

Abstract
The estrogen receptors (ER) bind with high affinity to many structurally diverse ligands by significantly distorting the contours of their ligand-binding pockets. This raises a question: To what degree is ER able to distinguish between structurally related regioisomers and enantiomers? We have explored the structural compliance and specificity of ERα with a set of ligands having a 7-oxa-bicyclo[2.2.1]hept-5-ene sulfonate core and basic side chains typical of selective ER modulators (SERMs). These ligands have two regioisomers, each of which is a racemate of enantiomers. Using orthogonal protecting groups and chiral HPLC, we isolated all 4 isomers and assigned their absolute stereochemistry by X-ray analysis. The 1S,2R,4S isomer has a 80–170-fold higher affinity for ERα than the others, and it profiles as a partial agonist/antagonist in cellular reporter gene assays and in suppressing proliferation of MCF-7 breast cancer cells with subnanomolar potency, far exceeding that of the other isomers. It is the only isomer found bound to ERα by X-ray analysis after crystallization with 4-isomer mixtures of closely related analogs. Thus, despite the general compliance of this receptor for binding a large variety of ligand structures, ER demonstrates marked structural- and stereo-specificity by selecting a single component from a mixture of structurally related isomers to drive ER-regulated cellular activity. Our findings lay the necessary groundwork for seeking unique ER-mediated pharmacological profiles by rational structural perturbations of two different types of side chains in this unprecedented class of ER ligands, which may prove useful in developing more effective endocrine therapies for breast cancer.
Graphical Abstract

INTRODUCTION
The activation of receptors for hormones and drugs generally displays rather strict dependence on the precise structure of the activating ligand, discriminating sharply among structural and regioisomers, as well as diastereomers and enantiomers. The estrogen receptor (ER), however, provides somewhat of a conundrum: In terms of molecules that can activate it, the ER is capable of binding and responding to a very wide variety of both steroidal and non-steroidal ligands, all with subnanomolar potency, including those having carbocyclic and heterocyclic core systems, and embellished with a variety of side chains. In addition, the large number of crystallographic structures of the ligand binding domain (LBD) of the ERα subtype of this receptor illustrate that the ligand binding pocket (LBP) undergoes a variety of distortions of secondary and tertiary structural elements as this protein interacts with ligands of diverse structure. For example, a ligand with a particularly large substituent that exceeds the capacity of any known LBP shape and appears destined not to bind, actually binds with higher affinity than its smaller congener; it accomplishes this by inducing the full unwinding of a helix to form a new pocket that neatly accommodates the large, “offending” group.1 In another case with the homologous ERβ subtype, a large substituent causes a major disorientation the last 25 residues of the C-terminus of the LBD.2
The terms “plasticity of the ligand binding pocket” or an “eclectic structure-activity relationship” have been used to describe this type of structural compliancy,1, 3–5 but the accommodating behavior of the ER LBD might be even more profound than this. Denaturation titration studies suggest that rather than undergoing distortions to accept structurally diverse ligands, the LBP does not in fact even exist in the absence of ligand, with the LBD being an intrinsically disordered domain prior to ligand binding.4 Such disordered domains are frequently found in signaling proteins, and they use this disorder to make efficient searches for interaction partners, in this case, a ligand, by a process termed “fly casting”.6 The issues of the compliance of the ER LBD in accommodating different ligands and its capacity to exert regio- and stereochemical discrimination arose in the context of our recent work to develop potent antagonists of ER action in breast cancer.
ERα, as a driver of proliferation in many breast cancers, is the target of endocrine therapies using aromatase inhibitors such as letrozole to deprive the cancer of endogenous estrogens, mixed agonist-antagonists such as tamoxifen to inhibit ERα proliferative signaling (typically referred to as selective ER modulators or SERMs), or full antagonists such as fulvestrant to also downregulate ERα levels (typically referred to as selective ER downregulators or SERDs). The favorable initial response of ERα-positive cancers to these endocrine therapies, however, is frequently followed by recurrence of the cancer in a more aggressive form that becomes progressively resistant to subsequent endocrine therapies.7 Hence, there is a need for the development of more efficacious and durable antagonists of ERα activity.
Efforts to improve SERMs, both in terms of their therapeutic efficacy in breast cancer and their on-target effect profiles, have been hampered, however, by widespread use of a uniform structure-based design strategy. In the active conformation of the ERα ligand-binding domain (LBD), helix 12 (h12) docks against h3 and h11 to form one side of the coactivator-binding surface, called activation function-2 (AF-2) (Figure 1a–b), and coactivator binding to AF-2 activates ERα-regulated transcription. To make a SERM, one of a small number of structurally similar basic side chains (BSCs) is appended onto an agonist, generally a planar, often achiral heterocyclic scaffold. The BSC engages in hydrogen bonding with Asp351 on the surface and in doing so directly displaces h12, thus interfering with the recruitment of coactivators to AF-2 and modulating the transcriptional response (Figure 1c), a mechanism appropriately termed “direct antagonism”. By contrast, we discovered certain ligands that can also achieve partial agonist, full antagonist, or SERM-like activity by modulating h12 indirectly through distortion of the C-terminal end of h11 or the h11–h12 loop; we have termed this distinct regulatory mechanism “indirect antagonism”.8
Figure 1.

The ERα LBD adopts ligand-dependent conformations. (a) Crystal structure of the estradiol (E2)-bound ERα LBD (PDB 1GWR). A coactivator peptide (CoA, yellow) is docked at the AF-2 surface. (b) Closer view of the E2-bound LBD showing the positioning of h12 relative to h3 and h11. (c) In the raloxifene (RAL)-bound LBD (PDB 1ERR), the BSC protrudes out of the ligand-binding pocket and displaces h12 from its active conformation. (d) In the OBHS-bound LBD (PBD: 4ZN9), the phenyl sulfonate group dislocates h11 and thus indirectly destabilizes h12.
Recently, we reported on ERα ligands possessing an inherent three-dimensional topology based on a structurally novel bridged oxabicyclic core.9–11 The phenyl sulfonate derivative of these ligands (termed OBHS: 7-oxabicyclo[2.2.1]hept-5-ene sulfonate) profiled as a partial agonist that neither stimulated nor inhibited the proliferation of breast cancer cells, and thus was designated as non-proliferative. Interestingly, OBHS and some related derivatives in the same series were found to show anti-inflammatory effects in MCF-7 12 and endometrial cells 13 in vivo, through inhibition of NFκB-mediated signaling. We also developed a series of sulfoxide-bridged OBHS derivatives (S-OBHS) which profiled with greater agonist activity due to the endo-positioning of the phenyl group.11 A third series with a sulfonamide linker (OBHS-N) enabled attachment of two pendent groups, some of which reached a full antagonist profile comparable to fulvestrant on the wild type receptor.11, 14
Crystal structures of members of the three OBHS series reveal that their antagonism operates by the indirect mechanism, with the pendent group modulating activity by shifting h11 in different directions, altering indirectly the stability of h12 in the agonist conformer (Figure 1d).8 Binding was supported by one phenol mimicking the A-ring of estradiol and forming a tight h-bond network with Glu353 and Arg394, while the other phenol extended between h3 and h11 towards h12. All of these compounds were synthesized and tested as racemates, but recent structures reveal that one isomer was predominantly selected for binding.8, 14 Moving forward, we are interested in whether we could combine the indirect and direct antagonism mechanisms by attaching a BSC to members of the OBHS ligands and whether this might prove efficacious in treatment-resistant breast cancer or provide novel pharmacology for on-target side effects in other tissues.
The installation of a BSC on the OBHS core leads to 4 different stereo and regioisomers (OBHS-BSC: 2 regioisomers due to substitution at either of the two 4-hydroxyphenyl groups and 2 enantiomers corresponding to each of the chiral regioisomers, Figure 2). We chose to designate the two regioisomers “proximal” or “distal”, reflecting whether the BSC was on the same side as—or opposite to—the phenyl sulfonate group. Initially, these OBHS-BSC compounds were tested as this 4 isomer mixture, and some of these mixtures were found to have promising binding affinities and to have good potencies in suppressing ERα transcriptional activity and breast cancer MCF-7 cell proliferation. Given, however, the aforementioned tolerance of the ER LBD to accommodate ligands of different shapes and sizes, we wondered to what extent this receptor is able to discriminate among the four isomers in these mixtures and whether more than one of them might be contributing to the promising activity shown by the mixture.
Figure 2.

The four isomers of OBHS-BSC. One pair of enantiomers has the BSC disposed on the same side of the bicyclic ring as the phenyl sulfonate group (A, Proximal, top) and the other enantiomer pair place the BSC across the bicyclic system from the phenyl sulfonate group (B, Distal, bottom).
In this study, we have probed these stereo- and regiochemical issues in the OBHS-BSC series by developing an efficient means for separating the regioisomers and the enantiomers, and then determining their regiochemistry and their absolute stereochemistry. We then compared the ER binding affinity, and the transcriptional regulating and the antiproliferative activity and potency of each isomer, as well as that of the 4 isomer mixture, in cells. We also obtained new crystal structures of a number of members of the OBHS-BSC series in complexes with ERα. We were pleased to find that essentially all of the ER binding and cellular activities reside in only one of these 4 isomers, which is also the one found in most of the crystal structures. Thus, despite its structural compliancy, the ER is highly discriminating among the various OBHS-BSC regio and stereoisomers in terms of binding and cellular activity. Nevertheless, even within this OBHS-BSC ligand series, members having different BSC substituents induce different orientations of the ligand core within the LBP of ERα that affect the disposition and sites of interaction of the substituents, highlighting again that this receptor combines the attributes of ligand structural compliancy and specificity in an unusual, in fact rather eclectic manner.
RESULTS AND DISCUSSION
Chemical Synthesis and Stereochemistry
The OBHS core is synthesized via a Diels-Alder reaction of 3,4-bis-(4-hydroxyphenyl)furan with phenyl vinylsulfonate.10 Thus, a straightforward route for synthesis of OBHS-BSCs involved the installation of a BSC by alkylation of one of the two phenols on OBHS (exemplified by the 2-(N-piperidino) ethyl group found in the SERM Raloxifene) (Scheme 1). Unfortunately, the two mono-alkylated regioisomers (3 and 4) formed after the alkylation of OBHS using 2-(N-piperidino)ethyl chloride hydrochloride (BSC) (Scheme 1) proved inseparable on normal phase silica gel chromatography due to their high polarity and very similar Rf values. The use of HPLC purification of these highly polar compounds also seemed problematic because the acid additives in mobile phase might lead to epimerization of the pure, desired exo epimers. Thus, one of our goals was to develop an alternative synthetic route that would produce all 4 isomers, but would avoid an HPLC purification step after the installation of the BSC.
Scheme 1.

Synthesis of OBHS-BSC isomer mixture.
We envisioned that the above aim might be achieved if the two phenol moieties of OBHS could be protected with two substantially different non-polar protecting groups, giving a mixture of non-polar regioisomers that would first be separated chromatographically (illustrated in Scheme 2A). After regioisomer separation and partial deprotection under mutually orthogonal conditions, the individual monophenol regioisomers, each of which is a mixture of enantiomers, could then be resolved by chiral HPLC under neutral conditions, prior to adding the basic group, which makes chromatography difficult. Finally, monoalkylation of the one free phenol on each regio/stereoisomer with a BSC and cleavage of the remaining phenol protecting group would give the final four individual OBHS-BSC isomers as pre-separated species, without the need for any further challenging isomer separation. We hypothesized that the size of the orthogonal protecting groups on the phenols might also be crucial and that a sterically bulky protecting group on one of the phenols might help in the chromatographic separation at the regioisomer stage.15
Scheme 2.

A Synthesis and separation of regioisomers of monophenol protected OBHS derivatives. (Insets: ORTEP diagram of compound 10 as a racemate showing the distal arrangement between the benzyl ether and phenyl sulfonate. TLC analysis of the migration of the regioisomeric benzyl TBDPS bis-ethers, 7 and 8 as racemates.) B Separation of enantiomers by chiral HPLC. The HPLC traces of pure enantiomers are also shown (scale in minutes). The assignment of absolute stereochemistry to compounds 9a and 9b are made by analogy to 10a and 10b, respectively (see text for details). (Insets: ORTEP diagram of a pure enantiomer of OBHS regioisomer 10 (10b, with 1S,2R,4S stereochemistry). Preparative HPLC analysis of the elution of the enantiomers of the proximal regioisomers (9a and 9b) and the distal regioisomers (10a and 10b)).
Initially, the two phenols of 3,4-bis-(4-hydroxyphenyl)furan were protected as a benzyl ether and a sterically bulky heptafluorotoluene ether. The 4+2 cycloaddition of this di-protected furan with phenyl vinylsulfonate provided a mixture of regioisomers as predominantly exo-epimers (the exo preference of the readily reversible Diels-Alder reaction in the OBHS series is well precedented9), and the exo epimers of the two regioisomers could be separated from the small amounts of endo epimers by normal phase silica chromatography. The subsequent deprotection16 of the heptafluorotoluene group using sodium methoxide,15 however, led to epimerization of the sulfonate, giving some endo-isomer that was noted by TLC analysis. Therefore, we replaced heptafluorotoluene with tert-butyldiphenyl silyl (TBDPS), a protecting group that retains the sterically bulky feature but could be cleaved under milder conditions.16 This modification allowed effective separation of regioisomers (7 and 8) by standard flash chromatography and also avoided any epimerization during tetraammonium fluoride (TBAF)-promoted deprotection of TBDPS in the next step (Scheme 2A).
At this stage, we were able to obtain a crystal structure of one of the above mentioned monoprotected OBHS derivatives as a racemate, and this isomer was shown to possess the free phenol proximal to the phenyl sulfonate (10) (Scheme 2A). The next challenge was to develop HPLC conditions that could allow us to separate the benzyl-protected enantiomers of each of the regioisomers. After several unsuccessful attempts using normal phase Chiralpak columns, we shifted to the Whelk-O columns having Pirkle-type chiral stationary phases. Using this chromatographic medium, we were able to separate cleanly all four enantiomers using isopropanol/hexane as eluent (Scheme 2B). In addition, we were also able to obtain a crystal structure of one of the pure enantiomers (10b) derived from regioisomer (10) (Scheme 2B).
The absolute configuration of 10b was found to be (1S,2R,4S), and by extension we could assign the absolute configuration of the other enantiomer (10a) in this regioisomeric series (10ab), as well as that of the two OBHS-BSC analogs, 14ab, derived from 10ab, respectively. Notable, in this regioisomeric series the free phenol and ultimately the BSC are disposed proximal to the phenyl sulfonate.
To complete the synthesis, the BSC was installed on the free phenol of each of the enantiomers by alkylation with 2-(N-piperidino)ethyl chloride hydrochloride (Scheme 3). The final step that involved cleavage of benzyl protecting group from the phenol turned out to be more challenging than we anticipated.17 Our first attempts used hydrogenolysis with Pd/C, but this led to both debenzylation and an unexpected, undesired further reduction of the very hindered olefin in the oxabicyclic ring. An alternative debenzylation condition using FeCl318 led to opening of the oxabicyclic ring. However, a TMSI-mediated debenzylation,17 conducted in the presence of thiourea, allowed us avoid any side reactions and obtain the desired free phenol derivative (Scheme 3).
Scheme 3.

Installation of basic side chain on each enantiomer of OBHS regioisomer followed by debenzylation.
Despite our best efforts, we were unable to obtain crystals from any of these OBHS-BSC derivatives for X-ray structure elucidation. Consequently, while the two crystal structures we have obtained enable unambiguous assignment of the absolute configuration of the proximal regioisomeric series of proximal OBHS-BSC derivatives (10ab, 13ab, 14ab), assignment of absolute configurations of the distal series (9ab, 11ab, 12ab) has been made by analogy and comparison of their chromatographic behavior (HPLC).
Biological Results
Relative binding affinity assays
With the four isomers of OBHS-BSC in hand (12ab, 14ab), we examined the stereo- and regiochemical preferences for their binding to ERα and for any resultant modulation of ERα-dependent transcriptional and anti-proliferative activities. Binding affinities were determined by a radiometric competitive binding assay,19, 20 and were expressed as relative binding affinity (RBA) values, where estradiol has an RBA of 100%. To compare the role of the BSC, the RBAs of corresponding benzyl ethers (9–10) were also determined. The results of the above assays are summarized in Table 1.
Table 1.
Relative Binding Affinities (RBA values; estradiol RBA = 100) of OBHS analogs for the ERα and ERβ.a
| Entry | Compd. | ERα | ERβ | α/β ratio |
|---|---|---|---|---|
| 1 |
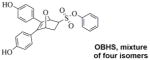
|
9.3 ± 0.6 | 1.7 ± 0.2 | 5.4 |
| 2 |
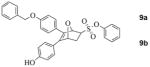
|
0.064 ± 1.5 | 0.096 ± 6.4 | 0.66 |
| 3 | 4.28 ± 0.35 | 0.55 ± 0.45 | 7.78 | |
| 4 |
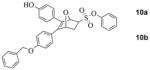
|
0.032 ± 4.0 | 0.114 ± 26 | 0.28 |
| 5 | 0.05 ± 1.9 | 0.08 ± 13 | 0.62 | |
| 6 |
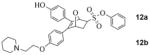
|
0.178 ± 0.04 | 0.0244 ± 0.01 | 7.3 |
| 7 | 0.307 ± 0.05 | 0.058 ± 0.01 | 5.3 | |
| 8 |
|
0.149 ± 0.02 | 0.096 ± 0.03 | 1.55 |
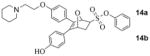
| 9 | 25.8 ± 7.1 | 1.21 ± 0.23 | 21.32 | |
|
|
8.24 ± 0.88 | 0.424 ± 0.04 | 19.4 |
For each of the pairs of structures in entries 2–9, only one enantiomer is shown. Structures of the individual isomers can be obtained by reference to prior figures.
The difference in the RBAs of the parent compound without the BSC, OBHS (entry 1, Table 1), and the four component regio/enantiomeric mixture of OBHS-BSC isomers (entry 10) was not significant. However, one of the pure OBHS-BSC enantiomers (14b, entry 9) binds ERα with an affinity that is 80–170 fold better than any of the 3 other isomers (entries 6–8); this isomer has the BSC disposed proximal to the phenyl sulfonate, as we had predicted (see below), and it is notable that the greatest difference in binding affinity (ca 170-fold) was between 14b and its enantiomer (14a, entry 8), more so than its regioisomer (12b, entry 7). Clearly, ERα, despite the structural compliance of its ligand-binding pocket,1 is exquisitely regio- and stereoselective in its preference for binding only one of the isomeric ligands in this series, the 1S,2R,4S proximal isomer (see Figure 2 for the actual structure).
Likewise, in the monobenzyl ether series, one enantiomer having the benzyl group disposed in a manner proximal to the sulfonate (9b, entry 3) also binds much better than the other 3 isomers (9a, 10ab, entries 2, 4 and 5). Overall, the binding affinity of each member of the monobenzyl series (9ab, 10ab) is ca 6-fold less than that of the corresponding member in the OBHS-BSC series (12ab, 14ab), but the dominance of the preferred isomer (9b, entry 3) over the other three is comparable to that in the BSC series.
We also determined the binding affinities of all of these compounds for ERβ, although this ER subtype does not appear to be a major driver of proliferation in estrogen target tissues or in breast cancer.21 All members of the BSC isomer series have lower binding affinity for ERβ than for ERα, with again the isomer 14b (entry 9) being the highest binder. Compared to the other three isomers (14a and 12ab, entries 6–8), it has less preferential binding; this results in isomer 14b having a ca. 20-fold binding preference for ERα vs. ERβ. In the benzyl-substituted isomer series (9ab, 10ab, entries 2–5), isomer 9b (entry 3) again has the highest affinity but it has a lower selectivity in binding to ERβ than to ERα. Except for 9b, all of the benzyl-substituted isomers show preferential affinity for ERβ.
It is particularly encouraging that the affinities of the mixtures are almost exactly what one would predict for a four-component system in which each component is making its contribution in proportion to its composition in the mixture. For example, the predicted affinity of the four equal component mixture of OBHS-BSC isomers is 6.61 ± 1.82 (the sum of the RBA values for entries 6–9 divided by 4) is not significantly different from the affinity measured directly for this 4-component mixture, 8.24 ± 0.88 (12ab and 14ab, entry 10).
Gene transcription and proliferation assays
The pure OBHS-BSC isomers as well as the 4-isomer mixture were tested for ERα-mediated transcriptional activity using a 3xERE-luciferase reporter gene co-transfected with an expression vector for ERα in human HepG2 liver cells, a cell line that is well suited for assaying the potency and efficacy of ER agonists and antagonists (See Figure 3a–b and summary in tabular data).11, 22 In agonist mode assays, cells were treated with increasing concentrations of estradiol or ligands, from which we were able to obtain measures of potency (as EC50 values) as well as efficacy values relative to estradiol (Eff; % of E2). The antagonist mode assays, cells were treated with an increasing concentration of ligands in the presence of 10 nM estradiol, and we obtained IC50 values and final efficacy values at the highest concentrations, again as Eff, % of E2; low numbers indicate more complete antagonism. In addition to comparing the activities of the four OBHS-BSC isomers among themselves, they were also compared with OBHS, hydroxytamoxifen (SERM) and fulvestrant (full antagonist).
Figure 3.

Transcriptional and anti-proliferative profiles of OBHS-BSC stereo-and regioisomers. a) Luciferase activity was measured in HepG2 cells transfected with 3X-ERE-driven luciferase reporter and expression vectors encoding ERα and treated in triplicate with increasing doses (up to 10−5 M) of the isomers and control compounds (E2, OBHS, fulvestrant and hydroxytamoxifen). Data shown as a percent activity relative to 10−5 M E2. b) Luciferase activity was measured in HepG2 cells transfected with 3X-ERE-driven luciferase reporter and expression vectors encoding ERα, and treated in triplicate with increasing doses (up to 10−5 M) of the isomers in the presence of 10 nM E2 and control compounds (OBHS, fulvestrant and hydroxytamoxifen). Data is shown as a percent activity relative to 10−5 M E2. c) Proliferation was measured in MCF-7 cells treated in triplicate with increasing doses (up to 10−5 M) of the isomers and control compounds (E2, OBHS, fulvestrant and hydroxytamoxifen). Data is shown as a percent activity relative to vehicle. In the tabular summary, aAverage efficacy (mean ± s.e.m.) and EC50 values were determined for experiments shown in Figure 3a. bAverage efficacy (mean ± s.e.m.) and IC50 values were determined for experiments shown in Figure 3b. cAverage efficacy (mean ± s.e.m.) and IC50 values were determined for experiments shown in Figure 3c. EC50 and IC50 values for some compounds could not be determined (−).
In agonist mode, OBHS-BSC 14b had the greatest potency, with an EC50 value of ~0.02 nM; the other three isomers activated ERα only at ca. 1,000 fold higher concentration (20–150 nM). Notably, among these 4 OBHS-BSC isomers, the potency dominance of 14b over the other three in regulating transcription parallels but exceeds that of their binding affinities. In the agonist mode, efficacy values ranged between 24 and 36% that of estradiol, indicating that they are partial agonists, but they are less agonistic than OBHS, which has an efficacy value of 64%. The efficacy of the highest affinity isomer closely matched with the efficacy of hydroxytamoxifen. Notable as well was the fact that the 4-isomer mixture had an EC50 value 8-fold higher than that of the best isomer (14b), which is within the range of the 4-fold higher value expected for the 4 component mixture.
When tested in the antagonist mode, the efficacies of these compounds matched to a considerable degree the efficacy values in the agonist mode, indicating that we are seeing close to the full activity of all 4 isomers and that they are all partial agonist/antagonists. Compound 14b and the four isomer mixture were found to have the lowest efficacy, ~33%, which is almost half that of OBHS, indicating that they are better antagonists than OBHS and have an antagonist efficacy similar to that of hydroxytamoxifen. Similar to the agonist mode assays, 14b had the greatest potency, with an IC50 value of ~6 nM, far greater than that of the low binding isomers, and the four isomer mix had an IC50 value 2-fold higher than that of 14b.
We also tested these pure OBHS-BSC isomers for their ability to inhibit ERα-mediated proliferation in MCF-7 cells, an estrogen-dependent breast cancer cell line. MCF-7 cells grown in normal serum were switched to charcoal-stripped serum and treated immediately with increasing concentrations of the ligands, allowing us to profile both the agonists and antagonists in the same assay (Figure 3c). In this assay all four of the OBHS-BSC isomers profiled as antagonists, with efficacies ranging from ~45% to ~ 13%, while OBHS actually has some low agonist activity. Notably, the efficacies of 14b and the 4 isomer mixture closely matched that of the SERM hydroxytamoxifen and the full antagonist fulvestrant (Figure 3 summary table), the other three compounds being less efficacious. The IC50 values from these assays paralleled that of the transcription assays, in which the highest affinity isomer 14b had the highest potency (IC50 = 0.3 nM), and the other three isomers had at least 200-fold lower potency. As expected, the 4-isomer mixture had a ~4 fold higher IC50 value than that of the high affinity isomer 14b. Hence, from these studies we were able to conclude that all of the cell-activity is driven by the highest affinity isomer 14b, which, due to the incorporation of BSC, behaves as a better antagonist than the parent OBHS both in terms of potency and efficacy.
Crystallography of ERα-OBHS Complexes
Based on the crystal structure of OBHS analogs (Figure 1), we had expected that the regioisomer having BSC proximal to the phenyl sulfonate group (A, Figure 2) might be the one best able to project the BSC towards helix-12, effectively forcing its transition from the agonist conformation to the SERM antagonist conformation, while at the same time ensuring that the free phenolic OH would be positioned properly for its important interactions with Glu353 and Arg394. It was not certain, however, that this would actually be the case; neither could one predict in advance which enantiomer of this proximal regioisomer (A) would be preferred for binding to the ER nor whether any of the four isomers would show enhanced anti-proliferative activity.
To determine the receptor-binding orientations of OBHS-BSC stereo/regio-isomers, we solved x-ray crystal structures of the ERα LBD in complexes with various OBHS-BSC analogs. To this end, we crystallized the LBD with a mixture containing all four OBHS-BSC stereo/regio-isomers, including complexes with related compounds having halide substitutions 23 on the sulfonate ester phenyl group. From these attempts, we were able to obtain the best structures of the OBHS analogs with p-bromophenyl sulfonate esters, each showing clear electron density for the isomer with regio- and stereochemistry corresponding to 14b. In the unsubstituted compound (Figure 4a), the phenyl ring attached to the oxabicyclic core of the ligand engaged in hydrogen bonding with Glu353, which drives high-affinity binding to the receptor. Like raloxifene, the OBHS-BSC ligand (Figure 4b) formed a hydrogen bond with Asp351 via its raloxifene-derived BSC, which protruded out of the pocket towards helix 12. The electron density of the 4-bromophenyl moiety was poor, however, suggesting that it adopted multiple conformations. The structure of an OBHS derivative with the same p-bromophenyl substitution and a tamoxifen-derived BSC (Figure 4c) showed unambiguous density for the isomer with stereochemistry corresponding with 14b, the high affinity isomer, although only two of the four monomers in its asymmetric unit had adequate electron density to resolve the 4-bromophenyl moiety.
Figure 4.

The SERM-derived BSC alters the OBHS-binding orientation. (a–c) Crystal structures of ERα LBDs showing the binding orientations of (a) an OBHS analog (PDB 4ZNW), and OBHS-BSC analogs with either (b) a raloxifene-derived side chain (PDB 5TN9), or (c) a tamoxifen-derived side chain (PDB 5TNB). The chemical structure of the bound ligand is shown above each panel. The 2Fo-Fc electron density and Fo-Fc difference maps of OBHS-BSC analogs were contoured at 1.0 σ and 3.0 σ, respectively. (d) The BSC-mediated h-bond with Asp351 rotates the oxabicyclic ligand core (curved red arrow). The OBHS- and OBHS-BSC-bound LBDs in panels a and b were superposed. (e–f). BSC-induced rotation of the ligand core repositions h11. The OBHS- and OBHS-BSC-bound LBDs in panels a–c were superposed on the E2-bound LBD (PDB 3UUD).
Curiously, instead of abutting against h11 as seen in the analog without a BSC (Figure 4a),8 the 4-bromophenyl group in these subunits was redirected into the ligand-binding pocket towards h8 (Figure 4c), similar to the binding orientation of the 17α-trifluoromethylphenylvinyl-substituted E2 structure.1 In both OBHS-BSC-bound structures, redirection of the p-bromophenyl group was driven by the BSC, which induced a shift in the ligand core to accommodate h-bond formation with Asp351 (Figure 4d). Also, this BSC-induced redirection of the 4-bromophenyl group led to the repositioning of h11 in both cases (Figure 4e–f). Thus, the combination of 7 structures of OBHS analogs, 3 of S-OBHS, and 8 of OBHS-N, identifies isomer preferences for binding,8 while our new structures with a BSC reveal preferential selection of binding with the additional BSC substituent on the phenols directed towards h12 in the parent compounds.
CONCLUSION
This work highlights the high regio- and stereospecificity of ERα in selecting a single compound from among a four-isomer mixture for binding and activation, despite the known ability of this receptor to markedly alter the shape of its ligand binding pocket to accommodate ligands of different size and shape, a behavior that might properly be termed eclectic, in the sense of being more particular than understandable.1, 3 Notably, the finding that only one OBHS-BSC isomer fully accounts for the binding and cellular activity of the 4-component mixture, suggests that initial screens of compounds in this series can be performed for convenience on 4-isomer mixtures, with the expectation that binding affinities and cellular potencies measured for the mixture will most likely be ca. one-quarter that of the highest activity isomer. Our structural studies on members of the OBHS-BSC ligand class also highlight the challenges of trying to optimize two distinct pendent groups targeting indirect and direct antagonism. While the A-ring mimetic phenol in OBHS is uniformly fixed by h-bonding, the rest of the three-dimensional core displays the ability to rotate as the external BSC and internal pendent group compete for binding interactions. Finally, the structure-affinity/activity profiles we have developed in this work lay the foundation for a structure-based strategy to increase the potency as well as optimize the anti-proliferative and anti-inflammatory properties of ERα ligands, aspects that will be the subject of future studies.
METHODS
Compound synthesis, separations, and characterizations, X-ray crystallographic and biological assay data are given as Supporting Information.
Supplementary Material
Acknowledgments
Support of this research through a grant from the Breast Cancer Research Foundation (BCRF 082854 to J.A.K.) and the National Institutes of Health (PHS 5R01DK015556 to J.A.K. and 5R01DK077085 to K.W.N.) is gratefully acknowledged. J.C.N. is supported by the Ballen Isles Men’s Golf Association. S.S. is supported by the Frenchman’s Creek Women for Cancer Research. X-ray diffraction data was collected at the Stanford Synchrotron Radiation Lightsource (SSRL) (beamline 11-1).
FUNDING SOURCES
Breast Cancer Research Foundation: BCRF 082854
National Institutes of Health: 5R01DK015556, 5R01DK077085
Ballen Isles Men’s Golf Association – no grant number
Frenchman’s Creek Women for Cancer Research – no grant number
Footnotes
Accession Codes. Coordinates for complexes of ERα with OBHS-BSCRAL (PDB ID: 5TN9) and with OBHS-BSCTAM (PDB ID: 5TNB) have been deposited in the Protein Data Bank (HPUB). Other previously deposited structures, identified in the legends for Figures 1 and 4, have PDB IDs: 1GWR, 1ERR, 3UUD, 4ZN9, 4ZNW.
The Supporting Information section contains procedural information of compound synthesis, separations, and characterizations, and X-ray crystallographic studies and biological assays.
Supporting Information Available: This material is available free of charge via the Internet.
References
- 1.Nettles KW, Bruning JB, Gil G, O’Neill EE, Nowak J, Guo Y, Kim Y, DeSombre ER, Dilis R, Hanson RN, Joachimiak A, Greene GL. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–568. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure (London, England: 1993) 2001;9:145–153. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 3.Katzenellenbogen JA. The 2010 Philip S. Portoghese Medicinal Chemistry Lectureship: addressing the “core issue” in the design of estrogen receptor ligands. J Med Chem. 2011;54:5271–5282. doi: 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gee AC, Katzenellenbogen JA. Probing conformational changes in the estrogen receptor: evidence for a partially unfolded intermediate facilitating ligand binding and release. Mol Endocrinol. 2001;15:421–428. doi: 10.1210/mend.15.3.0602. [DOI] [PubMed] [Google Scholar]
- 5.Katzenellenbogen JA, Muthyala R, Katzenellenbogen BS. The nature of the ligand-binding pocket of estrogen receptor alpha and beta: the search for subtype-selective ligands and implications for the prediction of estrogenic activity. Pure and Applied Chemistry. 2003;75:2397–2403. [Google Scholar]
- 6.Huang Y, Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J Mol Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Forbes JF, Cuzick J, Buzdar A, Howell A, Tobias JS, Baum M. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet OncolLancet Infect Dis. 2008;9:45–53. doi: 10.1016/S1470-2045(07)70385-6. [DOI] [PubMed] [Google Scholar]
- 8.Nwachukwu JC, Srinivasan S, Zheng Y, Wang S, Min J, Dong C, Liao Z, Nowak J, Wright NJ, Houtman R, Carlson KE, Josan JS, Elemento O, Katzenellenbogen JA, Zhou HB, Nettles KW. Predictive features of ligand-specific signaling through the estrogen receptor. Mol Syst Biol. 2016;12:864. doi: 10.15252/msb.20156701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou HB, Comninos JS, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. Synthesis and Evaluation of Estrogen Receptor Ligands with Bridged Oxabicyclic Cores Containing a Diarylethylene Motif: Estrogen Antagonists of Unusual Structure. J Am Chem Soc. 2005;48:7261–7274. doi: 10.1021/jm0506773. [DOI] [PubMed] [Google Scholar]
- 10.Zheng Y, Zhu M, Srinivasan S, Nwachukwu JC, Cavett V, Min J, Carlson KE, Wang P, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Development of Selective Estrogen Receptor Modulator (SERM)-Like Activity Through an Indirect Mechanism of Estrogen Receptor Antagonism: Defining the Binding Mode of 7-Oxabicyclo[2.2.1]hept-5-ene Scaffold Core Ligands. Chemmedchem. 2012;7:1094–1100. doi: 10.1002/cmdc.201200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang P, Min J, Nwachukwu JC, Cavett V, Carlson KE, Guo P, Zhu M, Zheng Y, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Identification and structure-activity relationships of a novel series of estrogen receptor ligands based on 7-thiabicyclo[2.2.1]hept-2-ene-7-oxide. J Med Chem. 2012;55:2324–2341. doi: 10.1021/jm201556r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, Kulp K, Hochberg RB, Zhou H, Katzenellenbogen JA, Katzenellenbogen BS, Kim Y, Joachimiak A, Greene GL. NF[kappa]B selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. 2008;4:241–247. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Gong P, Chen Y, Nwachukwu JC, Srinivasan S, Ko C, Bagchi MK, Taylor RN, Korach KS, Nettles KW, Katzenellenbogen JA, Katzenellenbogen BS. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Science Trans Med. 2015;7:271ra279. doi: 10.1126/scitranslmed.3010626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srinivasan S, Nwachukwu JC, Bruno NE, Dharmarajan V, Goswami D, Kastrati I, Novick S, Nowak J, Cavett V, Zhou HB, Boonmeun N, Zhao Y, Min J, Frasor J, Katzenellenbogen BS, Griffin PR, Katzenellenbogen JA, Nettles KW. Full antagonism of the estrogen receptor without a prototypical ligand side chain. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2236. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Detsi A, Koufaki M, Calogeropoulou T. Synthesis of (Z)-4-Hydroxytamoxifen and (Z)-2-[4-[1-(p-Hydroxyphenyl)-2-phenyl]-1butenyl]phenoxyacetic Acid. J Org Chem. 2002;67:4608–4611. doi: 10.1021/jo0255328. [DOI] [PubMed] [Google Scholar]
- 16.Wuts PGM, Greene TW. Greene’s Protective Groups in Organic Synthesis. 4. Wiley-Interscience; New Jersey: 2006. [Google Scholar]
- 17.Song ZJ, King AO, Waters MS, Lang F, Zewge D, Bio M, Leazer JL, Javadi G, Kassim A, Tschaen DM, Reamer RA, Rosner T, Chilenski JR, Mathre DJ, Volante RP, Tillyer R. An efficient asymmetric synthesis of an estrogen receptor modulator by sulfoxide-directed borane reduction. Proc Natl Acad Sci USA. 2004;101:5776–5781. doi: 10.1073/pnas.0307415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodebaugh R, Debenham JS, Fraser-Reid B. Debenzylation of complex oligosaccharides using ferric chloride. Tetrahedron Letters. 1996;37:5477–5478. [Google Scholar]
- 19.Katzenellenbogen JA, Johnson HJ, Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 20.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered Ligand Binding Properties and Enhanced Stability of a Constitutively Active Estrogen Receptor: Evidence That an Open Pocket Conformation Is Required for Ligand Interaction. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 21.Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol. 2003;17:203–208. doi: 10.1210/me.2002-0206. [DOI] [PubMed] [Google Scholar]
- 22.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 23.Srinivasan S, Nwachukwu JC, Parent AA, Cavett V, Nowak J, Hughes TS, Kojetin DJ, Katzenellenbogen JA, Nettles KW. Ligand-binding dynamics rewire cellular signaling via estrogen receptor-α. Nat Chem Biol. 2013;9:326–332. doi: 10.1038/nchembio.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.