeres are nucleo-protein structures consisting of tandem hexameric TTAGGG repeats that protect the ends of linear chromosomes. Ultraviolet light irradiation induces DNA photoproduct formation in telomeric repeat sequences that are then removed by nucleotide excision repair. The orange lines represent the covalent bonds formed between two adjacent pyrimidines after UV exposure. The hotspots for formation of pyrimidine (6-4) pyrimidine photoproducts (6-4 PP) and cyclobutane pyrimidine dimer (CPD) are shown.
INTRODUCTION
Telomere structure and function
Telomeres are nucleoprotein DNA structures that cap and protect the ends of linear chromosomes. They have been likened to the plastic caps at the end of shoelaces to prevent degradation and fraying. Telomeres preserve chromosome stability, promote cellular survival and proliferation, and prevent degenerative diseases and cancer (1, 2). Dysfunctional and uncapped telomeres drive aging-related pathologies and carcinogenesis (3, 4). In humans the telomeres consist of a tandem array of hexameric 5’-TTAGGG repeats that average about 10 kb long, and terminate in a 3’ single stranded tail consisting of 50 to 200 nucleotides of TTAGGG repeats (5). The tail can wrap around and invade the telomeric duplex to form a large lasso-like t-loop structure (6, 7). The structural rearrangement of telomeres requires a specialized complex of six proteins termed shelterin, that is essential for telomere function (reviewed in (8, 9)) (Figure 1). Telomeric repeat binding factors 1 and 2 (TRF1 and TRF2) bind the TTAGGG duplexes through an Myb DNA binding domain and form homodimers via their TRFH (TRF Homology) domains (10, 11). TRF1 and TRF2 do not interact directly, but are connected through their binding to a shared partner, the shelterin protein TIN2 (12). The protection of telomeres 1 (POT1) protein contains two OB fold domains that mediate binding to the telomeric 3’ tail and single stranded TTAGGG repeats (13, 14). TIN2 and POT1 are connected via their interaction with the fifth shelterin protein TPP1 (12, 15), thereby linking POT1 with the rest of shelterin. Finally, the last shelterin member, RAP1, interacts exclusively with TRF2 (16) (Figure 1). Shelterin proteins interact with enzymes in every known DNA repair pathway and regulate activity of these enzymes at telomeres (for recent reviews see (17, 18)). This review will focus on interactions with nucleotide excision repair (NER) factors.
Figure 1.

Telomere structure. Telomeres are nucleo-protein structures that protect the ends of linear chromosomes. The complex of six proteins called shelterin promotes the folding of the telomeric DNA into a t-loop (see text for more details). The magnifying glass highlights the region of 3’ single strand tail invasion into the duplex to form the t-loop structure.
Telomeres provide a solution to the problems of replicating the tips of linear chromosomes, and the ability of nucleases to degrade chromosome ends. The end replication problem arises from the inability of the DNA replication machinery to complete synthesis at the very tips of the lagging strand, which leads to chromosome shortening every cell division (Figure 2). In germ cells and in ~90% of cancer cells, the end replication problem is overcome by telomerase, a reverse transcriptase that uses an integral RNA template to add single stranded TTAGGG repeats to the telomeric tail (19–21). However, most human somatic cells are deficient in telomerase activity. Telomeres also solve the end protection problem, which refers to the inherent resemblance of chromosome ends to DNA double strand breaks (DSBs). Loss of shelterin causes the chromosome ends to be falsely recognized by the DNA damage response (DDR) machinery, which triggers p53-mediated cell senescence or apoptosis (9, 22) (Figure 2). If the p53 pathway is defective, then the uncapped telomeres will be fused by the non-homologous end joining (NHEJ) or alternative end joining (A-EJ) machineries, leading to chromosome end-to-end fusions (9). Alternatively, the uncapped telomeres will be inappropriately processed by the homologous recombination repair machinery, which leads to telomere alterations (9, 23). This DDR signaling at telomeres is normally suppressed by TRF2 which inhibits ATM kinase activation, or POT1 which inhibits ATR kinase (24). Thus, shelterin proteins can influence DNA repair at telomeres by regulating the DNA damage response and signaling proteins.
Figure 2.

The end replication and protection problems. In somatic cells each round of cell division leads to telomere shortening due to the inability of the replication machinery to complete replication of the lagging strand (end replication problem). When telomeres become critically short they are unable to form the protective t-loop which triggers a DNA damage response due to the telomere uncapping (end protection problem). In normal cells this results in cell senescence or cell death. However, in p53 defective cells telomere uncapping leads to chromosome end-to-end fusions and genomic instability and ultimately cell crisis. Survivors that overcome crisis do so by up-regulating telomerase (90% of cancer cells) or by activating the alternative lengthening of telomeres pathway (ALT, 10% of cancer cells) to maintain telomere lengths.
Overview of nucleotide excision repair at telomeres
Nucleotide excision repair (NER) is the main DNA repair pathway involved in the removal of bulky lesions. This flexible repair mechanism involves over 30 distinct polypeptides that coordinate a series of steps (reviewed in (25)). Damage recognition proteins initiate the global genome repair (GGR) and RNA polymerase blockage initiates the transcription coupled repair (TCR) pathways of NER. In GGR the lesion is recognized by XPC-hHR23B, which may be preceded by XPE/DDB2 protein. Strand separation around the lesion occurs by helicases XPB and XPD in the TFIIH complex generating a ~30 nucleotide bubble. XPA and RPA stabilize the bubble. Strand incision of the bubble initiates with endonuclease XPF-ERCC1 on the 5’ side, followed by XPG endonuclease on the 3’ side. This releases a lesion-containing oligonucleotide (NER product) that is 24–32 nucleotides in length. The Sancar lab discovered that these repair products function in cellular signaling (26). The final step of gap filling is achieved with DNA polymerases δ/ε and accessory proteins and the nick is sealed by DNA ligase (27).
Telomeres present a unique challenge to DNA damage processing compared to elsewhere in the genome, due to their highly repetitive G-rich sequence, which allows the formation of alternate structures such as G-quadruplexes and t-loops (17, 28, 29). They are also proposed to be highly sensitive to bulky and helix distorting DNA lesions that block DNA replication, because of a lack of origin firing within telomeres that might otherwise compensate for unreplicated regions (17, 30–32). Information regarding the impact of bulky DNA lesions on telomere biology and NER function at telomeres is largely limited to the study of damage caused by ultraviolet (UV) light irradiation.
UV PHOTOPRODUCT FORMATION AT TELOMERES
DNA exposure to UV light leads to the formation of UV photoproducts. If these lesions are not removed they can induce mutagenesis and cytotoxicity (25, 33). There are a wide variety of UV photoproducts, however the primary targets of UV light are the pyrimidine bases of DNA, resulting in the covalent linkage between the cyclobutane rings of two adjacent pyrimidines (34). The covalent linkage between the C5 and C6 of the neighboring pyrimidines produces cyclobutane pyrimidine dimers (CPDs), while the linkage between the C4 of one pyrimidine to the C6 of the adjacent base produces pyrimidine (6-4) pyrimidine photoproducts (6-4 PP). Although, pyrimidine dimers can arise at 5’TT, 5’CC, 5’CT and 5’TC sequences, the 5’TT and 5’CC sites are the preferred targets for CPD and 6-4 PP formation, respectively (35). Telomeres contain an abundance of di-pyrimidine sites in both the leading and lagging strands, making them a prime target for UV light-induced damage (Figure 3A). Given the importance of telomere preservation in genome stability, telomere sensitivity to UV light compared to the rest of the genome has been investigated. Compared to specific loci in the genome including regions of the p53 gene and 28S ribosomal DNA, the telomeric DNA isolated from UVC exposed primary fibroblasts appear to harbor more CPDs (36). However, when compared to the bulk genome, the telomeres isolated from UVC exposed human fibroblasts contain fewer CPD and 6-4 PP lesions (37). Interestingly, when purified telomeres are exposed to UVC in vitro they exhibit nearly equal amounts of UV photoproducts compared to those generated in purified bulk genomic DNA (37). These results strongly suggest that the presence of telomeric shelterin proteins partly protects telomeres from UV-induced DNA damage in vivo (Figure 3B). Consistent with this, the pre-incubation of purified telomeric DNA fragments with purified telomeric TRF1 protein in vitro reduces the amount of UVC-induced CPDs and 6-4PPs, compared to naked DNA and non-telomeric fragments (37). The shelterin protein mediated shielding of telomeric DNA highlights another important role for this complex in preserving telomere integrity.
Figure 3.

Telomere sequences are hot spots for UV photoproduct formation (A) but are partially shield by the shelterin protein complex (B). Orange lines represent the covalent bonds formed between two adjacent pyrimidines after UV exposure. The red bases show the preferred targets for CPDs and 6-4 PPs.
NUCLEOTIDE EXCISION REPAIR OF TELOMERIC PHOTOPRODUCTS
Studies of the formation and removal of UV-induced bulky lesions at telomeres gave rise to conflicting results, therefore, whether NER functions at telomeres had been a matter of debate. On the one hand, telomeres were shown to be refractory to CPD repair when compared with specific genome loci, such as regions of the p53 gene, when a PCR-based assay was used to identify and quantify damaged DNA isolated from UVC irradiated fibroblasts (37). On the other hand, an earlier study showed CPDs were repaired in telomeric restriction fragments isolated from UVC irradiated fibroblasts when T4 endonuclease V was used to identify and measure telomeric lesions (38). More recently, our laboratory demonstrated the repair of both CPDs and 6-4 PPs in highly purified telomeres from UVC exposed human fibroblasts by using a direct and quantitative detection method of immuno-staining telomeres for both lesions (37). These conflicting observations are potentially explained by the use of different cell lines, and different techniques for quantifying lesions, but may also be due to the comparison of telomere repair to specific genomic regions (36) versus the bulk genome (37). Interestingly, we found that the rate of 6-4 PPs removal is similar at telomeres compared to the bulk genome, but that CPD removal at telomeres is 1.5-fold faster (37). Our study also provided the first direct evidence for NER function at telomeres by demonstrating the persistence of both CPD and 6-4 PP lesions at the telomeres from UV-exposed XPA deficient cells derived from a patient with xeroderma pigmentosum (XP) syndrome (37). These cells lack NER activity. Collectively, studies on UV photoproduct formation and repair at telomeres give rise to interesting new questions, especially related to the role of the NER pathway in telomere preservation upon DNA damage. For example, the presence of a CPD lesion within a telomeric fragment prevents TRF1 binding to the DNA (37). Since TRF1 loss leads to telomere defects such as telomere fragility (32), this finding highlights the importance of UV photoproduct removal for preserving telomere integrity.
CAN UV IRRADIATION AND BULKY ADDUCTS CAUSE TELOMERE DYSFUNCTION?
Telomeres in sunlight or UVB exposed skin from humans and mice are shorter compared with unexposed skin, suggesting a link between UV exposure and telomere alterations. Using quantitative fluorescence in situ hybridization to measure telomeres in human skin, Ikeda and colleagues observed shorter telomeres in sun-exposed skin compared to non-exposed areas, and reported shorter telomeres in skin containing actinic keratosis lesions compared to skin lacking these sun-damage associated lesions (39). The application of sun screen containing photolyase enzymes prevented the appearance of UV-induced telomere shortening in preparations of skin biopsies obtained 24 hours after exposure (40). This suggests that the telomere shortening may be related to telomere damage rather than extensive cell turn over in the affected areas. In mice, chronic UVB exposure accelerates telomere shortening in exposed skin, and this shortening is exacerbated either in the absence of telomerase or NER factor XPC (41). This supports a model that the failure to remove UV photoproducts at telomeres may interfere with telomere replication leading to critically short telomeres that cannot be rescued if telomerase is absent. Consistent with this, we observed that the inability to accurately bypass CPDs during DNA replication due to loss of DNA polymerase η, caused an increase in UVC-induced fragile telomeres and telomere loss (30). A fragile telomere appears as multi-telomeric signals at a chromatid end and is thought to arise from unreplicated regions, and telomere loss appears as a chromatid end lacking telomere staining and is proposed to result from replication fork collapsed into a double strand break (32). However, the impact of UV damage on telomere structure and function in individuals with XP lacking NER activity is largely unknown.
The effect of other forms of bulky DNA lesions on telomere stability remains to be investigated. Interestingly, NER factors XPB, XPD and XPC have all been implicated in telomere preservation in response to oxidative stress. Cells derived from XP individuals with mutations in NER helicases XPB or XPD, showed elevated rates of hydrogen peroxide induced telomere loss and telomeric end-to-end fusions (42, 43). Cells derived from Xpc knock out mice show increased levels of fragile telomeres that could be suppressed by culturing the cells at 3% oxygen to reduce the oxidative stress (41). These results may be due to potential roles for these NER factors in base excision repair pathways for removing oxidatively generated base lesions, as shown for XPC (44). Telomeres are shown to be highly susceptible to damage caused by reactive oxygen species (45, 46). Thus, another intriguing possibility is that telomeres are susceptible to hydroxyl radical induced formation of 8,5’-cyclopurine deoxynucleotide damage. However, the biological relevance of these lesions is unclear due to their very low abundance (47). These bulky oxidatively induced adducts that strongly DNA block replication and transcription are repaired by NER, rather than base excision repair, and have been implicated in causing neurological disease in XP (48–50).
CANONICAL AND NON-CANONICAL ROLES FOR NER PROTEINS AT TELOMERES
XPF-ERCC1 Endonuclease
Endonuclease XPF-ERCC1 has been implicated in telomere maintenance both through canonical roles in NER, and non-canonical roles in processing at telomeric ends. These studies have also uncovered a potential role for shelterin protein TRF2 in regulating NER. The first indication that TRF2 may regulate XPF-ERCC1 activity derived from the observation that telomere fusions induced by TRF2 loss is prevented by also eliminating XPF-ERCC1. TRF2 inhibition causes loss of the telomeric 3’tail and subsequent chromosome end-to-end fusions, but not in XPF-ERCC1 deficient cells, suggesting that TRF2 prevents this endonuclease from cleaving the tail (51). This study was also the first to report that TRF2 interacts with XPF-ERCC1 in cell extracts (51). The endonuclease possesses a TRF2 docking motif (52), but a direct interaction between these proteins remains to be demonstrated. Other studies suggest that the scaffold protein SLX4 is required to bridge the interaction between TRF2 and XPF-ERCC1 (53, 54). The XPF-ERCC1 interaction with SLX4 functions during inter-strand cross link repair, but not during NER (54, 55). Further study of purified TRF2 and XPF-ERCC1 proteins will be required to determine whether they can interact directly, and whether TRF2 can regulate the endonucleases catalytic activity.
Whether TRF2 regulates XPF-ERCC1 activity in the context of NER at telomeres is currently unknown. Mice engineered to overexpress TRF2 in skin cells develop phenotypes that are reminiscent of XP in regions that are exposed to light (56). These mice are also more sensitive to UVB-induced skin cancer and showed higher levels of CPDs and 6-4PPs in genomic DNA one hour after UV irradiation, compared to wild type (56). The mechanism for increased light sensitivity and slower DNA photoproduct removal in the bulk genome is unknown. One possibility is that excess TRF2 may sequester XPF-ERCC1 at telomeres and interfere with NER elsewhere in the genome. Interestingly, about a third of human basal cell carcinomas and squamous cell carcinomas show elevated TRF2 expression, although it is not clear if these tumors are related to sun exposure (56).
Evidence in human cells and mice also suggest that the TRF2 and XPF-ERCC1 interaction has a non-canonical role in telomere length regulation that remains poorly understood. Mice overexpressing TRF2 in skin showed faster rates of telomere shortening and increased fragile telomeres in light exposed regions (56). The accelerated telomere shortening phenotype requires XPF-ERCC1 protein in both mice and human cells overexpressing TRF2, yet does not require XPF-ERCC1 endonuclease activity in the human cells (56, 57). However, XPF-ERCC1 interacts with endonucleases MUS81/EME1 and SLX1 through the SLX4 complex that also binds TRF2 (54). Perhaps the presence of XPF-ERCC1 influences the stability of the SLX4 complex at telomeres and its interaction with TRF2, and thus, indirectly influences the activity of other endonuclease that may degrade telomeres. Finally, evidence indicates the XPF-ERCC1 may protect against aberrant invasion of the 3’ telomeric overhang into interstitial telomeric sequences (51, 58). However XPF-ERCC1 does not regulate sister chromatid exchange between telomeres, at least in normal cells under non-genotoxic conditions (59). Many questions remain regarding the role of the XPF-ERCC1 interactions with TRF2 in both canonical repair pathways for preserving telomeric DNA, and non-canonical mechanisms of telomere length homeostasis.
CSB and transcription coupled repair
As described in the introduction, NER exists in two sub-pathways including GGR which removes bulky lesions from the entire genome, and TCR which removes lesions from the transcribed strand of actively transcribed regions (reviewed in (25)). Defects in TCR cause the neurological disorder Cockayne Syndrome (25). Whether TCR functions at telomeres is unknown, however the TCR factor CSB has been found to interact with shelterin protein TRF2 (60). CSB is recruited when the RNA polymerase is blocked at a lesion, and then further recruits TCR factor CSA and the subsequent NER machinery (25). The C-rich strand of telomeres is transcribed into long non-coding G-rich RNAs termed TERRA that initiate in the sub-telomere and function in telomere processing and telomere length homeostasis (reviewed in (61)). CPDs present on the transcribed C-rich strand may be repaired by telomeric TCR, and may explain the 1.5 fold faster rate of CPD removal in telomeric DNA, compared to the bulk genome, whereas 6-4 PP repair rates are similar (37). The more helix distorting 6-4 PPs lesions are rapidly removed by GGR, often obscuring TCR events (62). Whether UV photoproducts and failures in telomeric TCR or GGR alter telomeric transcription or TERRA levels is unknown.
Previous work has uncovered interesting roles for CSB protein in telomere maintenance that may be related to canonical roles in TCR, as well as poorly understood non-canonical roles. CSB localizes to a subset of telomeres in human cells and physically binds TRF2 (60), but whether TRF2 influences CSB’s activity or function in TCR is not known. CSB depletion in human cells leads to decreased TERRA, even in the absence of exogenous DNA damage (60). The mechanism remains to be determined, but may relate to failed removal of oxidative cyclopurine adducts at telomeres, since the cells in this study were likely cultured in standard 20% oxygen and were under oxidative stress. This same study found that CSB deficient cells show increased chromatid telomere loss and fragile telomeres, even in the absence of exogenous damage, suggesting that CSB and/or TCR may be required to promote telomere replication (60). Similar results were observed in XPC, XPB, and XPD deficient cells (see above). This result may be explained by CSB mediating TCR removal of oxidative lesions at telomeres (49, 63), and/or by CSB regulating R-loops at telomeres that also block replication. However, direct evidence for TCR of oxidative lesions is lacking. CSB has been implicated in regulating R-loop formation, stability and processing. The RNA-DNA hybrid generated during telomere transcription generates R-loops (64, 65), that may be stabilized at stalled RNA polymerases in the absence of TCR, and may persist at telomeres because the displaced strand can form a stable G-quadruplex structure (66). Even in the absence of exogenous DNA damage, R-loops can be converted into DNA double strand breaks in a process that requires CSB, and the NER endonucleases XPF and XPG (67). This may be problematic at telomeres based on evidence that DSB repair is inhibited at telomeres (68). Furthermore, RNA-DNA hybrids and associated R-loops regulate homologous recombination at telomeres (64). It will be important to determine whether TCR is active at telomeres, and whether this pathway and TCR factors CSA and CSB are required for defending telomeres against bulky DNA lesions and R-loops.
SIMILARITIES BETWEEN DISORDERS CAUSED BY DEFECTS IN TELOMERE MAINTENANCE AND NER
The intersection of telomere biology and NER has important implications for human health. Both UV irradiation and telomere shortening are associated with skin aging and increased skin cancer risk (Figure 4). Genetic mutations that compromise NER or the ability to accurately bypass DNA photoproducts during genome replication leads to a dramatic increase in sunlight-induced skin alterations and cancers in the XP disorder (69). Genetic mutations that compromise telomerase activity and telomere maintenance lead to a spectrum of inherited disorders commonly referred to as telomere syndromes or telomeropathies (1, 70). Individuals with telomere syndromes possess telomere lengths that are much shorter than age matched controls. One of the most common, Dyskeratosis congenita (DKC), is characterized by a triad of symptoms including nail dystrophy, oral mucosal leukoplakia and skin hyperpigmentation (poikiloderma) particularly in sun-exposed areas of the upper chest, neck and face (71, 72). Other symptoms include bone marrow failure, pulmonary fibrosis and liver fibrosis (72). The poikiloderma phenotype is shared with XP, and so is a predisposition to skin cancer (69). Skin squamous cell carcinoma is the second most frequent cancer in DKC, with head and neck squamous cell carcinomas being the most common (73). It is not clear whether DKC patients are more susceptible to UV-induced skin cancers compared to the general population, however, clinical care recommendations include limited sun exposure and use of sun screen (72). While individuals with DKC are clearly not as sensitive to sunlight as XP individuals, the overlap in symptoms suggest that critically short telomeres may exacerbate skin pathologies caused by DNA damage.
Figure 4.

Symptoms of disorders caused by defects in NER and telomere maintenance. Shown are distinct and overlapping symptoms that have been reported in individuals with xeroderma pigmentosum caused by defects in NER, and in dyskeratosis congenita caused by mutations in telomere maintenance genes including telomerase.
Studies in transgenic mice further support the model that critically short telomeres may contribute to skin pathologies that develop in the face of DNA damage. Chronic UVB irradiation of mice lacking NER factor XPC induces skin cancer in exposed mice, that is accelerated in a background of shortened telomeres in Xpc and telomerase double knock out mice (41). In another model, mice engineered to overexpress shelterin protein TRF2 in the skin develop phenotypes that are reminiscent of XP including premature skin atrophy, hyperpigmentation and increased skin cancer in regions that are exposed to light (56). TRF2 overexpression induces telomere shortening, and these mice are also more sensitive to UVB-induced skin cancer (56). Collectively, these studies suggest that telomere shortening and dysfunction may exacerbate skin pathologies and skin cancer triggered by DNA damage. Combined with evidence that UV irradiation can further accelerate telomere shortening (see above), this suggests that UV-induced telomere dysfunction may contribute to UV-induced skin carcinogenesis and skin aging.
CONCLUSIONS
The NER pathway is essential for removing a wide variety of DNA lesions generated by environmental and anti-cancer genotoxicants. Telomeres are susceptible to damage caused by UV light and environmental genotoxic metals, and when these lesions interfere with replication at telomeres they can induce telomere loss and telomere fragility (30, 37, 41, 56, 74). The impact of bulky lesions on telomere transcription and formation of telomeric R-loops at stalled RNA polymerases remains to be determined. Furthermore, oxidative lesions and UV photoproducts can interfere with shelterin TRF1 and TRF2 binding to telomeric DNA (37, 75), suggesting that an accumulation of unrepaired lesions may cause shelterin depletion at telomeres and compromise telomere function. These studies underscore the importance of DNA damage removal at telomeres for telomere preservation. In addition, while shelterin proteins have been found to interact with NER proteins (51, 60), whether shelterin regulates NER at telomeres remains to be determined. The evidence that UV exposed skin exhibits shortened telomeres compared to non-exposed regions (39), together with evidence that critically short telomeres exacerbate skin pathologies due to DNA damage (41), further support the important of NER at telomeres for human health. We are particularly delighted that the significance of Aziz Sancar’s work in NER mechanisms was recognized with the Nobel prize in the same year that we provided the first direct evidence that NER functions at telomeres by using XP-A patient cell lines (37). However, many questions remain unanswered regarding the biological consequences of DNA damage at telomeres, mechanisms of protecting telomeres from insult, and the efficiency of repairing telomeres from damage. A better understanding of how bulky DNA lesions are processed at telomeres will be valuable for advancing therapies and interventions that preserve telomere function to maintain healthy cells and ameliorate environmentally induced diseases associated with genotoxic exposures.
Acknowledgments
We apologize to those investigators whose work was not cited in the interest of preparing a concise review related to current knowledge on NER at telomeres. Research in the Opresko lab is supported by the National Institute of Environmental Health (ES R01ES022944, R21/33ES025606), and the National Institute of General Medical Sciences (R43GM108187).
Abbreviations
- 6-4 PP
pyrimidine (6-4) pyrimidine photoproducts
- CPD
cyclobutane pyrimidine dimer
- A-EJ
alternative end joining
- DDR
DNA damage response
- DSB
DNA double strand break
- GGR
global genome repair
- NER
nucleotide excision repair
- NHEJ
non-homologous end joining
- POT1
protection of telomeres 1
- TCR
transcription coupled repair
- TRF1
telomere repeat binding factor 1
- TRF2
telomere repeat binding factor 2
- TRFH
TRF homology
- UV
ultraviolet light
- XP
xeroderma pigmentosum
Footnotes
This article is part of the Special Issue highlighting Dr. Aziz Sancar's outstanding contributions to various aspects of the repair of DNA photodamage in honor of his recent Nobel Prize in Chemistry.
REFERENCES
- 1.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012 doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willeit P, Willeit J, Mayr A, Weger S, Oberhollenzer F, Brandstatter A, Kronenberg F, Kiechl S. Telomere length and risk of incident cancer and cancer mortality. JAMA. 2010;304:69–75. doi: 10.1001/jama.2010.897. [DOI] [PubMed] [Google Scholar]
- 4.Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13:397–404. doi: 10.1038/nrm3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88:657–666. doi: 10.1016/s0092-8674(00)81908-x. [DOI] [PubMed] [Google Scholar]
- 6.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 7.Doksani Y, Wu JY, de Lange T, Zhuang X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013;155:345–356. doi: 10.1016/j.cell.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 9.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bianchi A, Stansel RM, Fairall L, Griffith JD, Rhodes D, de Lange T. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999;18:5735–5744. doi: 10.1093/emboj/18.20.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 12.O'Connor MS, Safari A, Xin H, Liu D, Songyang Z. A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc Natl Acad Sci U S A. 2006;103:11874–11879. doi: 10.1073/pnas.0605303103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 14.Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11:1223–1229. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
- 15.Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J, Terns MP. TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol Cell Biol. 2010;30:2971–2982. doi: 10.1128/MCB.00240-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li B, Oestreich S, de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101:471–483. doi: 10.1016/s0092-8674(00)80858-2. [DOI] [PubMed] [Google Scholar]
- 17.Fouquerel E, Parikh D, Opresko P. DNA damage processing at telomeres: The ends justify the means. DNA Repair (Amst) 2016 doi: 10.1016/j.dnarep.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia P, Her C, Chai W. DNA excision repair at telomeres. DNA Repair (Amst) 2015;36:137–145. doi: 10.1016/j.dnarep.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt JC, Cech TR. Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015;29:1095–1105. doi: 10.1101/gad.263863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Sfeir AJ, Zou Y, Buseman CM, Chow TT, Shay JW, Wright WE. Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell. 2009;138:463–475. doi: 10.1016/j.cell.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 22.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 23.Doksani Y, de Lange T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb Perspect Biol. 2014;6:a016576. doi: 10.1101/cshperspect.a016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 25.Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst) 2015;36:13–18. doi: 10.1016/j.dnarep.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J, Choi JH, Gaddameedhi S, Kemp MG, Reardon JT, Sancar A. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem. 2013;288:20918–20926. doi: 10.1074/jbc.M113.482257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kemp MG, Gaddameedhi S, Choi JH, Hu J, Sancar A. DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair. J Biol Chem. 2014;289:26574–26583. doi: 10.1074/jbc.M114.597088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 29.Lormand JD, Buncher N, Murphy CT, Kaur P, Lee MY, Burgers P, Wang H, Kunkel TA, Opresko PL. DNA polymerase delta stalls on telomeric lagging strand templates independently from G-quadruplex formation. Nucleic Acids Res. 2013;41:10323–10333. doi: 10.1093/nar/gkt813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pope-Varsalona H, Liu FJ, Guzik L, Opresko PL. Polymerase eta suppresses telomere defects induced by DNA damaging agents. Nucleic Acids Res. 2014;42:13096–13109. doi: 10.1093/nar/gku1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drosopoulos WC, Kosiyatrakul ST, Yan Z, Calderano SG, Schildkraut CL. Human telomeres replicate using chromosome-specific, rather than universal, replication programs. J Cell Biol. 2012;197:253–266. doi: 10.1083/jcb.201112083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douki T, Reynaud-Angelin A, Cadet J, Sage E. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry. 2003;42:9221–9226. doi: 10.1021/bi034593c. [DOI] [PubMed] [Google Scholar]
- 34.Pfeifer GP. Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment. Photochem Photobiol. 1997;65:270–283. doi: 10.1111/j.1751-1097.1997.tb08560.x. [DOI] [PubMed] [Google Scholar]
- 35.Douki T, Cadet J. Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions. Biochemistry. 2001;40:2495–2501. doi: 10.1021/bi0022543. [DOI] [PubMed] [Google Scholar]
- 36.Rochette PJ, Brash DE. Human telomeres are hypersensitive to UV-induced DNA Damage and refractory to repair. PLoS Genet. 2010;6:e1000926. doi: 10.1371/journal.pgen.1000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parikh D, Fouquerel E, Murphy CT, Wang H, Opresko PL. Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat Commun. 2015;6:8214. doi: 10.1038/ncomms9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kruk PA, Rampino NJ, Bohr VA. DNA damage and repair in telomeres: relation to aging. Proc Natl Acad Sci U S A. 1995;92:258–262. doi: 10.1073/pnas.92.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikeda H, Aida J, Hatamochi A, Hamasaki Y, Izumiyama-Shimomura N, Nakamura K, Ishikawa N, Poon SS, Fujiwara M, Tomita K, Hiraishi N, Kuroiwa M, Matsuura M, Sanada Y, Kawano Y, Arai T, Takubo K. Quantitative fluorescence in situ hybridization measurement of telomere length in skin with/without sun exposure or actinic keratosis. Hum Pathol. 2014;45:473–480. doi: 10.1016/j.humpath.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 40.Berardesca E, Bertona M, Altabas K, Altabas V, Emanuele E. Reduced ultraviolet-induced DNA damage and apoptosis in human skin with topical application of a photolyase-containing DNA repair enzyme cream: clues to skin cancer prevention. Mol Med Report. 2012;5:570–574. doi: 10.3892/mmr.2011.673. [DOI] [PubMed] [Google Scholar]
- 41.Stout GJ, Blasco MA. Telomere length and telomerase activity impact the UV sensitivity syndrome xeroderma pigmentosum C. Cancer Res. 2013;73:1844–1854. doi: 10.1158/0008-5472.CAN-12-3125. [DOI] [PubMed] [Google Scholar]
- 42.Gopalakrishnan K, Low GK, Ting AP, Srikanth P, Slijepcevic P, Hande MP. Hydrogen peroxide induced genomic instability in nucleotide excision repair-deficient lymphoblastoid cells. Genome Integr. 2010;1:16. doi: 10.1186/2041-9414-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ting AP, Low GK, Gopalakrishnan K, Hande MP. Telomere attrition and genomic instability in xeroderma pigmentosum type-b deficient fibroblasts under oxidative stress. J Cell Mol Med. 2010;14:403–416. doi: 10.1111/j.1582-4934.2009.00945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D'Errico M, Parlanti E, Teson M, de Jesus BM, Degan P, Calcagnile A, Jaruga P, Bjoras M, Crescenzi M, Pedrini AM, Egly JM, Zambruno G, Stefanini M, Dizdaroglu M, Dogliotti E. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006;25:4305–4315. doi: 10.1038/sj.emboj.7601277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 46.Oikawa S, Tada-Oikawa S, Kawanishi S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry. 2001;40:4763–4768. doi: 10.1021/bi002721g. [DOI] [PubMed] [Google Scholar]
- 47.Belmadoui N, Boussicault F, Guerra M, Ravanat JL, Chatgilialoglu C, Cadet J. Radiation-induced formation of purine 5',8-cyclonucleosides in isolated and cellular DNA: high stereospecificity and modulating effect of oxygen. Org Biomol Chem. 2010;8:3211–3219. doi: 10.1039/c004531d. [DOI] [PubMed] [Google Scholar]
- 48.Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5'-(S)-cyclo-2'-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- 49.Brooks PJ. The 8,5'-cyclopurine-2'-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair (Amst) 2008;7:1168–1179. doi: 10.1016/j.dnarep.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5',8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci U S A. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu XD, Niedernhofer L, Kuster B, Mann M, Hoeijmakers JH, de Lange T. ERCC1/XPF removes the 3' overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol Cell. 2003;12:1489–1498. doi: 10.1016/s1097-2765(03)00478-7. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, Lei M. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008;319:1092–1096. doi: 10.1126/science.1151804. [DOI] [PubMed] [Google Scholar]
- 53.Wan B, Yin J, Horvath K, Sarkar J, Chen Y, Wu J, Wan K, Lu J, Gu P, Yu EY, Lue NF, Chang S, Liu Y, Lei M. SLX4 assembles a telomere maintenance toolkit by bridging multiple endonucleases with telomeres. Cell Rep. 2013;4:861–869. doi: 10.1016/j.celrep.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svendsen JM, Smogorzewska A, Sowa ME, O'Connell BC, Gygi SP, Elledge SJ, Harper JW. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Su Y, Orelli B, Madireddy A, Niedernhofer LJ, Scharer OD. Multiple DNA binding domains mediate the function of the ERCC1-XPF protein in nucleotide excision repair. J Biol Chem. 2012;287:21846–21855. doi: 10.1074/jbc.M111.337899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munoz P, Blanco R, Flores JM, Blasco MA. XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat Genet. 2005;37:1063–1071. doi: 10.1038/ng1633. [DOI] [PubMed] [Google Scholar]
- 57.Wu Y, Zacal NJ, Rainbow AJ, Zhu XD. XPF with mutations in its conserved nuclease domain is defective in DNA repair but functions in TRF2-mediated telomere shortening. DNA Repair (Amst) 2007;6:157–166. doi: 10.1016/j.dnarep.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Vannier JB, Depeiges A, White C, Gallego ME. ERCC1/XPF protects short telomeres from homologous recombination in Arabidopsis thaliana. PLoS Genet. 2009;5:e1000380. doi: 10.1371/journal.pgen.1000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagelstrom RT, Blagoev KB, Niedernhofer LJ, Goodwin EH, Bailey SM. Hyper telomere recombination accelerates replicative senescence and may promote premature aging. Proc Natl Acad Sci U S A. 2010;107:15768–15773. doi: 10.1073/pnas.1006338107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Batenburg NL, Mitchell TR, Leach DM, Rainbow AJ, Zhu XD. Cockayne Syndrome group B protein interacts with TRF2 and regulates telomere length and stability. Nucleic Acids Res. 2012;40:9661–9674. doi: 10.1093/nar/gks745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azzalin CM, Lingner J. Telomere functions grounding on TERRA firma. Trends Cell Biol. 2015;25:29–36. doi: 10.1016/j.tcb.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 62.Vreeswijk MP, van Hoffen A, Westland BE, Vrieling H, van Zeeland AA, Mullenders LH. Analysis of repair of cyclobutane pyrimidine dimers and pyrimidine 6-4 pyrimidone photoproducts in transcriptionally active and inactive genes in Chinese hamster cells. J Biol Chem. 1994;269:31858–31863. [PubMed] [Google Scholar]
- 63.Menoni H, Hoeijmakers JH, Vermeulen W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J Cell Biol. 2012;199:1037–1046. doi: 10.1083/jcb.201205149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun. 2014;5:5220. doi: 10.1038/ncomms6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balk B, Maicher A, Dees M, Klermund J, Luke-Glaser S, Bender K, Luke B. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat Struct Mol Biol. 2013;20:1199–1205. doi: 10.1038/nsmb.2662. [DOI] [PubMed] [Google Scholar]
- 66.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell. 2014;56:777–785. doi: 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, Herbig U, Longhese MP, d'Adda di Fagagna F. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012 doi: 10.1038/ncb2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Opresko PL, Shay JW. Telomere-associated aging disorders. Ageing Res Rev. 2016 doi: 10.1016/j.arr.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 72.Savage SA, Dokal I, Armanios M, Aubert G, Cowen EW, Domingo DL, Giri N, Greene MH, Orchard PJ, Tolar J, Tsilou E, Van Waes C, Wong JM, Young NS, Alter BP. Dyskeratosis congenita: the first NIH clinical research workshop. Pediatr Blood Cancer. 2009;53:520–523. doi: 10.1002/pbc.22061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu FJ, Barchowsky A, Opresko PL. The Werner syndrome protein functions in repair of Cr(VI)-induced replication-associated DNA damage. Toxicol Sci. 2009;110:307–318. doi: 10.1093/toxsci/kfp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Opresko PL, Fan J, Danzy S, Wilson DM, 3rd, Bohr VA. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005;33:1230–1239. doi: 10.1093/nar/gki273. [DOI] [PMC free article] [PubMed] [Google Scholar]