

This study gives novel insights concerning how a mild systemic inflammation impacts phrenic motor plasticity (pMF), particularly adenosine-dependent pMF. We suggest that since this adenosine-dependent pathway is insensitive to systemic inflammation, it represents an alternative or “backup” mechanism of pMF when other mechanisms are suppressed.
Keywords: inflammation, spinal plasticity, respiratory plasticity, adenosine, acute intermittent hypoxia
Abstract
Phrenic motor facilitation (pMF), a form of respiratory plasticity, can be elicited by acute intermittent hypoxia (i.e., phrenic long-term facilitation, pLTF) or direct application of drugs to the cervical spinal cord. Moderate acute intermittent hypoxia (mAIH; 3 × 5-min episodes of 35–50 mmHg arterial Po2, 5-min normoxic intervals) induces pLTF by a serotonin-dependent mechanism; mAIH-induced pLTF is abolished by mild systemic inflammation induced by a low dose of lipopolysaccharide (LPS; 100 μg/kg ip). In contrast, severe acute intermittent hypoxia (sAIH; 3 × 5-min episodes of 25–30 mmHg arterial Po2, 5-min normoxic intervals) elicits pLTF by a distinct, adenosine-dependent mechanism. Since it is not known if systemic LPS blocks the mechanism giving rise to sAIH-induced pLTF, we tested the hypothesis that sAIH-induced pLTF and adenosine 2A (A2A) receptor-induced pMF are insensitive to mild systemic inflammation elicited by the same low dose of LPS. In agreement with our hypothesis, neither sAIH-induced pLTF nor cervical intrathecal A2A receptor agonist (CGS-21680; 200 μM, 10 μl × 3)-induced pMF were affected 24 h post-LPS. Pretreatment with intrathecal A2A receptor antagonist injections (MSX-3; 10 μM, 12 μl) blocked sAIH-induced pLTF 24 h post LPS, confirming that pLTF was adenosine dependent. Our results give insights concerning the differential impact of systemic inflammation and the functional significance of multiple cascades capable of giving rise to phrenic motor plasticity. The relative resistance of adenosine-dependent pMF to inflammation suggests that it provides a “backup” system in animals lacking serotonin-dependent pMF due to ongoing inflammation associated with systemic infections and/or neural injury.
NEW & NOTEWORTHY This study gives novel insights concerning how a mild systemic inflammation impacts phrenic motor plasticity (pMF), particularly adenosine-dependent pMF. We suggest that since this adenosine-dependent pathway is insensitive to systemic inflammation, it represents an alternative or “backup” mechanism of pMF when other mechanisms are suppressed.
many clinical disorders challenge the neural control of breathing, including chronic obstructive lung disease (Castellucci et al. 2015; Wills-Karp 1999), neuromuscular disorders (e.g., spinal injury, motor neuron disease; Dale-Nagle et al. 2010b; Golder and Mitchell 2005), and obstructive sleep apnea (Harper et al. 2014). Although most clinical disorders that compromise breathing are associated with systemic inflammation, there is limited research concerning how inflammation affects the neural system controlling breathing. However, recent studies have demonstrated that inflammation impairs at least one key aspect of the neural system controlling breathing, respiratory motor plasticity (Huxtable et al. 2011, 2013; Vinit et al. 2011).
Plasticity is a hallmark of neural systems (Abel et al. 2013; Allen and Barres 2005; Andero et al. 2014), and the neural system controlling breathing is no exception (Dale-Nagle et al. 2010a; Xing et al. 2013). One form of respiratory motor plasticity, phrenic motor facilitation (pMF), represents a persistent increase in the neural output of the phrenic nerve following the initiating stimulus. If the stimulus to pMF is acute intermittent hypoxia (AIH), the resulting pMF is known as phrenic long-term facilitation (pLTF). If the stimulus is pharmacological, plasticity is referred to by a more general term, phrenic motor facilitation (pMF).
Moderate AIH elicits pLTF by a serotonin- and brain-derived neurotrophic factor (BDNF)-dependent mechanism (Bach and Mitchell 1996; Baker-Herman et al. 2004; Baker-Herman and Mitchell 2002; MacFarlane et al. 2011) known as the Q pathway to pMF, since it is induced by Gq protein-coupled metabotropic receptors (Dale-Nagle et al. 2010a). When pLTF is elicited by severe AIH, pLTF results from an adenosine-dependent mechanism (Golder et al. 2008; Nichols et al. 2012). Adenosine 2A (A2A) receptor-induced pMF requires new synthesis of TrkB vs. BDNF (Golder et al. 2008); this BDNF-independent pathway is known as the S pathway, since it is induced by Gs protein-coupled metabotropic receptors.
Even mild systemic inflammation blocks mAIH-induced pLTF (Huxtable et al. 2011, 2013). However, no data are available concerning the impact of inflammation on sAIH-induced pLTF. Thus we tested the impact of mild inflammation on sAIH-induced pLTF and A2A receptor-induced pMF. Our model of inflammation was the same low dose of lipopolysaccharide (LPS) known to undermine the Q pathway to pMF. LPS is a toll-like receptor 4 agonist (Lu et al. 2008; Triantafilou and Triantafilou 2002) frequently used to study systemic inflammation (Huxtable et al. 2011; Lu et al. 2008; Min et al. 2009; Triantafilou and Triantafilou 2002; Vinit et al. 2011).
MATERIALS AND METHODS
Animals
Experiments were performed on male adult (3–4 mo) Sprague-Dawley rats (colony 211; Harlan, Indianapolis, IN). Rats were housed in a controlled environment (12:12-h light-dark cycle, daily humidity and temperature monitoring) with food and water ad libitum. The University of Wisconsin Institutional Animal Care and Use committee approved all protocols.
Experimental Groups
To test the effects of systemic inflammation, rats received intraperitoneal injections of LPS (E. coli 0111:B4; 100 μg/kg) or vehicle (sterile saline) 24 h before an experiment began (Huxtable et al. 2013). All rats were anesthetized lightly and intraperitoneal injections were made in the lower right quadrant of the abdomen to minimize organ compromise.
In the first experiment, we tested the hypothesis that pLTF elicited by sAIH is insensitive to mild LPS-induced systemic inflammation (24 h). Rats were assigned to four groups: 1) saline + sAIH (n = 8), 2) LPS + sAIH (n = 8), 3) saline without sAIH (time control, n = 4), and 4) LPS without sAIH (time control, n = 4).
In the second experiment, we tested the hypothesis that sAIH-induced pLTF still requires spinal A2A receptor activation during a systemic inflammation. To test this hypothesis, rats were arranged in four groups: 1) saline + A2A antagonist (MSX-3) + sAIH (n = 7), 2) LPS + MSX-3 + sAIH (n = 6), 3) saline + artificial cerebrospinal fluid (aCSF; MSX-3 vehicle) + sAIH (n = 4), and 4) LPS + aCSF + sAIH (n = 3).
For the third experimental series, we tested the hypothesis that pMF elicited by spinal A2A receptor agonist administration (CGS-21680) is insensitive to systemic inflammation. To test this hypothesis, rats were arranged in four groups: 1) saline + CGS-21680 (n = 6), 2) LPS + CGS-21680 (n = 6), 3) saline + aCSF (CGS-21680 vehicle) (n = 3), and 4) LPS + aCSF (n = 3).
Experimental Preparation
Rats were anesthetized in a closed chamber with isoflurane and moved to a temperature-regulated table. On the table, anesthesia was maintained with isoflurane delivered through a nose cone (3.5% isoflurane in 50% O2, balance N2). Body temperature was monitored with a rectal probe and kept between 36 and 38°C. A catheter was placed in the tail vein (24G × 3/4 in.; Surflo) or in the femoral vein (polyethylene PE50; Intramedic), and rats were infused (rate of infusion 0.5–3 ml·kg−1·h−1) throughout the surgery and protocol with a 1:2:0.13 solution of HesPan (6% Hetastarch in 0.9% NaCl), lactated Ringer, and 8.4% sodium bicarbonate to maintain fluid and acid-base balance. Rats were tracheotomized to enable artificial ventilation (Rodent Respirator model 683; Harvard Apparatus, Holliston, MA; tidal volume ∼2.5 ml). The lungs were hyperinflated for two breaths approximately once per hour to offset alveolar collapse. End-tidal Pco2 was monitored with a flow-thorough CO2 analyzer (Capnogard; Novametrix, Wallingford, CT); throughout the surgical preparation, end-tidal Pco2 was maintained between 40 and 45mmHg.
A midcervical, bilateral vagotomy was performed to prevent phrenic entrainment with the ventilator. A femoral arterial catheter (polyethylene PE50; Intramedic) was placed to monitor blood pressure and draw arterial blood samples for blood gas analysis (ABL-800 Flex; Radiometer, Westlake, OH). Blood pressure was monitored with a pressure transducer (Gould P23ID), and blood samples (0.2–0.4 ml) were drawn for blood gas analysis. Blood gases were assessed during baseline before and after drug delivery, during each of the three hypoxic episodes, and at 15, 30, 60, and 90 min posttreatment. The left phrenic nerve was isolated (dorsal approach), cut distally, desheathed, and covered with a saline-soaked cotton ball until protocols began.
Laminectomy was performed at cervical level 2 (C2) for all rats except those exposed to sAIH without drugs. A small incision was made in the dura, and a soft silicone catheter (2-French; Access Technologies, Skokie, IL) was inserted caudally 4–5 mm until the tip rested approximately over the C3–C4 segments to deliver drugs near the phrenic motor nucleus. The catheter was attached to a 50-μl Hamilton syringe filled with drug or vehicle solutions. Once surgery was complete, the animals were converted to urethane anesthesia (1.85 g/kg) with a slow infusion in the intravenous catheter over 15–20 min. Adequate anesthetic depth was assessed by lack of blood pressure response and respiratory nerve responses to a toe pinch using a hemostat.
Once rats were converted to urethane anesthesia, a minimum of 1 h was allowed before the experimental protocol commenced. After the neurophysiological protocol was complete, all animals were euthanized by urethane overdose.
Neurophysiological Measurements
Rats were paralyzed using pancuronium bromide for neuromuscular blockade (2.5 mg/kg iv). The left phrenic nerve was desheathed and placed on a bipolar silver electrode to record nerve activity; the nerve and electrode were immersed in mineral oil. Phrenic nerve activity was amplified (gain of 10,000×), bandpass filtered (100–10,000 Hz; model 1800; A–M Systems, Carlsborg, WA), rectified, and integrated (Paynter filter, time constant 50 ms, MA-821; CWE, Ardmore, PA). Integrated phrenic nerve bursts were digitized (8 kHz) and analyzed using WINDAQ data-acquisition systems (DATAQ Instruments, Akron, OH).
To start the protocol, the apneic threshold was determined by lowering the end-tidal CO2 until phrenic nerve activity ceased for ∼1 min. The recruitment threshold was then determined by slowly increasing the end-tidal CO2 until phrenic nerve activity resumed. The end-tidal CO2 was then raised ∼2 mmHg above the recruitment threshold, and ∼15–20 min were allowed to establish stable neural activity (i.e., baseline). Severe AIH was achieved by delivering three 5-min episodes of isocapnic hypoxia separated by 5-min intervals of baseline conditions (arterial Po2 ≥ 150 mmHg). Severe AIH was 25–30 mmHg as verified by blood gas analysis during each hypoxic episode. After the third hypoxic episode, all animals were returned to baseline inspired O2 levels and baseline conditions were maintained for the duration of the experiment. Time control experiments, which account for time-related variations in phrenic nerve activity, were performed in groups 1 and 3. For group 1, the animals received either saline or LPS without hypoxia. For group 3, the animals received either saline or LPS + intrathecal drug vehicle injections. An arterial blood sample was drawn 5–10 min after drug delivery and throughout the protocol; arterial Pco2 (PaCO2) was maintained ±2 mmHg of baseline levels by adjusting inspired CO2 or ventilator rate.
Drugs
CGS-21680 [2-p-(2-carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride, catalog no. C141; Sigma-Aldrich, St. Louis, MO] is a selective A2A receptor agonist. MSX-3 (catalog no. M3568; Sigma-Aldrich) is a selective A2A receptor antagonist. Drugs were stored at −20°C, diluted in dimethyl sulfoxide (DMSO), and stored in stock solutions of CGS-21680 (100 mM stock, 2-μl vial) and MSX-3 (2 mM stock, 5-μl vial).
During experiments, stock solutions were diluted to 200 μM CGS-21680 (198 μl of DMSO and 800 μl of aCSF, 3 × 10-μl bolus every 5 min) and 10 μM MSX-3 (995 μl of aCSF, 12-μl bolus). CGS-21680 was used at a higher dose than in a previous study (Golder et al. 2008) on the basis of preliminary experiments (Fig. 1); in the present experiments, we selected the minimal dose required to achieve consistent phrenic motor facilitation without hypoglossal facilitation. We cannot explain the reason for different concentration effects, but our previous study was done with rats from a different colony (Harlan SD 217 used in Golder et al. 2008 vs Harlan SD 211 used in present study). The MSX-3 dose is the same as used previously (Golder et al. 2008; Hoffman et al. 2010; Nichols et al. 2012).
Fig. 1.
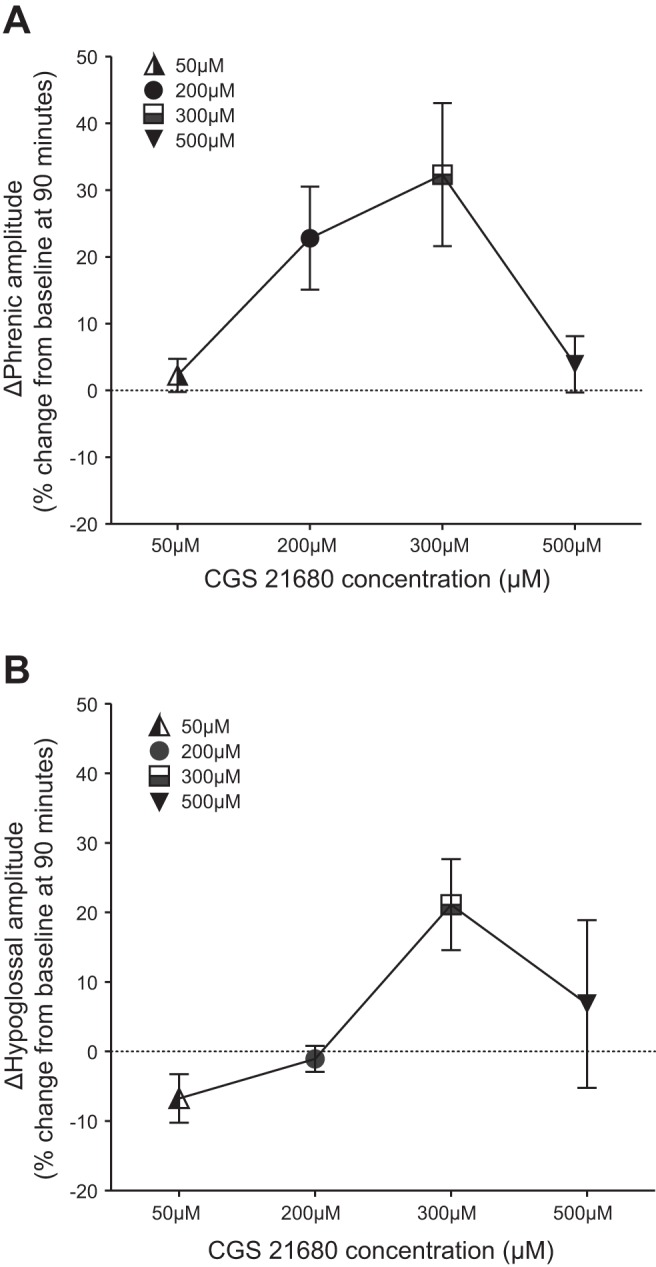
Preliminary dose-response study of phrenic and hypoglossal nerve amplitude changes 90 min after A2A agonist (CGS-21680) injections. A: integrated phrenic amplitude 90 min post-CGS-21680. B: integrated hypoglossal amplitude 90 min post-CGS-21680. The dose chosen was 200 μM since the phrenic response was robust (A; n = 5) but without significant response in the hypoglossal burst amplitude (B; n = 3). The lack of hypoglossal response assures us that the drug is not reaching the brain stem at an effective dose.
Intrathecal CGS-21680 was delivered in three 10-μl boluses every 5 min, and MSX-3, which has no effect on baseline phrenic nerve activity, was delivered slowly over 1 min, 5 min before the first hypoxic episode (Golder et al. 2008; Hoffman et al. 2010). Control groups received aCSF (in mM: 120 NaCl, 3 KCl, 2 CaCl, 2 MgCl, 23 NaHCO3, and 10 glucose bubbled with 95%O2-5%CO2) and DMSO. CGS-21680 controls received 800 μl of aCSF and 200 μl of DMSO (3 × 10 μl every 5 min), and MSX-3 controls received 995 μl of aCSF and 5 μl of DMSO (12-μl bolus).
Statistical Analyses
Integrated phrenic burst amplitude was normalized as percent change from baseline nerve output. Respiratory frequency was normalized to baseline nerve output and expressed as an absolute difference in burst per minute. Statistical analysis for the short-term hypoxic phrenic response was done with a one-way ANOVA (Prism 6; GraphPad Software). For statistical comparisons within and between treatment groups for mean arterial pressure (MAP) and blood gases (baseline and 90-min time points), a two-way ANOVA with a repeated-measures design was used (Prism 6). Statistical analysis of time controls was done by two-way repeated-measures ANOVA to detect significant differences between saline- and LPS-injected time control groups (Prism 6). Since there were no significant differences in phrenic burst amplitude at any point between time controls receiving saline vs. LPS (baseline, 15, 30, 60, or 90 min), the data were combined into a single time control group for subsequent analyses. Statistical comparisons within and between treatment groups for phrenic nerve amplitude and frequency (baseline, 15, 30, 60, and 90 min) were made with a two-way ANOVA with a repeated-measures design; individual post hoc comparisons were made using Tukey's post hoc test. For all analyses, the significance level was set to 0.05 (Prism 6). Results are presented as means ± SE.
RESULTS
Blood Gases and Mean Arterial Pressure
Baseline and 90-min posthypoxia.
In all groups, PaCO2 was regulated within ∼2 mmHg of baseline values throughout protocol and PaO2 was maintained above 150 mmHg with hypoxic episodes as the exception (Table 1). MAP, temperature, and pH were similar among groups at baseline and at 90 min posthypoxia (Table 1). Thus differences in PaCO2, PaO2, MAP, temperature, or pH regulation among groups cannot account for differential pMF responses.
Table 1.
PaCO2, PaO2, MAP, temperature, and pH at baseline and 90 min for experimental groups
| Experimental Groups | PaCO2, mmHg | PaO2, mmHg | MAP, mmHg | Temperature, °C | pH |
|---|---|---|---|---|---|
| Saline + sAIH | |||||
| Baseline | 47.3 ± 1.5 | 299.1 ± 8.2 | 111.8 ± 6.5 | 36.9 ± 0 | 7.4 ± 0 |
| 90 min | 47.7 ± 1.3 | 280.6 ± 7.5 | 98.7.0 ± 5.8 | 37.2 ± 0.1 | 7.4 ± 0 |
| LPS + sAIH | |||||
| Baseline | 46.0 ± 1.4 | 308.8 ± 7.2 | 119.3 ± 7.6 | 37.4 ± 0.1 | 7.4 ± 0 |
| 90 min | 46.2 ± 1.4 | 283.7 ± 5.2 | 95.2 ± 6.0 | 37.5 ± 0.1 | 7.4 ± 0 |
| Time control | |||||
| Baseline | 48.6 ± 0.9 | 283.1 ± 15.2 | 112.0 ± 5.1 | 36.9 ± 0.1 | 7.4 ± 0 |
| 90 min | 49.1 ± 1.2 | 271.0 ± 17.1 | 107.2 ± 5.5 | 37.1 ± 0.1 | 7.4 ± 0 |
| Saline + MSX-3 + sAIH | |||||
| Baseline | 46.2 ± 0.9 | 318.6 ± 6.2 | 108.3 ± 4.2 | 37.5 ± 0.1 | 7.4 ± 0 |
| 90 min | 45.8 ± 1.3 | 324.0 ± 9.2 | 86.5 ± 5.4 | 37.8 ± 0.1 | 7.4 ± 0 |
| LPS + MSX-3 + sAIH | |||||
| Baseline | 48.9 ± 1.5 | 343.3 ± 9.7 | 122.4 ± 13.0 | 37.2 ± 0.1 | 7.4 ± 0 |
| 90 min | 47.9 ± 1.2 | 333.0 ± 3.5 | 92.3 ± 12.5 | 37.4 ± 0.1 | 7.4 ± 0 |
| aCSF + sAIH | |||||
| Baseline | 47.4 ± 0.7 | 350.7 ± 15.2 | 105.5 ± 8.9 | 37.5 ± 0.1 | 7.4 ± 0 |
| 90 min | 47.9 ± 1.1 | 346.1 ± 9.1 | 81.5 ± 7.7 | 37.8 ± 0.2 | 7.4 ± 0 |
| Saline + CGS-21680 | |||||
| Baseline | 48.1 ± 1.7 | 300.5 ± 13.1 | 82.1 ± 6.9 | 37.3 ± 0.1 | 7.4 ± 0 |
| 90 min | 48.9 ± 1.7 | 282.1 ± 10.1 | 80.8 ± 3.9 | 37.3 ± 0.1 | 7.4 ± 0 |
| LPS + CGS-21680 | |||||
| Baseline | 47.1 ± 1.7 | 316.0 ± 12.4 | 92.6 ± 2.0 | 37.5 ± 0.1 | 7.4 ± 0 |
| 90 min | 47.7 ± 1.7 | 316.8 ± 12.2 | 78.2 ± 6.1 | 37.5 ± 0.1 | 7.4 ± 0 |
| Time control (aCSF) | |||||
| Baseline | 48.1 ± 0.9 | 311.0 ± 13.2 | 97.2 ± 4.4 | 37.3 ± 0.1 | 7.4 ± 0 |
| 90 min | 47.8 ± 1.0 | 296.6 ± 16.5 | 94.4 ± 2.5 | 37.4 ± 0.1 | 7.4 ± 0 |
All values are means ± SE. Data were collected for PaCO2, PaO2, MAP, temperature, and pH measured at baseline and 90 min. Animals were treated with either LPS (100 μg/kg) or saline and with sAIH only, A2A antagonist MSX-3 + sAIH, A2A agonist CGS-21680, or aCSF (MSX-3 and CGS-21680 vehicle) with and without sAIH. There were no significant differences in any group at baseline vs. 90 min.
Hypoxic episodes.
PaCO2 was regulated within ∼ 2 mmHg of its baseline value in all groups (Table 2). The sAIH target range was between 25 and 30 mmHg (Nichols et al. 2012); the only group that was not consistently in this range was sAIH with aCSF during hypoxic episode 1 (HX1); the average exposure value in this group was 35 mmHg (Table 2). MAP did not differ between groups, but MAP was significantly lower at sAIH vs. time control values. Temperature did not differ between groups, but temperature was significantly lower at sAIH + MSX-3 + saline vs. sAIH + MSX-3 + LPS at hypoxic episode 3 (HX3). pH did not differ between groups (Table 2).
Table 2.
PaCO2, PaO2, MAP, temperature, and pH during hypoxic episodes
| Experimental Groups | PaCO2, mmHg | PaO2, mmHg | MAP, mmHg | Temperature, °C | pH |
|---|---|---|---|---|---|
| Saline + sAIH | |||||
| HX1 | 45.5 ± 1.5 | 28.1 ± 1.1* | 64.0 ± 7.8* | 36.9 ± 0.0 | 7.4 ± 0 |
| HX2 | 46.3 ± 1.3 | 26.3 ± 0.9* | 51.0 ± 5.8* | 36.9 ± 0.1 | 7.4 ± 0 |
| HX3 | 45.7 ± 1.4 | 27.0 ± 0.7* | 51.8 ± 6.8* | 36.9 ± 0.1 | 7.4 ± 0 |
| LPS + sAIH | |||||
| HX1 | 44.9 ± 1.7 | 28.0 ± 1.4* | 81.9.5 ± 10.4* | 37.3 ± 0.1 | 7.4 ± 0 |
| HX2 | 47.4 ± 1.7 | 26.0 ± 0.7* | 65.5 ± 7.7* | 37.3 ± 0.1 | 7.4 ± 0 |
| HX3 | 44.1 ± 1.8 | 26.2 ± 0.4* | 57.0 ± 9.2* | 37.2 ± 0.1 | 7.4 ± 0 |
| Time control | |||||
| HX1 | 49.6 ± 0.7 | 285.0 ± 16.5 | 110.8 ± 6.8 | 37.0 ± 0.1 | 7.4 ± 0 |
| HX2 | 48.7 ± 0.8 | 281.8 ± 17.6 | 112.1 ± 5.3 | 37.0 ± 0.1 | 7.4 ± 0 |
| HX3 | 49.0 ± 0.9 | 277.3 ± 16.5 | 112.7 ± 5.8 | 37.2 ± 0.1 | 7.4 ± 0 |
| Saline + MSX-3 + sAIH | |||||
| HX1 | 45.8 ± 1.1 | 31.6 ± 1.6* | 42.3 ± 6.6 | 37.4 ± 0.1 | 7.4 ± 0 |
| HX2 | 45.0 ± 1.5 | 29.5 ± 0.7* | 60.9 ± 16.3 | 37.3 ± 0.1 | 7.4 ± 0 |
| HX3 | 44.7 ± 1.6 | 29.4 ± 0.7* | 45.2 ± 8.1 | 37.3 ± 0.1† | 7.4 ± 0 |
| LPS + MSX-3 + sAIH | |||||
| HX1 | 48.4 ± 1.7 | 30.7 ± 0.5* | 46.4 ± 9.9 | 37.1 ± 0.1 | 7.4 ± 0 |
| HX2 | 46.5 ± 1.9 | 30.1 ± 1.0* | 46.4 ± 10.1 | 37.0 ± 0.1 | 7.4 ± 0 |
| HX3 | 46.6 ± 1.6 | 29.5 ± 1.1* | 40.3 ± 5.1 | 36.9 ± 0.1† | 7.4 ± 0 |
| aCSF + sAIH | |||||
| HX1 | 47.1 ± 0.9 | 35.5 ± 2.5* | 69.11 ± 11.4 | 37.4 ± 0.1 | 7.4 ± 0 |
| HX2 | 47.6 ± 1.4 | 29.7 ± 1.5* | 52.8 ± 5.4 | 37.2 ± 0.1 | 7.4 ± 0 |
| HX3 | 47.4 ± 1.5 | 30.1 ± 1.3* | 54.0 ± 11.0 | 37.2 ± 0.1 | 7.4 ± 0 |
All values are means ± SE. HX1, HX2, and HX3 are the severe intermittent hypoxia (sAIH) episodes, and data were collected at each episode for PaCO2, PaO2, MAP, temperature, and pH. All experimental groups were pretreated with either saline or LPS, and after 24 h, the groups were treated with sAIH, normoxia (time control), A2A receptor antagonist MSX-3, or vehicle aCSF.
P ≤ 0.05 indicates a significant difference for saline + sAIH or LPS + sAIH vs. time control (O2 and MAP). †P ≤ 0.05 indicates a significant difference for saline + sAIH + MSX-3 vs. LPS + sAIH + MSX-3 (temperature, HX3).
Progressive Augmentation of the Short-Term Hypoxic Response During sAIH
Progressive augmentation of the short-term hypoxic response during sAIH (Nichols et al. 2012) is defined as an increase in the magnitude of the short-term phrenic response from the initial episode to the final episode of the intermittent hypoxia protocol (Powell et al. 1998). In all groups treated with sAIH, progressive augmentation was apparent because the average hypoxic phrenic response was increased in hypoxic episode 2 (HX2) and HX3 vs. HX1 (Fig. 2); one mitigating concerning factor is that there was a tendency toward more severe hypoxemia in HX2 and HX3 vs. HX1 (Table 2). This apparent progressive augmentation was independent of whether the animals had been pretreated with LPS or saline, or if they had had intrathecal drug pretreatments.
Fig. 2.
Change in the integrated phrenic burst activity within hypoxic episodes (i.e., the short-term hypoxic phrenic response) 24 h post-LPS (100 μg/kg). The numbers on the X-axis represent the hypoxic episode (1–3) within the severe AIH (sAIH) protocol. A: LPS + sAIH and saline + sAIH groups: progressive augmentation of the short-term hypoxic phrenic response between hypoxic episodes was unaffected by LPS (black bars) vs. saline (light gray bars). There were no changes in time controls. B: LPS + MSX-3 + sAIH (black bars), saline + MSX-3 + sAIH (light gray bars), and aCSF + sAIH groups (dark gray bars): LPS had no significant effect on short-term hypoxic responses. (LPS + aCSF + sAIH and saline + aCSF + sAIH groups were combined because the groups were not significantly different from each other). Intrathecal MSX-3 also did not affect progressive augmentation of the hypoxic phrenic response. Values are means ± SE. #P ≤ 0.05 indicates a significant difference from episode 1; %P ≤ 0.05 indicates a significant difference from episode 2; $P ≤ 0.05 indicates a significant difference from episode 3.
sAIH-Induced pLTF is Unaffected By LPS-Induced Inflammation
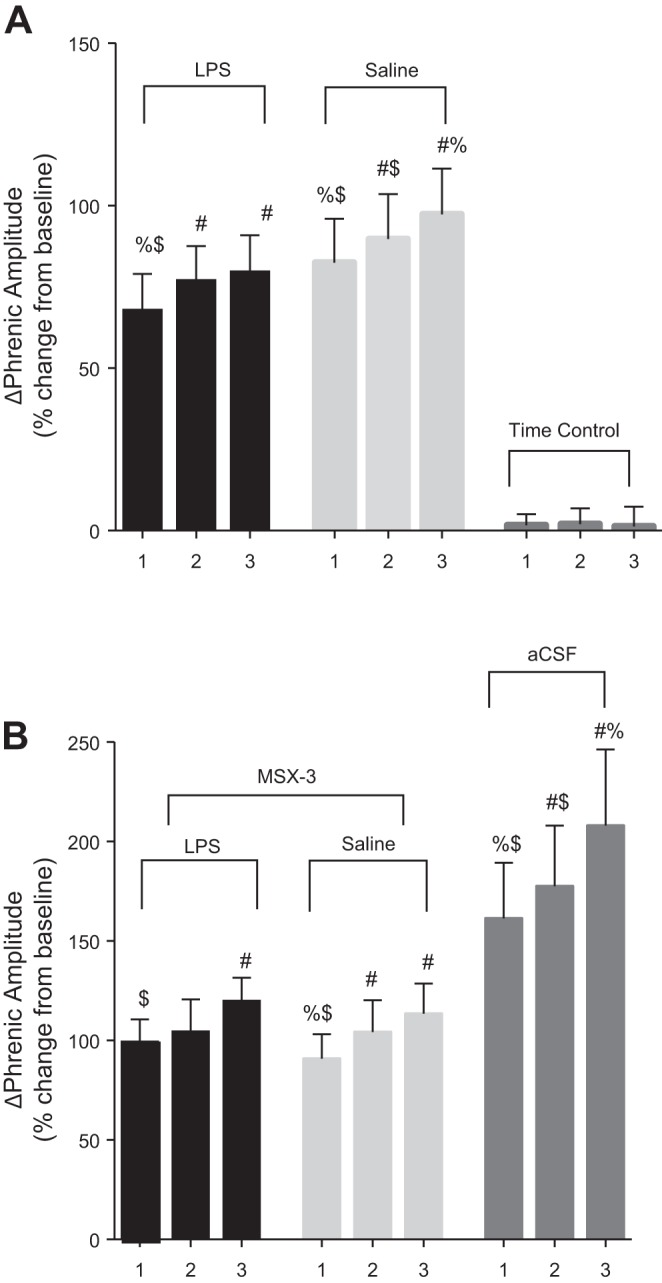
Systemic LPS inhibits mAIH-induced pLTF (35–55 mmHg) (Huxtable et al. 2013; Vinit et al. 2011). We have now demonstrated that sAIH-induced pLTF is not affected 24 h post-LPS. Following LPS and saline treatments, pLTF was significant compared with baseline 90 min post-sAIH (LPS: 53 ± 6%, n = 8 and saline: 59 ± 9%, n = 9; P ≤ 0.05; Fig. 3, A, B, and D). Time control experiments without AIH did not exhibit pLTF (4 ± 2%, n = 8; Fig. 3, C and D).
Fig. 3.

LPS-induced systemic inflammation (100 μg/kg; 24 h prior) does not affect pLTF elicited by sAIH. A–C: representative traces of compressed integrated phrenic neurograms during sAIH protocol. A: LPS treatment exposed to sAIH shows increased amplitude from baseline (BL). B: saline treatment exposed to sAIH shows increased amplitude from BL. C: time control animals without sAIH do not exhibit pLTF. D: group data for phrenic amplitude as a %change from BL. LPS + sAIH (circles; n = 8) and saline + sAIH (squares; n = 8) are compared with time control protocols (triangles; n = 8). After sAIH, there was a rapid increase from BL values and the increase persisted for 90 min. There was no difference between LPS + sAIH vs. saline + sAIH at any time (BL, 15, 30, 60, or 90 min; P > 0.05). #P > 0.05 indicates a significant difference from time control. E: group data for phrenic burst frequency (bursts/min). LPS + sAIH and saline + sAIH are compared with time controls. The LPS + sAIH group was significantly different from time control only at 30 min and significantly different from saline + sAIH at 15 and 60 min. The saline + sAIH group was significantly different from time control at 15, 30, and 60 min. #P > 0.05 indicates a significant difference from time control. %P > 0.05 indicates a significant difference from LPS + sAIH.
LPS had no significant effects on frequency pLTF following sAIH (LPS: 3 ± 1%, n = 8 vs. saline: 3 ± 2%, n = 8; P ≥ 0.05; Fig. 3E). Time controls without AIH did not exhibit changes in frequency at any time (1 ± 1%, n = 8; P ≥ 0.05; Fig. 3E).
After LPS, sAIH-Induced pLTF is Still A2A Receptor Dependent
Severe AIH elicits pLTF by an A2A receptor-dependent mechanism, and sAIH-induced pLTF is blocked by pretreatment of A2A receptor antagonist MSX-3 (Golder et al. 2008; Nichols et al. 2012). We confirmed that this same A2A receptor-dependent mechanism underlies persistent sAIH-induced pLTF following LPS by pretreating LPS-treated rats with MSX-3 before sAIH (Fig. 4). At 90 min post-sAIH in LPS and saline treatment groups, pLTF was significantly reduced compared with no MSX-3 pretreatment (LPS + MSX-3 + sAIH: 23 ± 8%, n = 6 and saline + MSX-3 + sAIH: 14 ± 4%, n = 7 vs. aCSF + sAIH: 106 ± 13%, n = 7; P ≤ 0.05; Fig. 4, A–D). Since there was no significant difference between the saline + aCSF + sAIH vs. LPS + aCSF + sAIH groups, they were combined into a single group, the aCSF + sAIH group mentioned above.
Fig. 4.

pLTF following sAIH remains A2A receptor dependent following LPS. A–C: representative traces of compressed integrated phrenic neurograms. A: LPS treatment + intrathecal MSX-3 + sAIH (5 min after MSX-3 administration); there was no increase in amplitude vs. BL. B: saline treatment + intrathecal MSX-3 + sAIH (5 min after MSX-3 administration); there was no increase in amplitude vs. BL. C: MSX-3 vehicle intrathecal administration (20% DMSO, 80% aCSF) and sAIH (5 min after vehicle administration); there was a marked increase in amplitude from BL. D: group data for phrenic amplitude as a %change from BL. LPS + MSX-3 + sAIH (circles; n = 6) and saline + MSX-3 + sAIH (squares; n = 7) are compared with aCSF + sAIH (triangles; n = 7). After sAIH, burst amplitude was increased from BL, but the increase did not persist after 30 min; there were significant differences for aCSF + sAIH vs. LPS + MSX-3 + sAIH (%P < 0.05) and saline + MSX-3 + sAIH ($P < 0.05) at 30, 60, and 90 min post-sAIH. There was no significant difference between LPS + MSX-3 + sAIH and saline + MSX-3 + sAIH at any time. E: group data for phrenic burst frequency (bursts/min). LPS + MSX-3 + sAIH and saline + MSX-3 + sAIH are compared with aCSF + sAIH. There were significant differences for aCSF + sAIH vs. saline + MSX-3 + sAIH ($P < 0.05) at 60 and 90 min.
Systemic LPS had no significant effects on frequency at any time (LPS: 1 ± 1%, n = 6 vs. saline: −1 ± 2%, n = 7; P > 0.05; Fig. 4E). In the combined aCSF + sAIH group, there was a significantly greater frequency at 60 and 90 min post-sAIH compared with either MSX-3-treated group (4 ± 1%, n = 7; P ≤ 0.05; Fig. 4E).
LPS Has No Effect on A2A Agonist-induced pMF
Spinal CGS-21680 elicits pMF, and this effect is blocked by the A2A antagonist MSX-3 (Golder et al. 2008). We tested if pMF elicited by CGS-21680 was blocked after LPS treatment (Fig. 5). LPS had no effect on pMF elicited by CGS-21680 vs. saline + CGS-21680 (LPS: 68 ± 9%, n = 6 vs. saline: 68 ± 6%, n = 6; P ≥ 0.05; Fig. 5, A, B, and D). Time controls show no pMF compared with CGS-21680-treated groups (6 ± 4%, n = 6; P ≤ 0.05; Fig. 5, C and D).
Fig. 5.

A2A receptor agonist-induced pMF is unaffected by LPS. A–C: representative traces of compressed integrated phrenic neurograms following intrathecal CGS-21680. A: LPS treatment and intrathecal CGS-21680 shows increases in burst amplitude vs. BL. B: saline treatment and intrathecal CGS-21680 shows increased amplitude vs. BL. C: the time control group given intrathecal vehicle, aCSF, did not show any time-dependent increase in phrenic burst amplitude. D: group data for phrenic amplitude as a %change from BL. LPS + CGS-21680 (circles; n = 6) and saline + CGS-21680 (squares; n = 6) are compared with aCSF-treated rats (triangle; n = 10). After intrathecal CGS-21680, pMF persisted at least 90 min. There was a significant difference from time control (#P < 0.05) for LPS + CGS-21680 and saline + CGS-21680 vs. aCSF at 30, 60, and 90 min. There was no significant difference between LPS + CGS-21680 and saline + CGS-21680 at any time. E: group data for phrenic burst frequency (bursts/min). LPS + CGS-21680 and saline + CGS-21680 are compared with aCSF (time control) protocols. The LPS + CGS-21680 group was significantly different from time control (#P < 0.05) at 90 min. The saline + CGS-21680 group was significantly different from time Control (#P < 0.05) at 30 min.
CGS-21680 did not elicit frequency pMF at any time, and LPS had no significant effect on this outcome (LPS: 3 ± 1%, n = 6 vs. saline: 2 ± 0%, n = 6; P ≥ 0.05; Fig. 5E). Time controls that did not receive CGS-21680 did not exhibit frequency changes at any time (1 ± 0%, n = 6; P ≥ 0.05; Fig. 5E).
DISCUSSION
We demonstrated that LPS (24 h post) does not affect adenosine-dependent phrenic motor facilitation elicited by severe AIH (S pathway) or spinal drug-induced A2A receptor activation. This observation is in direct contrast to the impact of the same mild LPS dose on serotonin-dependent pLTF following moderate AIH (Q pathway) (Huxtable et al. 2013; Vinit et al. 2011). Because of differences in the impact of mild inflammation on these distinct mechanisms of phrenic motor plasticity, the adenosine-dependent pathway may serve as a “backup” when the serotonin-dependent pathway can no longer be induced.
A2A receptors are Gs protein-coupled metabotropic receptors that elicit the S pathway to pMF (Dale-Nagle et al. 2010a). The S pathway can be elicited by severe AIH (Nichols et al. 2012) and direct spinal injection of A2A (Golder et al. 2008) or 5-HT7 receptor agonists (Hoffman and Mitchell 2011), or exchange protein activated by cyclic AMP activators (Fields et al. 2015). During severe AIH, we postulated that greater ATP release and extracellular adenosine accumulation (Gourine et al. 2005; Martin et al. 2007; Phillis et al. 1993) shifts the balance from the serotonin-dependent Q pathway predominant with moderate AIH toward reliance on the adenosine-dependent S pathway (Nichols et al. 2012). Once initiated, the S pathway is BDNF synthesis independent and relies on new synthesis of an immature TrkB isoform (Golder et al. 2008). The S pathway requires EPAC (Fields et al. 2015), Akt (Golder et al. 2008; Hoffman et al. 2010), and mTOR signaling (Dougherty et al. 2015); in contrast, the Q pathway requires ERK (Hoffman et al. 2012) and PKCθ (Devinney et al. 2015), but not mTOR signaling (Dougherty et al. 2015). Thus the Q and S pathways to pMF are completely distinct yet result in phenotypically similar pMF. In contrast to the inflammation insensitivity of the adenosine-induced S pathway, even a low LPS dose (delivered 3 or 24 h prior) abolishes the Q pathway-dependent pLTF elicited by moderate AIH (Huxtable et al. 2013; Vinit et al. 2011).
LPS-Induced Inflammation
Although LPS is a common model to investigate how inflammation affects the central nervous system (CNS), it does not cross the blood-brain barrier (Hoogland et al. 2015; Ming et al. 2015). Nevertheless, we (and others) have documented that LPS at the same dose, route, and timing of administration elicits spinal inflammation (Huxtable et al. 2013). Peripheral LPS-TLR4 signaling can elicit CNS inflammation because of circulating cytokines (which do cross the blood-brain barrier) or vagal feedback, which is known to elicit inflammatory CNS responses (Blatteis and Li 2000; Goehler et al. 1999; Laflamme et al. 1999; Maier et al. 1998; Rivest 2001; Schnydrig et al. 2007). Three hours after LPS administration, spinal homogenates exhibited transient increases in inflammatory gene expression; however, by 24 h after LPS, these changes were no longer significant (Huxtable et al. 2013). Despite the lack of persistent neuroinflammation, pLTF elicited by moderate AIH was completely abolished, and this effect could be reversed by high doses of the nonsteroidal anti-inflammatory drug ketoprofen (Huxtable et al. 2015). Despite the same dose, route, and timing of LPS administration, severe AIH-induced pLTF (Fig. 3) and pMF elicited by spinal A2A receptor activation (Fig. 5) were unaffected. We speculate that in situations where mild inflammation abolishes the Q pathway to pMF, the S pathway remains a viable backup mechanism.
AIH-induced frequency LTF is small and variable in this experimental preparation (Baker-Herman and Mitchell 2008). Even the small changes in frequency following severe AIH were unaffected by LPS, similar to phrenic burst amplitude pLTF (Fig. 2E). Changes in frequency with LPS and saline controls following severe AIH are consistent with our previous report (Nichols et al. 2012).
The A2A receptor antagonist MSX-3 blocks pLTF elicited by severe AIH (Nichols et al. 2012) and pMF elicited by intrathecal A2A agonist injections (Golder et al. 2008). Since intrathecal MSX-3 blocked severe AIH-induced pLTF with and without LPS, there is no evidence that an alternate mechanism elicited pLTF after LPS pretreatment. This conclusion is consistent with our finding that A2A agonist-induced pMF was still blocked by MSX-3 after LPS administration.
Conclusion
We conclude that mild systemic inflammation has no effect on adenosine-dependent phrenic motor facilitation, regardless of whether pMF was elicited by sAIH or intrathecal A2A receptor activation. Thus, in contrast to the Q pathway, the S pathway to pMF is insensitive to systemic inflammation and may represent an alternative means to elicit phrenic motor plasticity when primary mechanisms are suppressed. A detailed understanding of how inflammation affects distinct forms of respiratory motor plasticity is needed to develop pLTF and pMF for therapeutic advantage in devastating clinical disorders that compromise breathing capacity, such as spinal injury and/or motor neuron disease (ALS).
GRANTS
Support was provided by National Heart, Lung, and Blood Institute (NHLBI) Grants HL111598 and HL69064. I. M. Agosto-Marlin was supported by a supplement to NHLBI Grant HL111598. N. L. Nichols was supported by Francis Families Foundation and NHLBI Grant K99/R00 HL119606.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.M.A.-M., N.L.N., and G.S.M. conceived and designed research; I.M.A.-M. and N.L.N. performed experiments; I.M.A.-M. analyzed data; I.M.A.-M., N.L.N., and G.S.M. interpreted results of experiments; I.M.A.-M. prepared figures; I.M.A.-M. drafted manuscript; I.M.A.-M., N.L.N., and G.S.M. edited and revised manuscript; I.M.A.-M., N.L.N., and G.S.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Bradley Wathen for expert technical assistance.
Present address of I. M. Agosto-Marlin: Center for Integrative Brain Research, Seattle Children's Research Institute, Seattle, WA 98101.
Present address of N. L. Nichols: Department of Biomedical Sciences, University of Missouri, Columbia, MO 65211.
Present address of G. S. Mitchell: Center for Respiratory Research and Rehabilitation, Dept. of Physical Therapy and McKnight Brain Institute, University of Florida, Gainesville, FL 32610.
REFERENCES
- Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol 23: R774–R788, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol 15: 542–548, 2005. [DOI] [PubMed] [Google Scholar]
- Andero R, Choi D, Ressler K. BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog Mol Biol Transl Sci 122: 169–192, 2014. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol 104: 251–260, 1996. [DOI] [PubMed] [Google Scholar]
- Baker-Herman T, Fuller D, Bavis R, Zabka A, Golder F, Doperalski N, Johnson R, Watters J, Mitchell G. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci 7: 48–55, 2004. [DOI] [PubMed] [Google Scholar]
- Baker-Herman T, Mitchell G. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol 162: 8–17, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci 22: 6239–6246, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatteis CM, Li S. Pyrogenic signaling via vagal afferents: what stimulates their receptors? Auton Neurosci 85: 66–71, 2000. [DOI] [PubMed] [Google Scholar]
- Castellucci M, Rossato M, Calzetti F, Tamassia N, Zeminian S, Cassatella MA, Bazzoni F. IL-10 disrupts the Brd4-docking sites to inhibit LPS-induced CXCL8 and TNF-alpha expression in monocytes: implications for chronic obstructive pulmonary disease. J Allergy Clin Immunol 136: 781–791, 2015. [DOI] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol 669: 225–230, 2010a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Satriotomo I, Lovett-Barr MR, Vinit S, Mitchell GS. Spinal plasticity following intermittent hypoxia: implications for spinal injury. Ann NY Acad Sci 1198: 252–259, 2010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinney MJ, Fields DP, Huxtable AG, Peterson TJ, Dale EA, Mitchell GS. Phrenic long-term facilitation requires PKCtheta activity within phrenic motor neurons. J Neurosci 35: 8107–8117, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty BJ, Fields DP, Mitchell GS. Mammalian target of rapamycin is required for phrenic long-term facilitation following severe but not moderate acute intermittent hypoxia. J Neurophysiol 114: 1784–1791, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields DP, Springborn SR, Mitchell GS. Spinal 5-HT7 receptors induce phrenic motor facilitation via EPAC-mTORC1 signaling. J Neurophysiol 114: 2015–2022, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RP, Nguyen KT, Lee JE, Tilders FJ, Maier SF, Watkins LR. Interleukin-1beta in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? J Neurosci 19: 2799–2806, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Mitchell GS. Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J Neurosci 25: 2925–2932, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci 28: 2033–2042, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Llaudet E, Dale N, Spyer KM. Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci 25: 1211–1218, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper RM, Kumar R, Macey PM, Woo MA, Ogren JA. Affective brain areas and sleep-disordered breathing. Prog Brain Res 209: 275–293, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Golder FJ, Mahamed S, Mitchell GS. Spinal adenosine A2A receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J Physiol 588: 255–266, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J Physiol 589: 1397–1407, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Nichols NL, Macfarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J Appl Physiol 113: 1184–1193, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12: 114, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Peterson TJ, Watters JJ, Mitchell GS. Intermittent hypoxia-induced spinal inflammation impairs respiratory motor plasticity by a spinal p38 MAP kinase-dependent mechanism. J Neurosci 35: 6871–6880, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Vinit S, Watters JJ, Mitchell GS. Systemic LPS induces spinal inflammatory gene expression and impairs phrenic long-term facilitation following acute intermittent hypoxia. J Appl Physiol 114: 879–887, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Vinit S, Windelborn JA, Crader SM, Guenther CH, Watters JJ, Mitchell GS. Systemic inflammation impairs respiratory chemoreflexes and plasticity. Respir Physiol Neurobiol 178: 482–489, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-κB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci 19: 10923–10930, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 42: 145–151, 2008. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience 178: 45–55, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, Goehler LE, Fleshner M, Watkins LR. The role of the vagus nerve in cytokine-to-brain communication. Ann NY Acad Sci 840: 289–300, 1998. [DOI] [PubMed] [Google Scholar]
- Martin ED, Fernandez M, Perea G, Pascual O, Haydon PG, Araque A, Cena V. Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55: 36–45, 2007. [DOI] [PubMed] [Google Scholar]
- Min SS, Quan HY, Ma J, Han JS, Jeon BH, Seol GH. Chronic brain inflammation impairs two forms of long-term potentiation in the rat hippocampal CA1 area. Neurosci Lett 456: 20–24, 2009. [DOI] [PubMed] [Google Scholar]
- Ming Z, Sawicki G, Bekar LK. Acute systemic LPS-mediated inflammation induces lasting changes in mouse cortical neuromodulation and behavior. Neurosci Lett 590C: 96–100, 2015. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Dale EA, Mitchell GS. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol 112: 1678–1688, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillis JW, O'Regan MH, Perkins LM. Adenosine 5′-triphosphate release from the normoxic and hypoxic in vivo rat cerebral cortex. Neurosci Lett 151: 94–96, 1993. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol 112: 123–134, 1998. [DOI] [PubMed] [Google Scholar]
- Rivest S. How circulating cytokines trigger the neural circuits that control the hypothalamic-pituitary-adrenal axis. Psychoneuroendocrinology 26: 761–788, 2001. [DOI] [PubMed] [Google Scholar]
- Schnydrig S, Korner L, Landweer S, Ernst B, Walker G, Otten U, Kunz D. Peripheral lipopolysaccharide administration transiently affects expression of brain-derived neurotrophic factor, corticotropin and proopiomelanocortin in mouse brain. Neurosci Lett 429: 69–73, 2007. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Triantafilou K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol 23: 301–304, 2002. [DOI] [PubMed] [Google Scholar]
- Vinit S, Windelborn JA, Mitchell GS. Lipopolysaccharide attenuates phrenic long-term facilitation following acute intermittent hypoxia. Respir Physiol Neurobiol 176: 130–135, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol 17: 255–281, 1999. [DOI] [PubMed] [Google Scholar]
- Xing T, Fong AY, Bautista TG, Pilowsky PM. Acute intermittent hypoxia induced neural plasticity in respiratory motor control. Clin Exp Pharmacol Physiol 40: 602–609, 2013. [DOI] [PubMed] [Google Scholar]