

Abstract
C-Met is a receptor tyrosine kinase with multiple functions throughout embryonic development, organogenesis and wound healing and is expressed in various epithelia. The ligand of c-Met is Hepatocyte Growth Factor (HGF) which is secreted among others by mesenchymal stroma/stem (MSC) cells.
Physiological c-Met functions are centred around processes that underly cellular motility and invasive growth. Aberrant c-Met expression and activity is observed in numerous cancers and makes major contributions to cell malignancy. Importantly, HGF/c-Met signaling is crucial in the context of communication between cancer cells and the the tumor stroma.
Here, we review recent findings on roles of dysregulated c-Met in urogenital tumors such as cancers of the urinary bladder, prostate, and ovary. We put emphasis on novel aspects of cancer-associated c-Met expression regulation on both, HGF-dependent and HGF-independent non-canonical mechanisms. Moreover, this review focusses on c-Met-triggered signalling with potential relevance for urogenital oncogenesis, and on strategies to specifically inhibit c-Met activity.
Keywords: c-Met; HGF/SF signalling; c-Met inhibitors; MSC; Bladder, prostate and ovarian cancer
Background
c-Met (mesenchymal epithelial transition factor) is a multifunctional transmembrane tyrosine kinase and acts as a receptor for hepatocyte growth factor/Scatter factor (HGF/SF) [1]. It is expressed in various epithelial tissues (liver, pancreas, prostate, kidney, muscle, bone-marrow) during embryogenesis [2] and is also found on the cell surface of numerous tumorous cell populations. Shortly after its discovery, multiple oncogenetic properties of c-Met were described, including the stimulation of cell dissociation, migration, motility, and invasion of extracellular matrix [3–6]. Formation of mature c-Met is achieved by proteolytic cleavage of a precursor in a post-Golgi compartment, resulting in a small alpha and large beta polypeptide which then associate into a heterodimer. A disulfide bridge connects the small alpha unit and the extracellular segment of the membrane spanning beta subunit [7]. The extracellular part of the beta subunit is composed of an N-terminal sematophorin (sema) domain (essential for receptor activation) followed by a cysteine-rich portion (plexin sematophorin domain) and four IPT (immunoglobulin like plexins transcription factor) domains. A transmembrane helix connects the extracellular domain of c-Met to its intracellular section which can be divided into a juxtamembrane domain, a tyrosine kinase domain and the C-terminal region [2] (Figs. 1 and 2).
Fig. 1.
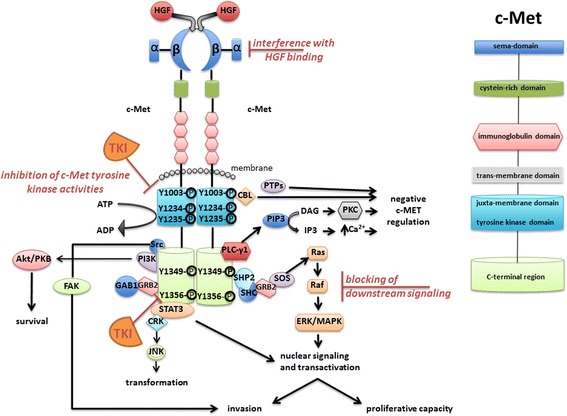
HGF/SF-mediated activation of c-Met and relayed downstream signalling. The c-Met receptor can be structured into distinct domains, including sema, cysteine-rich, immunoglobulin, trans-membrane, juxta-membrane, tyrosine kinase, and C-terminal region. Pharmacological intervention with activated c-Met signalling includes: (i) competitive interference with HGF/c-Met interaction, (ii) inhibition of the tyrosine kinase activity of c-Met with the use of tyrosine kinase inhibitors (TKI), or (iii) blocking of activated c-Met downstream signaling mediators. Accordingly, cell fate and development such as survival, transformation, cell motility, and proliferative capacity can be affected. This figure was adapted from Organ and Tsao, 2011 [2]
Fig. 2.
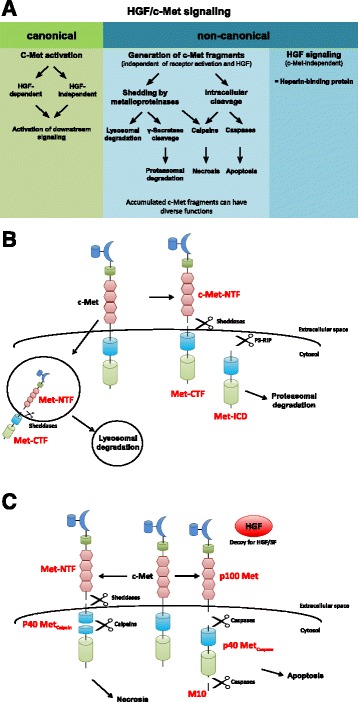
Pathways of c-Met signaling. a Overview of HGF/c-Met signaling via the canonical and non-canonical pathway. Canonical or “classical” HGF/Met signaling involves ligand-dependent and independent receptor activation which leads to the induction of downstream signaling cascades (left). Non-canonical HGF/c-Met signaling is independent of receptor activation. Generation of c-Met receptor fragments takes place under various cellular conditions such as apoptotic and necrotic stimuli as well as in the context of specific physiological circumstances. HGF is also able to exert signals independently of c-Met, e.g. upon interactions triggered by its heparin-binding domain. b Generation of c-Met fragments via shedding and cleavage by γ-secretase: Sheddases or metalloproteinases cleave full-length c-Met within its extracellular domain, resulting in different in a soluble extracellular N-terminal fragment (Met-NTF) and a membrane-associated C-terminal fragment (Met-CTF). Met-CTF can be further processed by the γ-secretase complex by presenilin-dependent intramembrane proteolysis (PS-RIP) into an intracellular domain (Met-ICD) which is routed to proteasomal degradation. Full-length membranous c-Met can also be internalized and cleaved by sheddases giving rise to Met-NTF and Met-CTF. These intracellularly generated c-Met fragments undergo lysosomal instead of proteasomal degradation. c Origin of c-Met fragments through intracellular cleavage by caspases and calpains: In response to apoptotic stimuli, c-Met is cleaved at two distinct sites in the intracellular domain by activated caspase-3, resulting in membrane-anchored p100 Met, a 40 kDa cytosolic p40 Met fragment and a small peptide (M10). Under necrotic conditions, c-Met is cleaved by metalloproteinases and further processed by calcium-independent proteases (calpains) instead of γ-secretase. The resulting product p40 Metcalpain differs from p40 Metcaspase by a few amino acid residues and is not able to promote apoptosis
Structure and function of c-Met
C-Met becomes activated through homo-dimerisation upon binding of its ligand HGF [8–12]. As a result of ligand-induced dimerization, the intracellular tyrosine kinase domains of the two receptor beta-subunits trans-phosphorylate each other at residues Tyr1234 and Tyr1235 within the catalytic loops [13]. This event fully unleashes the tyrosine kinases which subsequently catalyze further phosphorylation of the receptor chain at Tyr1003 in the juxtamembrane domain and Tyr1349/Tyr1356 within the C-terminal region (Fig. 1). These modifications relay further signals by generating specific contact interfaces (pTyr-containing motifs) with a variety of different src homology domain 2 (SH2) containing adaptor molecules to eventually regulate induction of cell motility, proliferation, invasiveness, and tubular morphogenesis [14–22].
C-Met receptor downstream signaling is mediated by recruitment of distinct adaptor and effector proteins such as src-homology-2- (SH2-), src-homology-3- (SH3-) domain-containing proteins and phospho-tyrosine-binding proteins (PTB) (Fig. 1). These adaptor proteins without enzymatic activity include Grb2 (growth factor receptor-bound protein 2), CRK ((CT10 (chicken tumor virus number 10) regulator of kinase)), and GAB1 (GRB2-associated binding protein 1). C-Met-binding proteins with enzymatic activity include Src kinases, PI3K (phosphatidylinositol 3-kinase), PLC-γ1 (phospholipase C-γ1), SHIP-2 (SH2 domain-containing 5’ inositol phosphatase), STAT-3 (signal transducer and activator of transcription), SOS (Ras guanine nucleotide exchange factor son-of-seven), PTPs (protein tyrosine phosphatases), e.g. SHP2 (SH2 domain-containing phosphatase-2) [14, 16–23]. HGF-stimulated c-Met activation can also phosphorylate downstream targets such as p44/42 MAP kinase (ERK) to mediate enhanced proliferation and cell cycle progression [2, 24, 25] (Fig. 1). In addition, Met can be activated even in the absence of HGF. In this context, Fan et al. described a novel HGF-indepedent mechanism in which non-receptor tyrosine kinase FER is involved. The upregulation of FER in ovarian cancers could trigger a signaling cascade downstream of c-Met in a HGF independent manner. Mechanistically, FER was shown to phosphorylate the tyrosine residue of the C-terminal SH2 domain docking site of c-Met, thus maintaining recruitment of signaling molecules such as GAB1 and Grb2 to the receptor. Most importantly, FER-mediated downstream signalling by c-Met could not be inhibited by a c-Met inhibitor alone [26].
Intra- and intercellular signaling triggered by c-Met in urogenital cancers
Activated c-Met in normal cells – predominantly in stromal cells including MSC - plays a major role in organogenesis and wound healing whereas in tumor cells, HGF-activated c-Met contributes to invasive and metastatic behavior [12, 27]. Various urogenital cancers like muscular-invasive urinary bladder cancer, prostate cancer and ovarian cancer express c-Met [28–30]. c-Met can be detected in about 70% of the different types of ovarian cancers such as low and high grade serous, endometrioid, clear cell and mucinous carcinomas, in about 30% of the tumors c-Met is expressed to a noticeably high degree [31–33]. C-Met over-expression is observed particularly in hyperactive tumors which also show elevated angiogenesis and neo-vascularization due to activity of vascular endothelial growth factor receptor-2 (VEGF-R2). These tumor properties correlate with poor prognosis [34–36].
In prostate cancer, it was shown that androgen deprivation is connected with a more aggressive phenotype and leads to an increased c-Met expression. In parallel, the androgen receptor, which is known as a negative regulator of c-Met, is downregulated [37–39]. Hence, c-Met signaling obviously has an important role in maintaining survival as well as proliferation in androgen receptor-independent prostate cancer cells [40]. In this context, evidence has been obtained for an association of c-Met activation with decreased expression of genes related to DNA repair. It was, thus, suggested that c-Met may contribute to the accumulation of DNA damage and mutations and consequently favor progression of castration-resistant prostate cancer [40].
A recent report shows that c-Met upregulation in castration-resistant prostate cancer tissue is associated with a different distribution pattern compared to biopsies from naïve prostate cancer tissue. Interestingly, a c-Met fragment lacking the c-terminal part of full-length membranous c-Met was found to accumulate in the nuclei of tumor cells preferentially in castration-resistant prostate cancer [41]. These results support the notion of a role for full-length and processed c-Met in the progression of androgen-independent prostate cancer.
Elevated c-Met expression in tumor cells also leads to enhanced c-Met activity within the tumor tissue, since profound HGF expression within the tumor stroma activates c-Met on cancer cells in a paracrine manner. HGF expression was shown to be clearly increased in urogenital cancers such as urinary bladder, prostate and ovarian cancer [42–45]. In prostate and bladder cancer, c-Met-related feedback activation crosstalk between carcinoma cells and tumor-associated fibroblasts was characterized: HGF secretion becomes upregulated in stromal fibroblasts co-cultured with tumor cells, which in turn enhances migratory and invasive properties of cancer cells [44, 46–51]. However, in normal ovary HGF is mainly detected in the ovarian surface epithelium on the outer surface of the ovary and to a lesser extend in ovarian stromal cells directly adjacent to the ovarian surface epithelium [45]. Moreover, an elevated HGF expression is detectable in epithelial cell components of ovarian tumors and in MSC suggesting an autocrine and/or paracrine stimulation of ovarian tumor growth including tumor cell protection [45, 52–54]. In other cancer entities such as gastric and colon cancer, HGF/c-Met signaling was shown to be operative in fibroblasts within the tumor microenvironment, thereby contributing to tumor progression [55, 56]. Moreover, immunohistochemical analysis showed c-Met expression on myofibroblasts mainly in the invasive area of lung adenocarcinoma [57]. c-Met expression on myofibroblasts in patients with small-sized lung adenocarcinoma was correlated with shortened patient survival [57]. It will be important to address in how far HGF-mediated fibroblast activation does also play a role in urogenital cancers.
Non-canonical signaling by c-Met
It is now well established that receptor tyrosine kinases can also act via signaling pathways other than the Ras/Raf cascade. “Canonical signaling” is currently used as a term for the signaling route extending from ligand binding to the cell surface exodomains of receptors resulting in dimerization/oligomerisation and receptor trans-/ autophosphorylation with subsequent intracellular signal release. Recently, “non-canonical signaling” has emerged as a concept to describe the specific generation of defined intracellular tyrosine kinase receptor fragments which have different functions [58]. The formation of these fragments is achieved by the activity of different proteases whose functions can be dependent or independent of receptor stimulation by ligand (Fig. 2).
Four cleavage products of c-Met with potential functions in signaling have as yet been described. The extracellular N-terminal fragment (Met-NTF) can be eliminated from the full-length receptor by metalloproteinases such as ADAM-10 and ADAM-17. Subsequent processing of the membrane-proximal C-terminal fragment (Met-CTF) by presenilin-dependent, regulated, intramembrane proteolysis (PS-RIP) results in an intracellular c-Met fragment (Met-ICD) which becomes subject to proteasomal degradation [59–62]. It is controversal as to whether the extracellular soluble domain of c-Met (NTF) exerts the physiological function of sequestering and, thus, antagonizing HGF/SF and is also a potent antagonist to the antagonist [63–65]. However, in mouse xenograft experiments with injected UOK261 bladder carcinoma cells, which show a high proteolytic cleavage rate of the full-length receptor, a linear relationship between soluble c-Met in the plasma or urine and total tumor volume was shown [63, 64]. Moreover, the level of soluble c-Met in urine from bladder cancer patients was shown to be a disease marker and to even allow for distinction between tumor stages as muscle no-invasive and invasive cancers [66]. These findings suggest that cleavage of c-Met plays a role in urinary bladder cancer progression.
By shedding the Met-NTF, the resulting fragments Met-NTF and-CTF can also be lysosomally degraded. Ancot et al. published a model describing the fate and subcellular location of the receptor-derived breakdown products post processing of the full-length cell surface receptor by PS-RIP and its proteasomal degradation upon removal of Met-NTF. However, if c-Met is membrane-bound and internalized, it can also undergo intracellular proteolytic cleavage. This process gives rise to Met-NTF and Met-CTF, which can subsequently be degraded lysosomally. Thus, fragment Met-CTF escapes processing by PS-RIP when the cleavage takes place intracellularly [67]. Both shedding-induced degradation pathways are independent of receptor activation and may serve the function of preventing overexpression and "over-activation" of c-Met [60, 61, 67].
Experimental overexpression of a segment comprising only the intracellular segment of c-Met devoid of the extracellular domain in NIH3 Ts fibroblasts showed an elevated oncogenic potential with enhanced proliferation and invasiveness compared to control fibroblasts [68]. Interestingly, constitutively active c-Met fragments were detected in cell nuclei of an aggressive breast carcinoma cell line. They showed trans-activating activity in an experimental Gal4-based transactivation assay and appear to have a function in connection with spontaneous migratory behaviour of this cell line [69]. However, the absence of tumor suppressor Wwox plays a role here. Wwox is believed to possess regulatory functions in both controlling the stability of full-length c-Met and in the inhibition of the transactivation function of nuclear Met-CTF [69]. As already mentioned above, the nuclear Met-ICD level is higher in castration-resistant than in naïve prostate cancer. Withdrawal of androgens upregulates nuclear Met-ICD which in turn activates transcription factor SOX9 and the β-catenin/ androgen receptor signaling pathway. In combination, this promotes cell transformation and three-dimensional tumor growth in castration-resistant prostate cancer [41]. These results suggest that proteolytically generated c-Met fragments that are not lysosomally or proteasomally degraded and thus, accumulate in the cell which can contribute to tumor progression. However, as yet little is known about the precise physiological or pathological mode of action and function of these fragments.
c-Met cleavage products are functional in the control of cell death and survival [70]. The receptor can be cleaved intracellularly at two distinct sites in response to stress stimuli. The resulting 40 kD cytosolic p40Met fragment, in contrast to the ligand-activated full-length receptor, acts pro-apoptotically via the mitochondrial permeabilization process of the intrinsic apoptotic pathway. Caspase-3 was identified as the responsible protease for the cleavage of c-Met [71–73].
Cleavage of c-Met yields the intracellular p40Met fragment, a membrane-bound p100Met fragment and, in addition, a peptide ten amino acid residues in length designated M10 and corresponding to C-terminal end of the receptor protein [74, 75]. The p100Met fragment encompassing the entire extracellular domain, is still membrane-bound and can act as a scavenger of HGF/SF, thereby serving as an inhibitor of the HGF/c-MET signal cascade [74]. M10 can block TGF-β-induced SMAD2 phosphorylation by interaction with SMAD2 and, thus, exerts antifibrotic effects [75].
The ratio of full-length c-Met and caspase-3 appears to have an influence on the extent of apoptosis in hepatocytes: High abundance of c-Met can suppress caspase-3 activity and, thus, suppress apoptosis. Under conditions of a strong apoptotic stimulus, however, this inhibitory effect of c-Met on caspase-3 is overcome and cell death is no longer blocked [76].
Remarkably, caspase-3 is also active in non-apoptotic melanoma and glioblastoma cells as well as in invasive bladder cancer promoting tumor progression [77–79]. Mukai et al. published that cancer cells are able to react on apoptotic stimuli with a higher invasiveness through caspase activation [80]. Until now there is no evidence for a role of caspase-3 mediated cleavage of c-Met in this context. Moreover, c-Met also becomes cleaved in necrosis [81] whereby c-Met can be shed by metalloproteases as discussed above and instead of further processing by the γ-secretase complex, it becomes degraded by calcium-dependent proteases (calpains). This process gives rise to a 40 kDa Met fragment designated p40Met calpain which differs from the previously described p40Met caspase fragment in a few amino acids and its inability to promote apoptosis [81]. Moreover, the mutation R970C found in lung cancer within the juxtamembrane domain of c-Met promotes the calpain-dependent formation of a novel 45 kDa fragment p45Met, particularly in confluent cells [82]. Overexpression of p45Met in epithelial cells enhances HGF-mediated cell scattering and invasion without induction of tyrosine kinase activity of p45Met. This indicates an alternative mechanism besides activation of cell scattering by full length receptor [82]. In addition, ectopic expression of R970C variant of Met in the suspension cell line Ba/F3 did not show any transforming capacity, supporting that HGF treatment, proteolytic cleavage and cell confluence are probably necessary for cancer-related effects of this variant [83].
Analysis of Met-overexpressing tumor samples from patients with non-small cell lung cancer demonstrated the existence of all Met fragments suggesting a variety of cellular conditions within lung tumors [81]. However, little is known about the role of the different c-MET fragments in tumor progression so far.
Interestingly, not only c-Met can contribute to tumor progression in a ligand-independent manner. Tate et al. indicated a role for c-Met ligand HGF in prostate cancer in the absence of its receptor [84]. HGF induces cell adhesion and migration in a c-Met-negative prostate cancer cell line on the basis of a low-affinity interaction between its heparin-binding domain and nucleolin. Nucleolin is a nuclear protein which is known to localize also to the surface of cells. In this setting, it may link HGF and integrin function [84]. Low or high concentrations of HGF have been observed to exert distinct functions also on non-tumorigenic cells, leading to the postulation that HGF can elicit cellular effects in a c-Met-independent manner in parallel with or alternatively to “classical” c-Met/HGF signaling [85, 86].
Experimental and pharmacological interference with c-Met activity in urogenital cancer cells
The identification of prominent c-Met expression in certain urogenital tumors suggested that the HGF/SF – c-Met axis may serve as an attractive target to be included into chemotherapeutic regimens. Different approaches have been taken towards molecularly targeting c-Met and HGF such as (i) competitive interference with HGF/c-Met interaction, (ii) inhibition of the tyrosine kinase activity of c-Met, or (iii) blocking of activated c-Met downstream signaling mediators (Fig. 1). Accordingly, several preclinical studies indicated that inhibition of c-Met represents a promising therapeutic strategy paralleled by findings that c-Met overexpression has prognostic value in urogenital cancers, particularly in ovarian cancer.
Using an ovarian cancer mouse model, the orally available c-Met inhibitor PF-2341066 was successfully applied to reduce cell proliferation, adhesion and invasion as well as to induce apoptosis [87]. This small-molecule inhibitor shows specificity against c-Met and anaplastic lymphoma kinase (ALK) and reduced mouse tumor size paralleled by increased animal survival [88].
Another c-Met inhibitor, Crizotinib (PF-02341066), represents a designed tyrosine kinase inhibitor based on the core structure of the c-Met-inhibitor PHA 665752 in which some functional groups of the originating molecule (PHA 665752) were altered. As a result Crizotinib has stronger affinity for the ATP-binding pocket of the kinase domain within the c-Met receptor. Consequently, binding of ATP to the c-Met kinase domain and subsequent tyrosine phosphorylation of the receptor and signal mediators are blocked [89, 90]. Moreover, Crizotinib displays further inhibitory potential for the tyrosine kinase receptor ALK and ROS1 (ROS proto-oncogene 1, receptor tyrosine kinase) [91]. Crizotinib has, hence, been approved for the treatment of ALK-positive non-small cell lung carcinoma in the United States since August 2011. Studies employing a combination of Crizitonib together with PF-05212384 or gedatolisib were performed to additionally inhibit the phosphatidyl inositol-3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway in the tumor cells. Results demonstrated superior anti-tumor effects of this combination compared to the individual agents. However, this combined treatment was also associated with the development of a gedatolisib-resistant subline, indicating increased expression of the multi-drug-resistant-1 gene [92].
The orally administrable tyrosine kinase inhibitor Foretinib (XL880, GSK1363089) exhibits a certain specificity and high affinity binding for both c-Met (IC50: 0.4 nM) and vascular epithelial growth factor receptor-2 (VEGF-R2/KDR) (IC50: 0.9 nM) [93]. This drug can also bind to a variety of other growth factor receptors, however, with significantly reduced binding affinities: PDGFRα (IC50: 3,6 nM), PDGFRβ (IC50: 9.6 nM), RON (IC50: 3 nM), c-KIT (IC50: 6,7 nM), FLT-3 (IC50: 3,6 nM) und TIE-2 (IC50: 1,1 nM) and low affinities to FGFR1 (IC50: 660 nM), und EGFR (IC50: 2,9 μM) [93–95]. An association of Foretinib with receptor tyrosine kinases is accompanied by a conformational change within the kinase structure and deprivation of enzymatic activity. Notably, Foretinib binding to its target tyrosine kinases receptors remains stable for at least 24 h [95].
Foretinib has also been demonstrated to block MAP kinase signaling and cell cycle progression during in vitro studies, substantiating this drug as a multi-target tyrosine kinase inhibitor [96]. Promising in vivo studies with significant Foretinib-mediated reduction of tumor size were performed in NSCLC [91], in lung metastases [93], and in Hedgehog medulloblastoma [97].
Potential effects of Foretinib as a multi-target tyrosine kinase inhibitor besides its specificity for c-Met are also focused on inhibition of VEGF-R2 signaling as an important pathway during angiogenesis and subsequent neo-vascularization. Here, Foretinib exhibits properties which primarily target the tumor microenvironment, thereby displaying dual anti-tumorigenic strategies. Promising effects of Foretinib in targeting VEGF-R2 signaling were achieved by an approximately 10-fold reduction in tumor size of tumors of the rare and aggressive small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), following tumor initiation by the two model cell lines SCCOHT-1 and BIN-67, respectively. MSC within the tumor microenvironment were shown to contribute to this outcome [52, 53, 67, 98, 99]. Supportive evidence of Foretinib’s anti-tumor activity through inhibition of c-Met- and VEGFR-2-mediated signal transduction was also demonstrated in further tumor models e.g. hepatocellular carcinoma [100], kidney cell carcinoma [101] and gastric carcinoma [102].
The tumor microenvironment plays an pivotal role for therapeutic approaches [103–105]. The importance simultaneously targeting tumor cells and the tumor microenvironment of urogenital cancers by anti-cancer drugs in is also underscored by results with DCC-2701 as a c-Met/TIE-2/VEGF-R inhibitor [106]. In the course of stroma–cancer cell interactions, human ovarian fibroblasts and MSC produced and secreted HGF, leading to elevated growth and migration of ovarian cancer cells coinciding with c-Met phosphorylation at Tyr1349. Tumor treatment with DCC-2701 was able to efficiently reduce the tumor burden in vivo by inhibition of c-Met phosphorylation and c-Met-mediated signaling, for cell growth and migration [106].
Conclusions
In summary, the abundance of c-Met expression and intracellular signaling in urogenital cancers may provide a selective molecular target for tumor therapeutic interventions. Moreover, multi-target c-Met tyrosine kinase inhibitors with the property to simultaneously affect cancer cells and the tumor microenvironment represent efficient and focused drugs to target urogenital cancers. In this context, it is important to note that cancer growth and metastasis of c-Met-positive urogenital cancers involves further stimuli besides HGF-binding and subsequent c-Met signaling since no anti-tumor effect was detectable in an ovarian cancer clinical trial after using a humanized IgG2 antibody directed against HGF (AMG-102), even though AMG-102 prevents HGF binding to c-Met and subsequent c-Met activation [107]. It is likely that the above-mentioned and not entirely understood non-canonical c-Met signaling mechanisms as well as c-Met-independent HGF activity are involved in this phenomenon.
Acknowledgements
The authors declare no financial, personal, or professional conflict of interest. All authors have critically read and approved this work. YY is supported by a postdoctoral fellowship from the DAAD and CSC. This work was supported by a grant from the Erich und Gertrud Roggenbuck-Stiftung for Cancer Research to RH.
Funding
This work was supported by a grant from the Erich und Gertrud Roggenbuck-Stiftung for Cancer Research to RH.
Availability of data and materials
Not applicable.
Author’s contibutions
KF and RH drafted the manuscript. YY and SJ contributed literature search, detailed structure of the manuscript, and design of the figures. All authors critically read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
All authors have critically read and approved this work.
Ethical approval and consent to participate
Not applicable.
Abbreviations
- c-Met
Mesenchymal epithelial transition factor
- CRK
CT10 (chicken tumor virus number 10) regulator of kinase
- GAB1
Grb2-associated binding protein 1
- Grb2
Growth factor receptor-bound protein 2
- HGF
Hepatocyte growth factor
- IPT
Immunoglobulin-like plexins transcription factor
- MAPK
Mitogen activated protein kinase
- MSC
Mesenchymal stroma/stem cells
- mTOR
Mammalian target of rapamycin
- NFκB
Nuclear factor κB
- PI3K
Phosphatidylinositol 3-kinase
- PLC-γ1
Phospholipase C- γ1
- PTB
Phospho-tyrosine-binding proteins
- PTPs
Protein tyrosine phosphatases
- semaphoring
Sema
- SF
Scatter factor
- SH2
Src homology domain-2
- SH3
Src homology-3 domain
- SHIP-2
SH2 domain-containing 5’ inositol phosphatase
- SHP2
SH2 domain-containing phosphatase-2
- SOS
Ras guanine nucleotide exchange factor son-of-seven
- STAT-3
Signal transducer and activator of transcription
- VEGF-R2
Vascular endothelial growth factor receptor-2
Contributor Information
Ralf Hass, Email: hass.ralf@mh-hannover.de.
Susanne Jennek, Email: Susanne.Jennek@med.uni-jena.de.
Yuanyuan Yang, Email: kate0000yang@gmail.com.
Karlheinz Friedrich, Phone: +49 3641 9396405, Email: KarlHeinz.Friedrich@med.uni-jena.de.
References
- 1.Rubin JS, Bottaro DP, Aaronson SA. Hepatocyte growth factor/scatter factor and its receptor, the c-met proto-oncogene product. Biochim Biophys Acta. 1993;1155:357–71. doi: 10.1016/0304-419x(93)90015-5. [DOI] [PubMed] [Google Scholar]
- 2.Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7–19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rong S, Segal S, Anver M, Resau JH, Vande Woude GF. Invasiveness and metastasis of NIH 3 T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc Natl Acad Sci U S A. 1994;91:4731–5. doi: 10.1073/pnas.91.11.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeffers M, Rong S, Vande Woude GF. Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis. J Mol Med (Berl) 1996;74:505–13. doi: 10.1007/BF00204976. [DOI] [PubMed] [Google Scholar]
- 5.Sachs M, Weidner KM, Brinkmann V, Walther I, Obermeier A, Ullrich A, Birchmeier W. Motogenic and morphogenic activity of epithelial receptor tyrosine kinases. J Cell Biol. 1996;133:1095–107. doi: 10.1083/jcb.133.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otsuka T, Takayama H, Sharp R, Celli G, LaRochelle WJ, Bottaro DP, Ellmore N, Vieira W, Owens JW, Anver M, Merlino G. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998;58:5157–67. [PubMed] [Google Scholar]
- 7.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2:289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–9. doi: 10.1016/0006-291X(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 9.Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–42. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 10.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 11.Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, Narsimhan RP, Hartmann G, Zarnegar R, Michalopoulos GK, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10:2867–78. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues GA, Park M. Autophosphorylation modulates the kinase activity and oncogenic potential of the Met receptor tyrosine kinase. Oncogene. 1994;9:2019–27. [PubMed] [Google Scholar]
- 14.Weidner KM, Sachs M, Birchmeier W. The Met receptor tyrosine kinase transduces motility, proliferation, and morphogenic signals of scatter factor/hepatocyte growth factor in epithelial cells. J Cell Biol. 1993;121:145–54. doi: 10.1083/jcb.121.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graziani A, Gramaglia D, dalla Zonca P, Comoglio PM. Hepatocyte growth factor/scatter factor stimulates the Ras-guanine nucleotide exchanger. J Biol Chem. 1993;268:9165–8. [PubMed] [Google Scholar]
- 16.Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S, Graziani A, Panayotou G, Comoglio PM. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77:261–71. doi: 10.1016/0092-8674(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 17.Boccaccio C, Ando M, Tamagnone L, Bardelli A, Michieli P, Battistini C, Comoglio PM. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–8. doi: 10.1038/34657. [DOI] [PubMed] [Google Scholar]
- 18.Pelicci G, Giordano S, Zhen Z, Salcini AE, Lanfrancone L, Bardelli A, Panayotou G, Waterfield MD, Ponzetto C, Pelicci PG, et al. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene. 1995;10:1631–8. [PubMed] [Google Scholar]
- 19.Fixman ED, Fournier TM, Kamikura DM, Naujokas MA, Park M. Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J Biol Chem. 1996;271:13116–22. doi: 10.1074/jbc.271.22.13116. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Guzman M, Dolfi F, Zeh K, Vuori K. Met-induced JNK activation is mediated by the adapter protein Crk and correlates with the Gab1 - Crk signaling complex formation. Oncogene. 1999;18:7775–86. doi: 10.1038/sj.onc.1203198. [DOI] [PubMed] [Google Scholar]
- 21.Lock LS, Royal I, Naujokas MA, Park M. Identification of an atypical Grb2 carboxyl-terminal SH3 domain binding site in Gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyrosine kinases. J Biol Chem. 2000;275:31536–45. doi: 10.1074/jbc.M003597200. [DOI] [PubMed] [Google Scholar]
- 22.Sakkab D, Lewitzky M, Posern G, Schaeper U, Sachs M, Birchmeier W, Feller SM. Signaling of hepatocyte growth factor/scatter factor (HGF) to the small GTPase Rap1 via the large docking protein Gab1 and the adapter protein CRKL. J Biol Chem. 2000;275:10772–8. doi: 10.1074/jbc.275.15.10772. [DOI] [PubMed] [Google Scholar]
- 23.Matsumoto R, Tsuda M, Wang L, Maishi N, Abe T, Kimura T, Tanino M, Nishihara H, Hida K, Ohba Y, et al. Adaptor protein CRK induces epithelial-mesenchymal transition and metastasis of bladder cancer cells through HGF/c-Met feedback loop. Cancer Sci. 2015;106:709–17. doi: 10.1111/cas.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mhawech-Fauceglia P, Afkhami M, Pejovic T. MET/HGF signaling pathway in ovarian carcinoma: clinical implications and future direction. Patholog Res Int. 2012;2012:960327. doi: 10.1155/2012/960327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blumenschein GR, Jr, Mills GB, Gonzalez-Angulo AM. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J Clin Oncol. 2012;30:3287–96. doi: 10.1200/JCO.2011.40.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan G, Zhang S, Gao Y, Greer PA, Tonks NK. HGF-independent regulation of MET and GAB1 by nonreceptor tyrosine kinase FER potentiates metastasis in ovarian cancer. Genes Dev. 2016;30:1542–57. doi: 10.1101/gad.284166.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumoto K, Nakamura T. Hepatocyte growth factor and the Met system as a mediator of tumor-stromal interactions. Int J Cancer. 2006;119:477–83. doi: 10.1002/ijc.21808. [DOI] [PubMed] [Google Scholar]
- 28.Inui M, Nishi N, Yasumoto A, Takenaka I, Miyanaka H, Matsumoto K, Nakamura T, Wada F. Enhanced gene expression of transforming growth factor-alpha and c-met in rat urinary bladder cancer. Urol Res. 1996;24:55–60. doi: 10.1007/BF00296735. [DOI] [PubMed] [Google Scholar]
- 29.Pisters LL, Troncoso P, Zhau HE, Li W, von Eschenbach AC, Chung LW. c-met proto-oncogene expression in benign and malignant human prostate tissues. J Urol. 1995;154:293–8. doi: 10.1016/S0022-5347(01)67297-5. [DOI] [PubMed] [Google Scholar]
- 30.Cheng HL, Trink B, Tzai TS, Liu HS, Chan SH, Ho CL, Sidransky D, Chow NH. Overexpression of c-met as a prognostic indicator for transitional cell carcinoma of the urinary bladder: a comparison with p53 nuclear accumulation. J Clin Oncol. 2002;20:1544–50. doi: 10.1200/JCO.20.6.1544. [DOI] [PubMed] [Google Scholar]
- 31.Di Renzo MF, Narsimhan RP, Olivero M, Bretti S, Giordano S, Medico E, Gaglia P, Zara P, Comoglio PM. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene. 1991;6:1997–2003. [PubMed] [Google Scholar]
- 32.Huntsman D, Resau JH, Klineberg E, Auersperg N. Comparison of c-met expression in ovarian epithelial tumors and normal epithelia of the female reproductive tract by quantitative laser scan microscopy. Am J Pathol. 1999;155:343–8. doi: 10.1016/S0002-9440(10)65130-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong AS, Roskelley CD, Pelech S, Miller D, Leung PC, Auersperg N. Progressive changes in Met-dependent signaling in a human ovarian surface epithelial model of malignant transformation. Exp Cell Res. 2004;299:248–56. doi: 10.1016/j.yexcr.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Parr C, Jiang WG. Expression of hepatocyte growth factor/scatter factor, its activator, inhibitors and the c-Met receptor in human cancer cells. Int J Oncol. 2001;19:857–63. [PubMed] [Google Scholar]
- 35.Yamashita Y, Akatsuka S, Shinjo K, Yatabe Y, Kobayashi H, Seko H, Kajiyama H, Kikkawa F, Takahashi T, Toyokuni S. Met is the most frequently amplified gene in endometriosis-associated ovarian clear cell adenocarcinoma and correlates with worsened prognosis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0057724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang C, Jardim DL, Hong D. MET in ovarian cancer: metastasis and resistance? Cell Cycle. 2014;13:1220–1. doi: 10.4161/cc.28515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey PA, Zhu X, Zarnegar R, Swanson PE, Ratliff TL, Vollmer RT, Day ML. Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am J Pathol. 1995;147:386–96. [PMC free article] [PubMed] [Google Scholar]
- 38.Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res. 2007;67:967–75. doi: 10.1158/0008-5472.CAN-06-3552. [DOI] [PubMed] [Google Scholar]
- 39.Liu T, Mendes DE, Berkman CE. From AR to c-Met: androgen deprivation leads to a signaling pathway switch in prostate cancer cells. Int J Oncol. 2013;43:1125–30. doi: 10.3892/ijo.2013.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda A, Nakashiro K, Hara S, Sasaki T, Miwa Y, Tanji N, Yokoyama M, Hamakawa H, Oyasu R. Inactivation of AR activates HGF/c-Met system in human prostatic carcinoma cells. Biochem Biophys Res Commun. 2006;347:1158–65. doi: 10.1016/j.bbrc.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 41.Xie Y, Lu W, Liu S, Yang Q, Carver BS, Li E, Wang Y, Fazli L, Gleave M, Chen Z. Crosstalk between nuclear MET and SOX9/beta-catenin correlates with castration-resistant prostate cancer. Mol Endocrinol. 2014;28:1629–39. doi: 10.1210/me.2014-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joseph A, Weiss GH, Jin L, Fuchs A, Chowdhury S, O'Shaugnessy P, Goldberg ID, Rosen EM. Expression of scatter factor in human bladder carcinoma. J Natl Cancer Inst. 1995;87:372–7. doi: 10.1093/jnci/87.5.372. [DOI] [PubMed] [Google Scholar]
- 43.Rosen EM, Joseph A, Jin L, Yao Y, Chau MH, Fuchs A, Gomella L, Hastings H, Goldberg ID, Weiss GH. Urinary and tissue levels of scatter factor in transitional cell carcinoma of bladder. J Urol. 1997;157:72–8. doi: 10.1016/S0022-5347(01)65286-8. [DOI] [PubMed] [Google Scholar]
- 44.Zhu X, Humphrey PA. Overexpression and regulation of expression of scatter factor/hepatocyte growth factor in prostatic carcinoma. Urology. 2000;56:1071–4. doi: 10.1016/S0090-4295(00)00795-0. [DOI] [PubMed] [Google Scholar]
- 45.Parrott JA, Skinner MK. Expression and action of hepatocyte growth factor in human and bovine normal ovarian surface epithelium and ovarian cancer. Biol Reprod. 2000;62:491–500. doi: 10.1095/biolreprod62.3.491. [DOI] [PubMed] [Google Scholar]
- 46.Nishimura K, Kitamura M, Miura H, Nonomura N, Takada S, Takahara S, Matsumoto K, Nakamura T, Matsumiya K. Prostate stromal cell-derived hepatocyte growth factor induces invasion of prostate cancer cell line DU145 through tumor-stromal interaction. Prostate. 1999;41:145–53. doi: 10.1002/(SICI)1097-0045(19991101)41:3<145::AID-PROS1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 47.Nishimura K, Kitamura M, Takada S, Nonomura N, Tsujimura A, Matsumiya K, Miki T, Matsumoto K, Okuyama A. Regulation of invasive potential of human prostate cancer cell lines by hepatocyte growth factor. Int J Urol. 1998;5:276–81. doi: 10.1111/j.1442-2042.1998.tb00603.x. [DOI] [PubMed] [Google Scholar]
- 48.Tamatani T, Hattori K, Iyer A, Tamatani K, Oyasu R. Hepatocyte growth factor is an invasion/migration factor of rat urothelial carcinoma cells in vitro. Carcinogenesis. 1999;20:957–62. doi: 10.1093/carcin/20.6.957. [DOI] [PubMed] [Google Scholar]
- 49.Wang P, Nishitani MA, Tanimoto S, Kishimoto T, Fukumori T, Takahashi M, Kanayama HO. Bladder cancer cell invasion is enhanced by cross-talk with fibroblasts through hepatocyte growth factor. Urology. 2007;69:780–4. doi: 10.1016/j.urology.2007.01.063. [DOI] [PubMed] [Google Scholar]
- 50.Grimm S, Jennek S, Singh R, Enkelmann A, Junker K, Rippaus N, Berndt A, Friedrich K. Malignancy of bladder cancer cells is enhanced by tumor-associated fibroblasts through a multifaceted cytokine-chemokine loop. Exp Cell Res. 2015;335:1–11. [DOI] [PubMed]
- 51.Lee YH, Apolo AB, Agarwal PK, Bottaro DP. Characterization of HGF/Met signaling in cell lines derived from urothelial carcinoma of the bladder. Cancers (Basel) 2014;6:2313–29. doi: 10.3390/cancers6042313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otte A, Rauprich F, von der Ohe J, Yang Y, Kommoss F, Feuerhake F, Hillemanns P, Hass R. c-Met inhibitors attenuate tumor growth of small cell hypercalcemic ovarian carcinoma (SCCOHT) populations. Oncotarget. 2015;6:31640–58. doi: 10.18632/oncotarget.5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otte A, Yang Y, von der Ohe J, Melzer C, Hillemanns P, Feuerhake F, Hass R. SCCOHT tumors acquire chemoresistance and protection by interacting mesenchymal stroma/stem cells within the tumor microenvironment. Int J Oncol. 2016;49:2453–63. doi: 10.3892/ijo.2016.3735. [DOI] [PubMed] [Google Scholar]
- 54.Madrigal M, Rao KS, Riordan NH. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med. 2014;12:260. doi: 10.1186/s12967-014-0260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu X, Chen X, Zhou Q, Li P, Yu B, Li J, Qu Y, Yan J, Yu Y, Yan M, et al. Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer Lett. 2013;335:128–35. doi: 10.1016/j.canlet.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Li F, Zhu YT. HGF-activated colonic fibroblasts mediates carcinogenesis of colonic epithelial cancer cells via PKC-cMET-ERK1/2-COX-2 signaling. Cell Signal. 2015;27:860–6. doi: 10.1016/j.cellsig.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 57.Tokunou M, Niki T, Eguchi K, Iba S, Tsuda H, Yamada T, Matsuno Y, Kondo H, Saitoh Y, Imamura H, Hirohashi S. c-MET expression in myofibroblasts: role in autocrine activation and prognostic significance in lung adenocarcinoma. Am J Pathol. 2001;158:1451–63. doi: 10.1016/S0002-9440(10)64096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen MK, Hung MC. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015;282:3693–721. doi: 10.1111/febs.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galvani AP, Cristiani C, Carpinelli P, Landonio A, Bertolero F. Suramin modulates cellular levels of hepatocyte growth factor receptor by inducing shedding of a soluble form. Biochem Pharmacol. 1995;50:959–66. doi: 10.1016/0006-2952(95)00219-P. [DOI] [PubMed] [Google Scholar]
- 60.Jeffers M, Taylor GA, Weidner KM, Omura S, Vande Woude GF. Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol Cell Biol. 1997;17:799–808. doi: 10.1128/MCB.17.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Foveau B, Ancot F, Leroy C, Petrelli A, Reiss K, Vingtdeux V, Giordano S, Fafeur V, Tulasne D. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol Biol Cell. 2009;20:2495–507. doi: 10.1091/mbc.E08-09-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schirrmeister W, Gnad T, Wex T, Higashiyama S, Wolke C, Naumann M, Lendeckel U. Ectodomain shedding of E-cadherin and c-Met is induced by Helicobacter pylori infection. Exp Cell Res. 2009;315:3500–8. doi: 10.1016/j.yexcr.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 63.Wajih N, Walter J, Sane DC. Vascular origin of a soluble truncated form of the hepatocyte growth factor receptor (c-met) Circ Res. 2002;90:46–52. doi: 10.1161/hh0102.102756. [DOI] [PubMed] [Google Scholar]
- 64.Athauda G, Giubellino A, Coleman JA, Horak C, Steeg PS, Lee MJ, Trepel J, Wimberly J, Sun J, Coxon A, et al. c-Met ectodomain shedding rate correlates with malignant potential. Clin Cancer Res. 2006;12:4154–62. doi: 10.1158/1078-0432.CCR-06-0250. [DOI] [PubMed] [Google Scholar]
- 65.Petrelli A, Circosta P, Granziero L, Mazzone M, Pisacane A, Fenoglio S, Comoglio PM, Giordano S. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc Natl Acad Sci U S A. 2006;103:5090–5. doi: 10.1073/pnas.0508156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McNeil BK, Sorbellini M, Grubb RL, 3rd, Apolo A, Cecchi F, Athauda G, Cohen B, Giubellino A, Simpson H, Agarwal PK, et al. Preliminary evaluation of urinary soluble Met as a biomarker for urothelial carcinoma of the bladder. J Transl Med. 2014;12:199. doi: 10.1186/1479-5876-12-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ancot F, Leroy C, Muharram G, Lefebvre J, Vicogne J, Lemiere A, Kherrouche Z, Foveau B, Pourtier A, Melnyk O, et al. Shedding-generated Met receptor fragments can be routed to either the proteasomal or the lysosomal degradation pathway. Traffic. 2012;13:1261–72. doi: 10.1111/j.1600-0854.2012.01384.x. [DOI] [PubMed] [Google Scholar]
- 68.Merlin S, Pietronave S, Locarno D, Valente G, Follenzi A, Prat M. Deletion of the ectodomain unleashes the transforming, invasive, and tumorigenic potential of the MET oncogene. Cancer Sci. 2009;100:633–8. doi: 10.1111/j.1349-7006.2008.01079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matteucci E, Bendinelli P, Desiderio MA. Nuclear localization of active HGF receptor Met in aggressive MDA-MB231 breast carcinoma cells. Carcinogenesis. 2009;30:937–45. doi: 10.1093/carcin/bgp080. [DOI] [PubMed] [Google Scholar]
- 70.Tulasne D, Foveau B. The shadow of death on the MET tyrosine kinase receptor. Cell Death Differ. 2008;15:427–34. doi: 10.1038/sj.cdd.4402229. [DOI] [PubMed] [Google Scholar]
- 71.Tulasne D, Deheuninck J, Lourenco FC, Lamballe F, Ji Z, Leroy C, Puchois E, Moumen A, Maina F, Mehlen P, Fafeur V. Proapoptotic function of the MET tyrosine kinase receptor through caspase cleavage. Mol Cell Biol. 2004;24:10328–39. doi: 10.1128/MCB.24.23.10328-10339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Foveau B, Leroy C, Ancot F, Deheuninck J, Ji Z, Fafeur V, Tulasne D. Amplification of apoptosis through sequential caspase cleavage of the MET tyrosine kinase receptor. Cell Death Differ. 2007;14:752–64. doi: 10.1038/sj.cdd.4402080. [DOI] [PubMed] [Google Scholar]
- 73.Lefebvre J, Muharram G, Leroy C, Kherrouche Z, Montagne R, Ichim G, Tauszig-Delamasure S, Chotteau-Lelievre A, Brenner C, Mehlen P, Tulasne D. Caspase-generated fragment of the Met receptor favors apoptosis via the intrinsic pathway independently of its tyrosine kinase activity. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deheuninck J, Foveau B, Goormachtigh G, Leroy C, Ji Z, Tulasne D, Fafeur V. Caspase cleavage of the MET receptor generates an HGF interfering fragment. Biochem Biophys Res Commun. 2008;367:573–7. doi: 10.1016/j.bbrc.2007.12.177. [DOI] [PubMed] [Google Scholar]
- 75.Atanelishvili I, Shirai Y, Akter T, Buckner T, Noguchi A, Silver RM, Bogatkevich GS. M10, a caspase cleavage product of the hepatocyte growth factor receptor, interacts with Smad2 and demonstrates antifibrotic properties in vitro and in vivo. Transl Res. 2016;170:99–111. doi: 10.1016/j.trsl.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma J, Zou C, Guo L, Seneviratne DS, Tan X, Kwon YK, An J, Bowser R, DeFrances MC, Zarnegar R. Novel Death Defying Domain in Met entraps the active site of caspase-3 and blocks apoptosis in hepatocytes. Hepatology. 2014;59:2010–21. doi: 10.1002/hep.26769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burton PB, Anderson CJ, Corbishly CM. Caspase 3 and p27 as predictors of invasive bladder cancer. N Engl J Med. 2000;343:1418–20. doi: 10.1056/NEJM200011093431915. [DOI] [PubMed] [Google Scholar]
- 78.Gdynia G, Grund K, Eckert A, Bock BC, Funke B, Macher-Goeppinger S, Sieber S, Herold-Mende C, Wiestler B, Wiestler OD, Roth W. Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol Cancer Res. 2007;5:1232–40. doi: 10.1158/1541-7786.MCR-07-0343. [DOI] [PubMed] [Google Scholar]
- 79.Liu YR, Sun B, Zhao XL, Gu Q, Liu ZY, Dong XY, Che N, Mo J. Basal caspase-3 activity promotes migration, invasion, and vasculogenic mimicry formation of melanoma cells. Melanoma Res. 2013;23:243–53. doi: 10.1097/CMR.0b013e32835f28d8. [DOI] [PubMed] [Google Scholar]
- 80.Mukai M, Kusama T, Hamanaka Y, Koga T, Endo H, Tatsuta M, Inoue M. Cross talk between apoptosis and invasion signaling in cancer cells through caspase-3 activation. Cancer Res. 2005;65:9121–5. doi: 10.1158/0008-5472.CAN-04-4344. [DOI] [PubMed] [Google Scholar]
- 81.Montagne R, Berbon M, Doublet L, Debreuck N, Baranzelli A, Drobecq H, Leroy C, Delhem N, Porte H, Copin MC, et al. Necrosis- and apoptosis-related Met cleavages have divergent functional consequences. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Montagne R, Baranzelli A, Muharram G, Catherine L, Lesaffre M, Vinchent A, Kherrouche Z, Werkmeister E, Cortot AB, Tulasne D. MET receptor variant R970C favors calpain-dependent generation of a fragment promoting epithelial cell scattering. Oncotarget. 2017. doi:10.18632/oncotarget.14499. [DOI] [PMC free article] [PubMed]
- 83.Tyner JW, Fletcher LB, Wang EQ, Yang WF, Rutenberg-Schoenberg ML, Beadling C, Mori M, Heinrich MC, Deininger MW, Druker BJ, Loriaux MM. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 2010;70:6233–7. doi: 10.1158/0008-5472.CAN-10-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tate A, Isotani S, Bradley MJ, Sikes RA, Davis R, Chung LW, Edlund M. Met-independent hepatocyte growth factor-mediated regulation of cell adhesion in human prostate cancer cells. BMC Cancer. 2006;6:197. doi: 10.1186/1471-2407-6-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pollack AL, Apodaca G, Mostov KE. Hepatocyte growth factor induces MDCK cell morphogenesis without causing loss of tight junction functional integrity. Am J Physiol Cell Physiol. 2004;286:C482–94. doi: 10.1152/ajpcell.00377.2003. [DOI] [PubMed] [Google Scholar]
- 86.Yamada M, Tatsumi R, Yamanouchi K, Hosoyama T, Shiratsuchi S, Sato A, Mizunoya W, Ikeuchi Y, Furuse M, Allen RE. High concentrations of HGF inhibit skeletal muscle satellite cell proliferation in vitro by inducing expression of myostatin: a possible mechanism for reestablishing satellite cell quiescence in vivo. Am J Physiol Cell Physiol. 2010;298:C465–76. doi: 10.1152/ajpcell.00449.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zillhardt M, Christensen JG, Lengyel E. An orally available small-molecule inhibitor of c-Met, PF-2341066, reduces tumor burden and metastasis in a preclinical model of ovarian cancer metastasis. Neoplasia. 2010;12:1–10. doi: 10.1593/neo.09948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 89.Bang YJ. The potential for crizotinib in non-small cell lung cancer: a perspective review. Ther Adv Med Oncol. 2011;3:279–91. doi: 10.1177/1758834011419002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cui JJ, Tran-Dube M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54:6342–63. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 91.Davare MA, Saborowski A, Eide CA, Tognon C, Smith RL, Elferich J, Agarwal A, Tyner JW, Shinde UP, Lowe SW, Druker BJ. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A. 2013;110:19519–24. doi: 10.1073/pnas.1319583110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Iezzi A, Caiola E, Broggini M. Activity of pan-class I isoform PI3K/mTOR Inhibitor PF-05212384 in combination with crizotinib in ovarian cancer xenografts and PDX. Transl Oncol. 2016;9:458–65. doi: 10.1016/j.tranon.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69:8009–16. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 94.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eder JP, Shapiro GI, Appleman LJ, Zhu AX, Miles D, Keer H, Cancilla B, Chu F, Hitchcock-Bryan S, Sherman L, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16:3507–16. doi: 10.1158/1078-0432.CCR-10-0574. [DOI] [PubMed] [Google Scholar]
- 96.Zillhardt M, Park SM, Romero IL, Sawada K, Montag A, Krausz T, Yamada SD, Peter ME, Lengyel E. Foretinib (GSK1363089), an orally available multikinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. Clin Cancer Res. 2011;17:4042–51. doi: 10.1158/1078-0432.CCR-10-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Faria CC, Golbourn BJ, Dubuc AM, Remke M, Diaz RJ, Agnihotri S, Luck A, Sabha N, Olsen S, Wu X, et al. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res. 2015;75:134–46. doi: 10.1158/0008-5472.CAN-13-3629. [DOI] [PubMed] [Google Scholar]
- 98.Otte A, Rauprich F, Hillemanns P, Park-Simon TW, von der Ohe J, Hass R. In vitro and in vivo therapeutic approach for a small cell carcinoma of the ovary hypercalcaemic type using a SCCOHT-1 cellular model. Orphanet J Rare Dis. 2014;9:126. doi: 10.1186/s13023-014-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang Y, Bucan V, Baehre H, von der Ohe J, Otte A, Hass R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int J Oncol. 2015;47:244–52. doi: 10.3892/ijo.2015.3001. [DOI] [PubMed] [Google Scholar]
- 100.Huynh H, Ong R, Soo KC. Foretinib demonstrates anti-tumor activity and improves overall survival in preclinical models of hepatocellular carcinoma. Angiogenesis. 2012;15:59–70. doi: 10.1007/s10456-011-9243-z. [DOI] [PubMed] [Google Scholar]
- 101.Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31:181–6. doi: 10.1200/JCO.2012.43.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shah MA, Wainberg ZA, Catenacci DV, Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe DC, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8 doi: 10.1371/journal.pone.0054014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mandel K, Yang Y, Schambach A, Glage S, Otte A, Hass R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013;22:3114–27. doi: 10.1089/scd.2013.0249. [DOI] [PubMed] [Google Scholar]
- 104.Yang Y, Otte A, Hass R. Human mesenchymal stroma/stem cells exchange membrane proteins and alter functionality during interaction with different tumor cell lines. Stem Cells Dev. 2015;24:1205–22. doi: 10.1089/scd.2014.0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Melzer C, Yang Y, Hass R. Interaction of MSC with tumor cells. Cell Commun Signal. 2016;14:20. doi: 10.1186/s12964-016-0143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kwon Y, Smith BD, Zhou Y, Kaufman MD, Godwin AK. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene. 2015;34:144–53. doi: 10.1038/onc.2013.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.