

Abstract
Lantibiotics are ribosomally synthesized and post-translationally modified antimicrobial peptides containing thioether rings. In addition to these crosslinks, the clinical candidate lantibiotic NAI-107 also possesses a C-terminal S-[(Z)-2-aminovinyl]-D-cysteine (AviCys) and a unique 5-chloro-L-tryptophan (ClTrp) moiety linked to its potent bioactivity. Bioinformatic and genetic analyses on the NAI-107 biosynthetic gene cluster identified mibH and mibD encoding flavoenzymes responsible for the formation of ClTrp and AviCys, respectively. The biochemical basis for the installation of these modifications on NAI-107 and the substrate specificity of either enzyme is currently unknown. Using a combination of mass spectrometry, liquid chromatography, and bioinformatic analyses we demonstrate that MibD is an FAD-dependent Cys decarboxylase and that MibH is an FADH2-dependent Trp halogenase. Most FADH2-dependent Trp halogenases halogenate free Trp, but MibH was only active when Trp was embedded within its cognate peptide substrate deschloro NAI-107. Structural comparison of the 1.85-Å resolution crystal structure of MibH with other flavin-dependent Trp halogenases revealed that subtle amino acid differences within the MibH substrate binding site generates a solvent exposed crevice presumably involved in determining its unusual substrate specificity.
INTRODUCTION
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a growing class of natural products containing a wide array of unique pharmacophores.1 These moieties are introduced on a ribosomally synthesized precursor peptide by dedicated biosynthetic enzymes,2 and bestow upon these molecules their biological activities. As such, characterization of RiPP biosynthetic enzymes provides an avenue to harness the potential of these catalysts as tools for the development of chemoenzymatic methodologies to generate novel therapeutic peptides.
Within RiPPs, lanthipeptides (lanthionine-containing peptides) encompass one of the most abundant and well-studied subfamilies to date.1 The key structural features defining lanthipeptides are thioether rings formed by the bis-amino acids lanthionine (Lan) and/or methyllanthionine (MeLan) (Figure 1a). These crosslinks are introduced enzymatically via dehydration of select Ser/Thr residues to 2,3-dehydroalanine (Dha) and (Z)-2,3-dehydrobutyrine (Dhb), respectively, within the C-terminal core region of a precursor peptide (LanA), followed by subsequent Michael-type additions of Cys thiols onto the newly generated dehydroamino acids (Figure 1a).3, 4 The enzymes responsible for catalyzing both modifications require an N-terminal leader peptide sequence within the precursor peptide for efficient processing.5–7 After thioether ring formation, one or more proteases remove the N-terminal leader sequence from the precursor peptide releasing the mature natural product. Lanthipeptides exhibiting antimicrobial activity are known as lantibiotics (lanthionine-containing antibiotics).
Figure 1.

Overview of NAI-107 biosynthesis. (a) Scheme for thioether ring formation during lanthipeptide biosynthesis. The enzymes that catalyze each step during NAI-107 biosynthesis are shown in parentheses. (Xn) connecting peptide (b) Structure of NAI-107. The amino acid sequence of the precursor peptide MibA is shown. Negative numbers represent the position of amino acids within the leader peptide (underlined) with respect to the first amino acid in the core region. Dehydroamino acids, lanthionine, and methyllanthionine are shown in purple, red, and blue, respectively. The aminovinyl cysteine and 5-chlorotryptophan are colored green and orange. NAI-107 also contains a 3,4-dihydroxyproline moiety shown in cyan. (c) NAI-107 biosynthetic gene cluster. (d,e) Proposed reactions catalyzed by the enzymes (d) MibD and (e) MibH/MibS. Dhx, dehydroalanine or dehydrobutyrine; AviMeCys, S-[(Z)-2-aminovinyl]-(3S)-3-methyl-d-cysteine.
In addition to these common post-translational modifications, some lanthipeptides contain additional modified residues installed by tailoring enzymes. These modifications in many cases increase the stability and biological activity of the peptides. For instance, epilancin 15X contains an N-terminal lactate that serves to protect it against the action of aminopeptidases.8 Similarly, the lantibiotic NAI-107 (also known as microbisporicin) contains a C-terminal AviCys predicted to arise from the oxidative decarboxylation of a Cys residue within the NAI-107 precursor peptide (Figure 1b).9 Presumably, this modification protects the peptide from carboxypeptidases and removes a negative charge, which has proven beneficial for bioactivity in semisynthetic lanthipeptide analogs.10 In addition, NAI-107 contains a 3,4-dihydroxyproline (diHPro) and a unique ClTrp responsible for increasing its bioactivity two-fold compared to its deschloro counterpart (Figure 1b).9, 11 The benefits these additional modifications confer to the stability and bioactivity of lantibiotics prompted biochemical characterization of the tailoring enzymes responsible for installing AviCys and ClTrp during NAI-107 biosynthesis.
NAI-107 is a produced by the actinomycete Microbispora sp. 107891.9, 12 NAI-107 acts as a peptidoglycan biosynthesis inhibitor by sequestering the cell wall precursor lipid II and is currently in preclinical trials for the treatment of multidrug resistant Gram-positive bacterial infections.13–15 NAI-107 biosynthesis commences with the translation of the precursor peptide MibA (Figure 1b).12, 16 Then, MibB dehydrates seven Ser/Thr residues within the core region of MibA in a glutamyl-tRNAGlu-dependent manner, and, the lanthipeptide cyclase MibC catalyzes the nucleophilic addition of Cys residues onto five dehydroamino acids.17 The biosynthetic details regarding AviCys, ClTrp, and diHPro installation during NAI-107 biosynthesis are not known.
Previous bioinformatic analyses of the NAI-107 biosynthetic gene cluster identified two genes coding for enzymes predicted to be responsible for the decarboxylation and halogenation (Figure 1c).12 One of the open reading frames (ORFs), mibD, encodes an enzyme that has sequence similarity to lanthipeptide decarboxylases (LanD).18, 19 These enzymes together with (R)-4’-phospho-N-pantothenoylcysteine (PPC) decarboxylases belong to the homo-oligomeric flavin-containing Cys decarboxylase (HFCD) superfamily that employs a flavin cofactor to catalyze the oxidative decarboxylation of C-terminal Cys-containing peptides (Figure 1d).18–20 Decarboxylation proceeds via oxidation of a Cys thiol to a thioaldehyde that subsequently serves to delocalize the negative charge developed at the former Cα upon decarboxylation.21–25 Nucleophilic addition of the C-terminal aminoenethiolate to a nearby dehydroamino acid within the core peptide yields the AviCys moiety (Figure 1d).25 At present it is unclear whether this latter reaction is catalyzed by lanthipeptide decarboxylases, lanthipeptide cyclases or if it proceeds non-enzymatically.
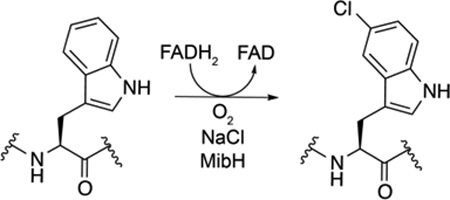
A second ORF, mibH, shows homology to flavin-dependent Trp halogenases.12 Members of this enzyme class (Figure 1e) use reduced flavin to activate molecular oxygen.26 A halide ion is believed to perform a nucleophilic attack onto the distal oxygen atom of a 4a–flavin hydroperoxide forming a hypohalite that subsequently transfers the halogen to a nearby conserved Lys residue (Lys102 in MibH). This chloramine intermediate is then believed to halogenate Trp.27–29 Within the NAI-107 biosynthetic gene cluster, mibS encodes a protein that shows sequence similarity to flavin reductases and is presumably responsible for providing reduced flavin to MibH (Figure 1e).12
In this study we reconstituted the activity of MibD, MibS, and MibH in vitro. Decarboxylation assays revealed MibD to be specific for the C-terminal sequence in the precursor peptide MibA and to not require the N-terminal leader peptide for activity. MibH and MibS form an FAD-dependent halogenase system. Interestingly, halogenation assays with a panel of different substrate analogs revealed MibH to be highly specific for the modified MibA core peptide carrying thioether rings suggesting halogenation to occur in one of the last steps of NAI-107 biosynthesis. Lastly, the 1.85-Å resolution crystal structure of holo MibH provides a structural basis for its unusual substrate specificity.
RESULTS AND DISCUSSION
In vitro reconstitution of MibD as a lanthipeptide Cys decarboxylase
The gene encoding MibD was heterologously expressed in Escherichia coli as a hexahistidine (His6) tagged protein and purified via immobilized metal affinity chromatography. To identify the type of flavin, the purified protein was heat-denatured, the flavin was isolated via reversed phase solid phase extraction, and analyzed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) revealing a mass of 782.81 Da, consistent with FAD (Supplementary Figure S1). The isolation of FAD from His6-MibD suggests the cofactor is not covalently linked.
To reconstitute MibD activity in vitro, enzyme assays were performed with the linear precursor peptide His6-MibA as substrate, resulting in the appearance of a lower molecular weight product as observed by MALDI-TOF MS (Figure 2a). To determine if the MibA leader peptide was required for MibD activity, enzyme assays were performed using the MibA core peptide as substrate resulting again in the formation of a new product displaying a loss of 47 Da (Figure 2b). Tandem MS/MS analysis of the product localized the modification to the C-terminus of the core peptide (Figure 2c) in agreement with the formation of an aminoenethiolate moiety from a C-terminal Cys residue.18, 19, 25, 30 Thus, in contrast to lanthipeptide dehydratases and cyclases, MibD activity is not dependent on a leader peptide. The absence of starting material following the activity assays (Figure 2a–b) suggests that MibD activity is not coupled to an oxidoreductase responsible for oxidizing FADH2. Instead, the results imply that the reduced flavin generated upon oxidative decarboxylation must have been reoxidized by O2. Finally, the observation that MibD is capable of utilizing the precursor peptide MibA lacking dehydrations and thioether rings as substrate suggests that these post-translational modifications are not important for mediating substrate recognition by MibD and that, in vivo, MibD may act first. The observation that MibD efficiently decarboxylates linear MibA is similar to reports on EpiD and MrsD involved in oxidative decarboxylation during the biosynthesis of epidermin and mersacidin, respectively.19, 31, 32 To unambiguously test whether the LanD proteins act first, kinetic comparisons would be needed between linear peptide and intermediates with various post-translational modifications installed, which are currently inaccessible. Some indication that LanD proteins may act on partially modified peptides in vivo comes from a study on MutD, the LanD enzyme involved in mutacin 1140 biosynthesis. This enzyme was reported to be less efficient in vivo when the B-ring was disrupted.33
Figure 2.

In vitro reconstitution of His6-MibD activity. (a–b) MALDI-TOF MS analysis of oxidative decarboxylation reactions after incubation with His6-MibD and (a) His6-MibA, or (b) MibA core peptide. Top mass spectra represent a control reaction performed in the absence of His6-MibD. Bottom mass spectra represent a reaction containing His6-MibD. SM, starting material; P, product. (c) ESI-Q/TOF tandem MS fragmentation analysis of MibA core peptide after modification by His6-MibD. Cys modified by MibD is shown in red. The y ions carrying a difference of 46 Da suggests modification occurs at the C-terminus. For (a) SM, average mass calc. [M+H]+ 8013, obs. 8022; P, average mass calc. [M+H]+ 7967, obs. 7971. For (b) SM, average mass calc. [M+H]+ 2355, obs. 2356; P, average mass calc. [M+H]+ 2309, obs. 2309. For (c) precursor parent ion monoisotopic mass calc. [M+H]+ 2307.90, obs. 2307.93.
Insights into the substrate specificity of lanthipeptide decarboxylases
The successful in vitro reconstitution of MibD activity prompted us to investigate its potential utility for decarboxylation of non-cognate lanthipeptide precursor peptides containing C-terminal Cys residues (Supplementary Table S1).34 Under a range of experimental conditions, His6-MibD was only active on its cognate substrate His6-MibA and did not act on other lanthipeptide precursor peptides ending in FTINVC (ProcA1.1), LVGKMC (ProcA1.7), and YWEGEC (ProcA2.8; Supplementary Table S1).
To obtain further insights into the substrate specificity, a sequence similarity network of the entire flavoprotein superfamily to which MibD belongs (PFAM02441) was built at an expected (E)-value cutoff of 10−31.35 Within the HFCD superfamily cluster (Figure 3a, green) lanthipeptide decarboxylases formed a separate subgroup (Figure 3a, light green) from PPC-decarboxylases, which are involved in the decarboxylation of PPC during coenzyme A biosynthesis.20 Interestingly, in addition to lanthipeptide decarboxylases, this subcluster also contained enzymes involved in the biosynthesis of the cysteine-derived sulfur-containing antibiotics holomycin and tropodithietic acid (Figure 3a, red). For both cases, these HFCD enzymes are predicted to catalyze key biosynthetic oxidative sulfur transformations.36, 37
Figure 3.

Substrate sequences of lanthipeptide decarboxylases. (a) Sequence similarity network of PFAM 02441 at a 60% sequence identity for each node. The HFCD superfamily cluster is shown in green. The LanD subcluster is colored light green. HFCD enzymes involved in the biosynthesis of holomycin and tropodithietic are shown in red. Other subfamilies are colored light blue. (b) Sequence alignment of select lanthipeptide precursor peptides from gene clusters containing lanthipeptide decarboxylases. Dehydratable residues are shown in blue and Cys residues are shown in red. Putative ring topologies are drawn on top of each group based on the topology of characterized lanthipeptides.
Close inspection of the precursor peptides associated with each lanthipeptide decarboxylase revealed that these LanAs can be classified into three different groups based on their putative ring topologies (Figure 3b). Irrespective of the group, with the exception of mersacidin, each substrate peptide has a similar C-terminal ring topology with the consensus sequence S-(F/W/N)-(N/C)-S-(F/Y/W/N)-C-C (Figure 3b). The conservation within this sequence suggests that lanthipeptide decarboxylases have evolved to recognize this motif and not only the C-terminal Cys. The non-cognate substrates investigated in this study that failed to be decarboxylated do not contain this motif (Supplementary Table S1), and the observed lack of activity with MibD supports its importance. Two additional lanthipeptide decarboxylases have been structurally and biochemically characterized in vitro, EpiD and MrsD.18, 19, 23, 24, 30, 38 Similar to MibD, MrsD did not demonstrate appreciable substrate tolerance.19 In contrast, EpiD was capable of decarboxylating a variety of substrates containing the C-terminal sequence (V/I/L/M/F/Y/W)-(A/S/V/T/C/I/L)-C.32 Hence, EpiD (or its homolog GdmD)39 appears to be a better prospect for synthetic biology applications.
MibS is an NADH-dependent flavin reductase
To probe whether MibS is responsible for supplying the FADH2 required for NAI-107 halogenation, mibS was heterologously expressed as a His6-tagged construct in E. coli and reconstituted in vitro. Initial expression attempts of the 20.9-kDa reductase resulted in insoluble protein. Co-expression of mibS together with the E. coli chaperones groES/EL afforded soluble protein.40 Upon purification, the His6-MibS UV/Vis absorption spectrum revealed two absorption maxima at 380 and 452 nm, characteristic of oxidized flavoproteins (Supplementary Figure S2). The bound flavin cofactor was isolated and identified as FAD via liquid chromatography electrospray ionization quadrupole time-of-flight mass spectrometry (LC/ESI-Q/TOF MS) (Figure S2). NAD(P)H oxidation assays showed His6-MibS to be highly specific for NADH as the electron donor with an apparent KM of 80 µM and apparent kcat of 198 min−1 (Figure S2). The successful reconstitution of MibS as an NADH-dependent FAD reductase allowed the development of enzyme assays to probe the chlorination reaction presumably catalyzed by MibH.
MibH is an FADH2-dependent halogenase responsible for ClTrp formation
Similar to His6-MibS, soluble protein was only obtained after co-expression of mibH with the chaperones groES/EL. Flavin was not present following downstream purification suggesting the enzyme has a weak binding affinity towards its cofactor. Similar results have been observed for other FADH2-dependent Trp halogenases.26, 27
Previous transcriptomic analysis of the NAI-107 native producer Microbispora corallina NRRL 30420 indicated that NAI-107 biosynthesis commences with the production of deschloro-deshydroxyl NAI-107.16 Deschloro-deshydroxyl NAI-107 activates a dedicated sigma factor needed for the transcription of additional NAI-107 biosynthetic genes including mibH.16 Thus MibH presumably acts at a later stage of NAI-107 biosynthesis, chlorinating the cyclized MibA core. Initial MibH activity assays were therefore carried out using deschloro NAI-107 (1) as a substrate. LC/ESI-Q/TOF MS analysis of halogenation assays performed with MibH, MibS, FAD, NADH, NaCl and 1 revealed the formation of a product peak containing the characteristic mass and isotopic distribution of chlorine-containing compounds (Figure 4a and Figure S3). No chlorinated product was observed when either MibS or MibH were omitted from the reaction assay (e.g. Figure 4a). Furthermore, tandem MS characterization of the chlorinated product revealed chlorination occurred at Trp4 (Figure 4b). Together, our results suggest MibH to be an unusual FADH2-dependent Trp halogenase capable of chlorinating Trp within peptides.
Figure 4.

In vitro reconstitution of His6-MibH activity. (a) LC/ESI-Q/TOF MS analysis of halogenation reactions catalyzed by MibH. (left) Total ion chromatogram (TIC) of halogenation reactions performed in the presence (bottom) or absence (top) of His6-MibH. The red number on top of each peak serves as an identifier. Starting material (peak 1). Expected chlorinated product (peak 2). Starting material was provided as a mixture of hydroxylated deschloro NAI-107 congeners.11 (right) ESI-Q/TOF MS analysis corresponding to each peak identified on the TIC. Black numbers on top of each family of peaks represent the [M+2H]2+ monoisotopic m/z values. Monohydroxylated, deschloro NAI-107 monoisotopic mass calc. [M+2H]2+ 1099.41; bishydroxylated deschloro NAI-107 monoisotopic mass calc. [M+2H]2+ 1107.41; monohydroxylated NAI-107, monoisotopic mass calc. [M+2H]2+ 1116.39; bishydroxylated NAI-107, monoisotopic mass calc. [M+2H]2+ 1124.39. (b) ESI-Q/TOF tandem MS/MS fragmentation analysis of deschloro NAI-107 after modification by His6-MibH. NAI-107 monoisotopic mass calc. [M+H]+ 2247.77, obs. 2248.78. Fragmentation inside the N-terminal ring has also been observed for the lantibiotic nisin, and the planosporicin-like family of lantibiotics.10 ClW; chlorotryptophan, diOHP; dihydroxyproline, EnCys; aminovinylcysteine. (c) Structural representation of substrates 3 and 4. Color scheme is the same as in Figure 1. Thioether rings are labeled with alphabet letters.
Characterization of the substrate specificity of MibH
All flavin-dependent halogenases characterized to date halogenate free Trp, Trp analogs, or substrates bound to coenzyme A or peptidyl carrier proteins (PCP).41–44 To determine the substrate specificity of MibH, halogenation assays were performed using free Trp. Regardless of the conditions tested, halogenation activity was not detected (Table 1). This result suggests that MibH only recognizes Trp within the context of a peptide backbone thus making this enzyme, to the best of our knowledge, the first characterized halogenase exhibiting specificity for Trp within a polypeptide.
Table 1.
Substrates tested for halogenation by MibH.
 | ||
|---|---|---|
| entry | substrate | activity* |
| Trp | − | |
| 1 | deschloro NAI-107 | + |
| 2 | linear His6-MibA precursor peptide | − |
| 3 | deschloro NAI-107Δ12–24 | − |
| 4 | mutacin 1140 | − |
| mutacin 1140 F1G | − | |
| mutacin 1140 K2A | − | |
active (+), inactive (−)
We next evaluated the potential of using MibH as a general peptide Trp halogenase. Its activity was tested using several NAI-107 analogs containing a varying degree of post-translational modifications, including the unmodified precursor peptide MibA (2) and deschloro NAI-107Δ12–24 (3), a synthetic NA1–107 variant composed of only the first two N-terminal rings (Figure 4c and Supplementary Figure S4). Surprisingly, given the substrate tolerance of other lanthipeptide tailoring enzymes, MibH was not capable of chlorinating either substrate under the conditions tested as observed by MALDI-TOF MS (Table 1). Intrigued by these results, we then investigated the capability of MibH to chlorinate mutacin 1140 (4), another antimicrobial lanthipeptide. Mutacin 1140 contains a Trp residue in the A-ring at the same position as 1 and shares an analogous overall structural topology with identical A and B rings (Figure 4c).33, 45, 46 Despite the high structural similarity to 1, no chlorination activity was detected with 4 (Table 1). MibH also did not act on the mutacin mutants F1G and K2A (Table 1).46 Collectively, our results indicate that the different post-translational modifications present in 1 as well as the overall structure of the cyclized compound including its C-terminal rings appear to determine the substrate specificity of MibH. The observation that the enzyme does not require the leader peptide on MibA is consistent with a growing number of RiPP tailoring enzymes that specifically recognize the macrocyclic scaffold and act late in a biosynthetic pathway. Chlorination after the rings are already installed is also consistent with the aforementioned observation wherein non-chlorinated NAI-107 produced earlier in the growth phase activates expression of mibH.16 These results also suggest that chlorination takes place after the removal of the leader peptide. This would require leader peptide cleavage in the cytosol since MibH does not contain any export signal. The protease(s) that removes the leader peptide during the biosynthesis of NAI-107 is currently not known.
Structural characterization of MibH
To determine the structural basis of such unusual peptide specificity the crystal structure of MibH bound to FAD was determined to 1.85-Å resolution (Figure 5a and Supplementary Table S2). MibH crystallized with four molecules in the asymmetric unit, and clear density corresponding to FAD and chloride ions was observed (Figure 5b) in three out of four of these molecules, whereas spurious electron density was observed for the fourth molecule. The overall structure of the MibH monomer consists of a large single domain consisting of a rectangular arrangement of several α-helices surrounding two β-sheets, with a triangular helical protuberance appended to the base of this rectangle. The FAD cofactor is positioned in a solvent exposed groove, roughly parallel to the main body of each monomer, with the adenosine moiety positioned near one set of β-strands and the isoalloxazine ring situated at the other set of β-strands. MibH contains two signatures motifs that are conserved among other FADH2-dependent halogenases. The WxWxIP (Trp283-Pro288) motif whose role in halogenation remains elusive,47 and the GxGxxG (Gly36-Gly41) flavin consensus sequence that is located in the vicinity of the FAD. A Lys/Glu pair that is catalytically relevant in other FADH2-dependent halogenases is also present in MibH (Lys102 and Glu355). In orthologous enzymes, the substrate Trp is located more than 10 Å away from the flavin and chlorination of the equivalent Lys facilitates halogenation via a covalent Lys chloramine intermediate.29 The conserved Glu may either facilitate proton abstraction from the Trp substrate following halogenation,26 or enhance the electrophilicity of the chloramine. Electron density corresponding to a chloride ion is located at a pocket at the Re face of the isoalloxazine, where it interacts with the backbone amides of Ser357 and Gly358. Pro353 forms the opposing edge of the binding pocket (Figure 5b). As in other FADH2-dependent halogenases, the size of the binding pocket likely precludes the binding of halides such as iodide but not bromide, consistent with the ability to purify brominated NAI-107 variants when the natural producer is supplemented with bromide sources.48
Figure 5.

Crystal structure of the lantibiotic halogenase MibH. (a) Overall cartoon representation of MibH.α helices are shown in purple, β sheets are shown in green, flexible loops are shown in wheat and the second protomer is shown gray. FAD and the Cl− ion are shown as sticks and as a cyan sphere respectively. (b) Simulated annealing omit difference Fourier map (Fo – Fc) contoured to 2.5σ of FAD and Cl−. The conserved Lys/Glu pair involved in chlorination is shown as sticks. The conserved WXWXIP motif is labeled in one amino acid code.
Comparison of MibH with other enzymes in the protein data bank (PDB) via the Dali server reveals a conserved global architecture shared amongst other Trp halogenases. The closest structural relatives to MibH include the 5-Trp halogenase PyrH involved in pyrroindomycin biosynthesis,49 the 7-Trp halogenases PrnA and RebH from pyrrolnitrin and rebeccamycin biosynthesis, respectively,26, 27 and the 6-Trp halogenase SttH from an uncharacterized NRPS cluster, all of which utilize free Trp as a substrate but exhibit different regioselectivity. Visual inspection of the proposed MibH active site based on conservation of important catalytic residues and the spatial arrangement of FAD and chloride reveals that MibH has a noticeable larger accessible surface area in comparison to free-tryptophan utilizing halogenases, which likely serves to accommodate its larger peptidic substrate (Figure 6a). Interestingly, the similar structural topology of MibH to other FADH2-dependent Trp halogenases suggests this larger crevice did not arise due to structural deletions. Instead, subtle amino acid variations within MibH are responsible for forming the cavity, as revealed by pairwise structural alignments of MibH with different members of this enzyme family.
Figure 6.

Insights into the substrate specificity of the 5-tryptophan halogenase MibH. (a) Cartoon representation of the overall structure of (left) MibH and (right) PyrH49 (PDB 2WET) with their respective substrate binding clefts shown as red surfaces. Color scheme is the same as in Figure 5. (b) Surface representation of the proposed Trp binding site (dashed white line) in (left) MibH and (right) PyrH colored by hydrophobicity.
In the absence of a substrate-bound structure, we propose this region to serve as the substrate binding cleft in MibH. The CASTp server was used to identify residues delineating the proposed peptide binding site,50 allowing the calculation of its solvent accessible surface area in PyMOL, which reveal this region to be 718.5 Å2 larger than that of other members of this enzyme family. In addition, the greater relative hydrophobicity of the MibH binding cleft also supports its peptide substrate preference over free tryptophan (Figure 6b). We next performed a docking analysis of MibH and its substrate deschloro NAI-107. Docking poses were obtained where the C-5 indole carbon is in closest proximity to the catalytic Lys102 with an 8.5-Å distance to its ε-nitrogen (Supplementary Figure S5). For catalysis to occur, we envision this distance to be reduced upon substrate binding.
To obtain further insights into the origins of the substrate specificity, a phylogenetic reconstruction using Maximum Likelihood methods (ML)51 of different FADH2-dependent halogenases that have been biochemically or structurally characterized was performed. Similar to recent results obtained in a phylogenetic study on the evolution of halogenases,52 the ML tree revealed two main clades showing a strong correlation between the distribution of halogenases and their substrate specificity (Figure 7). One clade is formed by proteins known to halogenate pyrrole, phenyl, alkynyl and alkyl groups of substrates tethered to a carrier (either a protein or coenzyme A) (Figure 7, green).41–44 These halogenases are present within assembly lines like non-ribosomal peptide synthetase (NRPS) pathways and are responsible for halogenating a tethered building block that is subsequently incorporated into a final molecule.41, 42 MibH belongs to the same clade as Trp halogenases that halogenate free Trp indicating it shares a closer evolutionary relationship with this type of enzymes. However, the early divergence of MibH from the other Trp halogenases perhaps reflects its ability to halogenate Trp embedded within a peptide substrate (Figure 7, blue). The phylogenetic inference displayed by these enzymes could prove to be helpful at predicting the substrate specificities of uncharacterized halogenases.
Figure 7.

Maximum likelihood phylogenetic unrooted tree of various characterized flavin-dependent halogenases. Support for each clade is indicated by bootstrap percentage values. The name of each terminal branch corresponds to the name of the halogenase. Tree is colored based on substrate specificity. Green, halogenases that act on tethered substrates; blue, halogenases that act on peptides; red, halogenases that act on free Trp. Two flavin-dependent oxidoreductases were used as outgroup.
CONCLUSION
Here we characterized two flavin-dependent enzymes involved in tailoring modifications during NAI-107 biosynthesis, the FAD-dependent Cys decarboxylase MibD, and the FADH2-dependent Trp halogenase MibH. MibD is shown to be specific for the C-terminal sequence of the precursor peptide MibA. Such specificity, coupled with the leader peptide-independent activity of this enzyme could be exploited for the utilization of MibD in RiPP combinatorial biosynthesis by incorporating the MibD recognition sequence within the C-terminus of target peptides.
Similar to MibD, the Trp halogenase MibH also exhibited high substrate specificity. Prior mechanistic studies of FADH2-dependent Trp halogenases provided evidence for the use of a covalent chloramine enzyme intermediate that can directly halogenate its substrate.29 The use of such a covalent intermediate is proposed to facilitate regioselectivity. The decoupling of the site where the active halogenating agent is generated from the substrate-binding site is proposed to assist in the evolution of catalysts with different substrate tolerance.49 Although MibH functions on a peptide, where extensive interactions between catalysts and substrate may already establish regioselectivity, the active site features including the Lys that is covalently modified, is largely conserved. These comparisons further support the theory that substrate specificities amongst different classes of FADH2-dependent halogenases are likely due to changes only at the substrate-binding site, rather than at the site where the active chlorinating agent is produced. This strategy allows for these enzymes to not only use free Trp and Trp bound to carrier proteins as substrates but also allows them to act on peptides as exemplified by the lantibiotic halogenase MibH.
SUPPORTING INFORMATION
For general methods and experimental details on the cloning, overexpression, and purification of the peptides and proteins used in this study, characterization of the cofactors bound to MibD and MibS, kinetic characterization of MibS, phylogenetic analysis of flavin dependent halogenases, isolation of 1, chemical synthesis of peptide 3, and generation of MibH-deschloro NAI-107 docking models see the Supporting Information. This material is free of charge via the Internet.
METHODS
In vitro reconstitution of MibD activity and characterization of its substrate specificity
His6-MibD (10 µM) was incubated with 150 µM linear His6-MibA at room temperature for 2 h in 100 µL of reaction buffer (20 mM Tris HCl pH 8.0, and 3 mM DTT). The reaction was purified using a C18 zip-tip concentrator to remove excess salts, mixed in a 1:1 ratio with α-cyano-4-hydroxycinnamic acid and analyzed by MALDI-TOF MS using a Voyager-DE STR in positive mode. To determine the importance of the N-terminal leader sequence for oxidative decarboxylation, 80 µM linear His6-MibA was incubated with trypsin in a 1:10 ratio (trypsin:peptide) in 100 µL of trypsin buffer (100 mM Tris HCl pH 8.0, 5 mM TCEP, 2 mM MgCl2) and incubated for 3 h at room temperature. Removal of the N-terminal leader sequence was monitored by MALDI-TOF MS analysis as described above. After complete N-terminal leader sequence removal, His6-MibD (2 µM) was added to the reaction and the solution was incubated for 15 min at room temperature. Analysis of the reaction was performed by MALDI-TOF MS as described above. To determine the localization of the post-transitional modification, an aliquot of 10 µL of the reaction mixture was injected into an Acquity UPLC Phenomenex Jupiter C18 column (5 µm, 0.1 × 150 mm) coupled to an electrospray ionization mass spectrometer (Q-TOF Synapt-G1 Waters in positive ion scan mode using manufacturer’s conditions for tandem MS/MS fragmentation). The sample was fractionated using a linear gradient from 2% (v/v) solvent A (0.1% (v/v) formic acid in acetonitrile) in 98% (v/v) solvent B (0.1% (v/v) formic acid in water) to 98% (v/v) solvent A over 20 min.
To characterize the substrate specificity of His6-MibD several lanthipeptide precursor peptides were used as substrates following the same reaction conditions as described above. Lanthipeptide precursor peptides ending in Cys (ProcA1.1, 1.7, and 2.8, Table S1) were obtained following previously described pocedures.53 Oxidative decarboxylation was monitored by MALDI-TOF MS analysis using a Voyager-DE STR.
Reconstitution of MibH activity in vitro and characterization of its substrate specificity
MibH activity assays were performed in 20 mM sodium phosphate buffer pH 7.4, 10 mM NaCl, 5 µM FAD, 5 µM His6-MibS, 5 µM His6-MibH, and 50 µM deschloromicrobisporicin. Reactions were initiated by the addition of 100 µM NADH. Halogenation assays were incubated at 30 °C for 2 h. After the incubation period, samples were centrifuged (13,000 × g, 10 min, 25 °C) to remove insoluble material and analyzed by LC/ESI-MS on an Acquity UPLC Phenomenex Jupiter C18 column (5 µm, 0.1 × 150 mm) coupled to an electrospray ionization Q-TOF Synapt-G1 mass spectrometer (Waters) in positive ion mode. LC/ESI-Q/TOF MS and tandem MS conditions used were similar to the ones already described above. To characterize the substrate specificity of MibH, several substrates (Table 1) were tested for halogenation activity using similar reaction conditions as described above. Samples were then centrifuged to remove insoluble material and desalted using a C18 zip-tip concentrator following standard procedures. An aliquot of the sample was mixed in a 1:1 ratio with 2, 5-dihydroxybenzoic acid matrix and the extent of halogenation was analyzed using a Bruker UltrafleXtreme MALDI TOF-TOF mass spectrometer in positive mode.
MibH structure determination
Following purification of MibH by nickel affinity chromatography as described in the SI, the protein was desalted by size exclusion chromatography using a HiLoad 16/600 Superdex 200 column (flow rate of 1 mL min−1) connected to an ÄKTA purifier at 4 °C (buffer of 20 mM HEPES pH 8.0 and 150 mM KCl). Eluted protein was then concentrated to 22 mg mL−1 in the presence of 2 mM FAD prior to crystallization. Crystals were obtained within 24 h in hanging drops containing 12.5% MPD, 12.5% PEG 3.35K, 0.1 M MES/imidazole pH 6.5, and 0.09 M NPS (1:1:1 NaNO3, Na2HPO4, (NH4)2SO4). Prior to data collection, crystals were transferred into the crystallization solution supplemented with 20% MPD and vitrified in liquid nitrogen. Data were collected at the Advanced Photon Source, Argonne National Lab using the Life-Science Collaborative Access Team (LS-CAT) 21-ID-G beam line. Raw data were integrated and scaled using the autoPROC software.54 The structure was determined by molecular replacement using the PHASER software55 and a modified model of PyrH (PDB: 2WET) with all non-identical residues truncated to Ala. The initial model was further improved using Buccaneer56 and interspersed with rounds of manual building using COOT.57 Interim models were routinely adjusted through rounds of crystallographic refinement using Refmac5.58 Refinement converged to the final model for which statistics are presented in Supplementary Table S2.
Accession codes
The structure factors and coordinates for the MibH structure have been deposited in the Protein Data Bank under the accession code 5UAO.
Supplementary Material
Acknowledgments
FUNDING SOURCES
This work was supported by the National Institutes of Health (R37 GM058822 to W.A.V., and R01 GM079038 to S.K.N.), and by a grant from the European Commission (contract no. 245066 for FP7-KBBE-2009-3) to M.S. M.A.O was supported by an NIGMS-NIH Chemistry-Biology Interface Training Grant (5T32-GM070421) and by the Ford Foundation. A Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer was purchased in part with a grant from the National Institutes of Health (S10 RR027109 A).
Footnotes
AUTHOR CONTRIBUTIONS
MAO and WAV designed experiments and analyzed biochemical data. MAO performed biochemical and bioinformatic experiments. SM synthesized and characterized NAI-107Δ12–24. DPC and SKN performed and analyzed structural experiments. NG and BL cloned mib genes. GNT provided prochlorosin precursor peptides. SD and MS provided Microbispora sp. 107891 and deschloromicrobisporicin. JE and LS provided mutacin 1140. MAO, WAV, DPC, and SKN wrote the manuscript.
REFERENCES
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortega MA, van der Donk WA. New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem. Biol. 2016;23:31–44. doi: 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knerr PJ, van der Donk WA. Discovery, biosynthesis, and engineering of lantipeptides. Annu. Rev. Biochem. 2012;81:479–505. doi: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- 4.Bierbaum G, Sahl HG. Lantibiotics: mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 2009;10:2–18. doi: 10.2174/138920109787048616. [DOI] [PubMed] [Google Scholar]
- 5.Plat A, Kuipers A, Rink R, Moll GN. Mechanistic aspects of lanthipeptide leaders. Curr. Protein Pept. Sci. 2013;14:85–96. doi: 10.2174/1389203711314020001. [DOI] [PubMed] [Google Scholar]
- 6.Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: new insights into the role of leader and core peptides during biosynthesis. Chem. Eur. J. 2013;19:7662–7677. doi: 10.1002/chem.201300401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velásquez JE, Zhang X, van der Donk WA. Biosynthesis of the antimicrobial peptide epilancin 15X and its unusual N-terminal lactate moiety. Chem. Biol. 2011;18:857–867. doi: 10.1016/j.chembiol.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castiglione F, Lazzarini A, Carrano L, Corti E, Ciciliato I, Gastaldo L, Candiani P, Losi D, Marinelli F, Selva E, Parenti F. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem. Biol. 2008;15:22–31. doi: 10.1016/j.chembiol.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Maffioli SI, Monciardini P, Catacchio B, Mazzetti C, Munch D, Brunati C, Sahl HG, Donadio S. Family of class I lantibiotics from actinomycetes and improvement of their antibacterial activities. ACS Chem. Biol. 2015;10:1034–1042. doi: 10.1021/cb500878h. [DOI] [PubMed] [Google Scholar]
- 11.Maffioli SI, Iorio M, Sosio M, Monciardini P, Gaspari E, Donadio S. Characterization of the congeners in the lantibiotic NAI-107 complex. J. Nat. Prod. 2014;77:79–84. doi: 10.1021/np400702t. [DOI] [PubMed] [Google Scholar]
- 12.Foulston LC, Bibb MJ. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. U. S. A. 2010;107:13461–13466. doi: 10.1073/pnas.1008285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Münch D, Müller A, Schneider T, Kohl B, Wenzel M, Bandow JE, Maffioli S, Sosio M, Donadio S, Wimmer R, Sahl HG. The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. J. Biol. Chem. 2014;289:12063–12076. doi: 10.1074/jbc.M113.537449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jabés D, Brunati C, Candiani G, Riva S, Romanó G, Donadio S. Efficacy of the new lantibiotic NAI-107 in experimental infections induced by multidrug-resistant Gram-positive pathogens. Antimicrob. Agents Chemother. 2011;55:1671–1676. doi: 10.1128/AAC.01288-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maffioli SI, Cruz JC, Monciardini P, Sosio M, Donadio S. Advancing cell wall inhibitors towards clinical applications. J. Ind. Microbiol. Biotechnol. 2016;43:177–184. doi: 10.1007/s10295-015-1703-9. [DOI] [PubMed] [Google Scholar]
- 16.Foulston L, Bibb M. Feed-forward regulation of microbisporicin biosynthesis in Microbispora corallina . J. Bacteriol. 2011;193:3064–3071. doi: 10.1128/JB.00250-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ortega MA, Hao Y, Walker MC, Donadio S, Sosio M, Nair SK, van der Donk WA. Structure and tRNA specificity of MibB, a lantibiotic dehydratase from Actinobacteria involved in NAI-107 biosynthesis. Cell Chem. Biol. 2016;23:370–380. doi: 10.1016/j.chembiol.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kupke T, Stevanovic S, Sahl HG, Götz F. Purification and characterization of EpiD, a flavoprotein involved in the biosynthesis of the lantibiotic epidermin. J. Bacteriol. 1992;174:5354–5361. doi: 10.1128/jb.174.16.5354-5361.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majer F, Schmid DG, Altena K, Bierbaum G, Kupke T. The flavoprotein MrsD catalyzes the oxidative decarboxylation reaction involved in formation of the peptidoglycan biosynthesis inhibitor mersacidin. J. Bacteriol. 2002;184:1234–1243. doi: 10.1128/JB.184.5.1234-1243.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kupke T, Uebele M, Schmid D, Jung G, Blaesse M, Steinbacher S. Molecular characterization of lantibiotic-synthesizing enzyme EpiD reveals a function for bacterial Dfp proteins in coenzyme A biosynthesis. J. Biol. Chem. 2000;275:31838–31846. doi: 10.1074/jbc.M004273200. [DOI] [PubMed] [Google Scholar]
- 21.Strauss E, Zhai H, Brand LA, McLafferty FW, Begley TP. Mechanistic studies on phosphopantothenoylcysteine decarboxylase: trapping of an enethiolate intermediate with a mechanism-based inactivating agent. Biochemistry. 2004;43:15520–15533. doi: 10.1021/bi048340a. [DOI] [PubMed] [Google Scholar]
- 22.Strauss E, Begley TP. Mechanistic studies on phosphopantothenoylcysteine decarboxylase. J. Am. Chem. Soc. 2001;123:6449–6450. doi: 10.1021/ja016020y. [DOI] [PubMed] [Google Scholar]
- 23.Blaesse M, Kupke T, Huber R, Steinbacher S. Crystal structure of the peptidyl-cysteine decarboxylase EpiD complexed with a pentapeptide substrate. Embo J. 2000;19:6299–6310. doi: 10.1093/emboj/19.23.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blaesse M, Kupke T, Huber R, Steinbacher S. Structure of MrsD, an FAD-binding protein of the HFCD family. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003;D59:1414–1421. doi: 10.1107/s0907444903011831. [DOI] [PubMed] [Google Scholar]
- 25.Kempter C, Kupke T, Kaiser D, Metzger JW, Jung G. Thioenols from peptidyl cysteines: oxidative decarboxylation of a 13C–labeled substrate. Angew. Chem. Intl. Ed. 1996;35:2104–2107. [Google Scholar]
- 26.Dong C, Flecks S, Unversucht S, Haupt C, van Pee KH, Naismith JH. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh E, Garneau S, Walsh CT. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2005;102:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeh E, Cole LJ, Barr EW, Bollinger JM, Jr, Ballou DP, Walsh CT. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry. 2006;45:7904–7912. doi: 10.1021/bi060607d. [DOI] [PubMed] [Google Scholar]
- 29.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 30.Kupke T, Götz F. The enethiolate anion reaction products of EpiD. pKa value of the enethiol side chain is lower than that of the thiol side chain of peptides. J. Biol. Chem. 1997;272:4759–4762. doi: 10.1074/jbc.272.8.4759. [DOI] [PubMed] [Google Scholar]
- 31.Kupke T, Kempter C, Gnau V, Jung G, Götz F. Mass spectroscopic analysis of a novel enzymatic reaction. Oxidative decarboxylation of the lantibiotic precursor peptide EpiA catalyzed by the flavoprotein EpiD. J. Biol. Chem. 1994;269:5653–5659. [PubMed] [Google Scholar]
- 32.Kupke T, Kempter C, Jung G, Götz F. Oxidative decarboxylation of peptides catalyzed by flavoprotein EpiD. Determination of substrate specificity using peptide libraries and neutral loss mass spectrometry. J. Biol. Chem. 1995;270:11282–11289. doi: 10.1074/jbc.270.19.11282. [DOI] [PubMed] [Google Scholar]
- 33.Escano J, Stauffer B, Brennan J, Bullock M, Smith L. Biosynthesis and transport of the lantibiotic mutacin 1140 produced by Streptococcus mutans . J. Bacteriol. 2015;197:1173–1184. doi: 10.1128/JB.02531-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA. Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10430–10435. doi: 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta. 2015;1854:1019–1037. doi: 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Walsh CT. Identification of the gene cluster for the dithiolopyrrolone antibiotic holomycin in Streptomyces clavuligerus . Proc. Natl. Acad. Sci. U. S. A. 2010;107:19731–19735. doi: 10.1073/pnas.1014140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brock NL, Nikolay A, Dickschat JS. Biosynthesis of the antibiotic tropodithietic acid by the marine bacterium Phaeobacter inhibens . Chem. Commun. 2014;50:5487–5489. doi: 10.1039/c4cc01924e. [DOI] [PubMed] [Google Scholar]
- 38.Schmid DG, Majer F, Kupke T, Jung G. Electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry to reveal the substrate specificity of the peptidyl-cysteine decarboxylase EpiD. Rapid Commun. Mass Spectrom. 2002;16:1779–1784. doi: 10.1002/rcm.780. [DOI] [PubMed] [Google Scholar]
- 39.van Heel AJ, Mu D, Montalban-Lopez M, Hendriks D, Kuipers OP. Designing and producing modified, new-to-nature peptides with antimicrobial activity by use of a combination of various lantibiotic modification enzymes. ACS Synth. Biol. 2013;2:397–404. doi: 10.1021/sb3001084. [DOI] [PubMed] [Google Scholar]
- 40.Payne JT, Andorfer MC, Lewis JC. Regioselective arene halogenation using the FAD-dependent halogenase RebH. Angew. Chem. Int. Ed. 2013;52:5271–5274. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podzelinska K, Latimer R, Bhattacharya A, Vining LC, Zechel DL, Jia Z. Chloramphenicol biosynthesis: the structure of CmlS, a flavin-dependent halogenase showing a covalent flavin-aspartate bond. J. Mol. Biol. 2010;397:316–331. doi: 10.1016/j.jmb.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, Moore BS. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014;10:640–647. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buedenbender S, Rachid S, Muller R, Schulz GE. Structure and action of the myxobacterial chondrochloren halogenase CndH: a new variant of FAD-dependent halogenases. J. Mol. Biol. 2009;385:520–530. doi: 10.1016/j.jmb.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 45.Smith L, Zachariah C, Thirumoorthy R, Rocca J, Novak J, Hillman JD, Edison AS. Structure and dynamics of the lantibiotic mutacin 1140. Biochemistry. 2003;42:10372–10384. doi: 10.1021/bi034490u. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Wilson-Stanford S, Cromwell W, Hillman JD, Guerrero A, Allen CA, Sorg JA, Smith L. Site-directed mutations in the lanthipeptide mutacin 1140. Appl. Environ. Microbiol. 2013;79:4015–4023. doi: 10.1128/AEM.00704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flecks S, Patallo EP, Zhu X, Ernyei AJ, Seifert G, Schneider A, Dong C, Naismith JH, van Pee KH. New insights into the mechanism of enzymatic chlorination of tryptophan. Angew. Chem. Int. Ed. 2008;47:9533–9536. doi: 10.1002/anie.200802466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cruz JC, Iorio M, Monciardini P, Simone M, Brunati C, Gaspari E, Maffioli SI, Wellington E, Sosio M, Donadio S. Brominated variant of the lantibiotic NAI-107 with enhanced antibacterial potency. J. Nat. Prod. 2015;78:2642–2647. doi: 10.1021/acs.jnatprod.5b00576. [DOI] [PubMed] [Google Scholar]
- 49.Zhu X, De Laurentis W, Leang K, Herrmann J, Ihlefeld K, van Pee KH, Naismith JH. Structural insights into regioselectivity in the enzymatic chlorination of tryptophan. J. Mol. Biol. 2009;391:74–85. doi: 10.1016/j.jmb.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- 52.Xu G, Wang BG. Independent evolution of six families of halogenating enzymes. PLoS One. 2016;11:e0154619. doi: 10.1371/journal.pone.0154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thibodeaux GN, van der Donk WA. An engineered lantipeptide synthetase serves as a general leader peptide-dependent kinase. Chem. Commun. 2012;48:10615–10617. doi: 10.1039/c2cc34138g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bunkoczi G, Echols N, McCoy AJ, Oeffner RD, Adams PD, Read RJ. Phaser.MRage: automated molecular replacement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013;69:2276–2286. doi: 10.1107/S0907444913022750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 57.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 58.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.