

Abstract
Background
The cellular and molecular mechanisms of chronic rejection of transplanted organs remain obscure. It is known however that macrophages play a critical role in the injury and repair of allografts. Among multiple factors influencing macrophage infiltration to allografts, the fractalkine CX3CL1/ CX3CR1 signaling pathway and actin cytoskeleton, which is regulated by a small GTPase RhoA, are of the utmost importance. To define the role of macrophage/RhoA pathway involvement in chronic rejection we generated mice with monocyte/macrophage specific deletion of RhoA.
Methods
Hearts from BALB/c (H-2d) donors were transplanted into RhoAflox/flox (no Cre) and heterozygous Lyz2Cre+/−RhoAflox/flox recipients treated with CTLA4-Ig to inhibit early T cell response. Allografts were assessed for chronic rejection and monocyte/macrophage functions.
Results
The deletion of RhoA inhibited macrophage infiltration, neointimal hyperplasia of vasculature and abrogated chronic rejection of the allografts. The RhoA deletion down-regulated G protein-coupled fractalkine receptor CX3CR1, which activates RhoA pathway and controls monocyte/macrophage trafficking into the vascular endothelium. This in turn promotes, through over-proliferation and differentiation of smooth muscle cells in the arterial walls, neointimal hyperplasia.
Conclusions
Our finding of co-dependence of chronic rejection on monocyte/macrophage CX3CR1/CX3CL1 and RhoA signaling pathways may lead to development of novel anti-chronic rejection therapies.
Keywords: RhoA, chronic rejection, fractalkine, CX3CR1, macrophage, actin
Introduction
The chronic rejection of transplanted organs is the major and still unresolved problem in transplantation. Although phenomenon of chronic rejection, by virtue of its complexity and multifactoriality is still not well understood, it is known that monocytes and macrophages play an important role in this process (1–2). Mammalian organs (including heart) contain various subsets of macrophages with different origin (embryonic yolk sack versus blood or bone marrow-derived) and functions (3–6). The injury or other organ/tissue stress, including transplantation, drives the production of bone marrow-derived monocytes, which via circulation enter target organ/tissue where they differentiate into dendritic cells and macrophages (7). The subset of these monocyte/macrophages, expressing high level of chemokine receptor CX3CR1 is specifically targeted to the endothelium of blood vessel walls, which produce high level of CX3CL1 chemokine (fractalkine; (7–8). Interestingly, one of the hallmarks of chronic rejection is the remodeling (through the over- proliferation and differentiation of smooth muscle cells) of the arterial walls, resulting in neointimal hyperplasia, vessel occlusion and eventual failure of the graft (9,10). The smooth muscle over-proliferation and differentiation during neointimal hyperplasia as well as development of atherosclerotic plaque depend selectively on CX3CR1 expressing monocytes and CX3CR1/CXC3L1 pathway (8, 11–13). The targeted deletion of CX3CR1 inhibits these processes and also prolongs cardiac allograft survival in mouse model system (14). In addition, it has been shown that CX3CR1/CX3CL1 response involves actin polymerization during cell movement (15, 16). The CX3CR1 belongs to the family of G protein-coupled receptors (17), which upon ligand binding undergo conformational changes into the guanine nucleotide exchange factors (GEFs; 18; Fig. 8). The GEFs, in turn, activate RhoA and thus, indirectly, regulate actin polymerization pathway (19). Previous studies on chronic rejection showed that RhoA/ROCK pathway interference (using Y-27632 inhibitor) disrupts actin and actin-related cell functions (20–23), affects macrophages and abrogates chronic rejection of mouse cardiac allografts (24). Recently, we also showed that RhoA deletion causes disregulation of RhoA pathway and changes actin organization in murine peritoneal macrophages (25). All these data prompted us to study how the targeted deletion of RhoA in the monocyte/macrophage lineage affects macrophages and chronic rejection of cardiac allografts in transgenic mouse model system. Here we show that deletion of RhoA down-regulates monocyte/macrophage CX3CR1/CX3CL1 signaling, inhibits macrophage infiltration and vessel occlusion, and abrogates chronic rejection of mouse cardiac allografts.
Figure 8. Hypothetical model of the effect of RhoA, actin and CX3CR1/CX3CL1 interactions on development of chronic rejection of the allograft.

The fractalkine (CX3CL1) is an inflammatory chemokine abundantly produced by vasculature endothelium. Fractalkine receptor (CX3CR1)-expressed on the surface of monocytes/macrophages is a G protein-coupled receptor. It is known that upon binding to fractalkine the CX3CR1 changes conformation and becomes GEF, which activates RhoA. This induces changes in actin polymerization allowing monocyte/macrophages to enter the allograft. Once in the allograft, they mount inflammatory response, neointimal hyperplasia and chronic rejection. Simultaneously, activation of RhoA and proper actin organization/dynamics control CX3CR1 recycling. Correct receptor recycling controls CX3CR1 mRNA and protein expression levels. Upon RhoA deletion (marked by green Δ) all above interactions and signaling pathways and their effects are disrupted (green Δ) leading to inhibition of chronic rejection.
Materials and Methods
Animals, generation of RhoA-deficient mice, and genotyping
Breeding and all experiments were performed according to The Methodist Hospital Research Institute’s animal care and use NIH standards in concordance with the “Guide for the Care and Use of Laboratory Animals” (DHHS publication No. (NIH) 85–23 Revised 1985), the PHS “Policy on Humane Care and Use of Laboratory Animals” and the NIH “Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training.”
RhoAflox/flox mice (gift from dr Richard A. Lang from UC Department of Pediatrics and UC Department of Ophthalmology, Cincinnati Children’s Hospital, Cincinnati, Ohio) were crossbred with B6.129P2-Lyz2tm1(cre)Ifo/J mice (JAX® Mice (Bar Harbor, Maine, USA) to generate Lyz2Cre+/− RhoAflox/flox mice and genotyped as described in Liu et al. (25–29). RhoA deletion in Lyz2Cre+/− RhoAflox/flox mice is based on the principle behind the Lyz Cre system of deletion of floxed genes, and the fact that the activity of Lyz2 promoter is specific for monocyte/macrophage lineage (26).
Heart Transplantations
Heart allografts from BALB/c (H-2d) donors were heterotopically transplanted into Lyz2Cre+/− RhoAflox/flox recipients (each experimental group consisted of 3–5 animals). Recipients received CTLA4-Ig (0.25 mg i.p. on day 2 and day 4 post- transplantation). Control groups consisted of RhoAflox/flox (no Cre) and heterozygous Lyz2Cre+/−RhoA+/flox recipients treated with CTLA4-Ig. Additional control groups (described in chapter 4 of the Result section (dealing with the level of CX3CR1 expression in blood and bone marrow monocytes) included wild type and RhoA-deleted naïve (pre-transplantation, untreated) mice.
Histology, Neointimal index, collagen deposition and immunostaining
The paraffin sections of transplanted hearts were stained with VVG (Verhoeff-Van Gieson) and Trichrome Masson and immunostaining as described in Liu et al. (25). The VVG stained sections were used to calculate neointimal index (vessel occlusion). For comparison of vessel occlusion we inspected 5–8 vessels from each mouse (3 mice per experimental group), and 20 vessels were used to calculate neointimal indexes. Actin, vinculin, transferrin receptor and CX3CR1 immunostaining were performed as in Liu et al (24, 25). The following antibodies against macrophage markers were used: F4/80 (BioLegend, USA or Serotec, USA), Mac-2 (Cedarlane Laboratories, USA). Transferrin Receptor / CD71 was from ThermoFisher Scientific, USA, CX3CR1 (H-70) from Santa Cruz, USA and secondary antibodies were from ThermoFisher Scientific. In immunostained slides, cells were counted in 3–12 independent samples and statistical significance of cell percentage differences was evaluated using student t test. Wild type and RhoA deleted mice (3–5 of each) were sacrificed after 30–50 days post-transplantation. The neointimal index and collagen occupied area in Masson stained samples was calculated (collagen/total area) using Image-pro plus 6.0 as decscribed previously (25).
Apoptosis assay
Fixed heart sections were stained with Tunel- DeadEnd™ Fluorometric TUNEL System (Promega, USA) according to the manufacturer protocol and immunostained with Mac-2 primary antibody and Alexa Fluor 546 conjugated goat anti rat IgG (Life Technologies, USA) secondary antibody.
Peritoneal macrophage apoptosis assay was performed as described in Liu et al. (25)
Monocyte isolation, flow cytometry, RTPCR and BRDU assay
Monocytes were isolated from peripheral blood collected form the mouse tails in the presence of heparin. For bone marrow monocyte isolation, mice were sacrificed and tibia and femur were removed. Bone marrow cells were flushed out bones with DMEM medium. Monocytes were used for flow cytometry with following antibodies (all from BioLegend, USA): anti- Cd45, CD11b, CD115, CX3CR1 and CCR2. Flow cytometry on blood and bone marrow monocytes and peritoneal macrophages and RTPCR were performed as described in Liu et al. (25). Flow cytometry on macrophages isolated from heart samples was performed as follows: Hearts were minced finely and digested with shaking in DMEM containing collagenase II for 30min at 37°C. The digested heart tissue was filtered through 70μM filters and pelleted by centrifugation in DPBS. For flow cytometry viable cells were stained using Zombie Aqua™ (Biolegend) for 30min at 4°C. Nonspecific binding of antibodies was blocked by incubation with CD16, after which cells were labeled with CD45, CD11b, F4/80, CD206, CX3CR1 and CCR2 (all from BioLegend, USA). Antibodies were washed off and the samples were fixed/permeabilized in cyto fix/cytoPerm BD Bioscience). For proliferation assay the BRDU labeled DNA was digested for 1 hour with DNase (Sigma) at 37°C, and then samples were stained with anti-BRDU antibody (BioLegend).
Results
1. Deletion of RhoA inhibits chronic rejection and macrophage infiltration of cardiac allograft
We previously showed that macrophages isolated from Lyz2Cre+/− RhoAflox/flox mice were deficient in expression of RhoA mRNA and its protein (29). Here we performed heterotopic heart transplantations into the Lyz2Cre+/− RhoAflox/flox recipients. We found that treatment of cardiac allograft recipients (RhoAflox/flox (no Cre; further in the text and figures called the wild type), and heterozygous Lyz2Cre+/−RhoAflox/flox (further called the RhoA KO) with CTLA4-Ig (0.25 mg i.p. on day 2 and 4 post- transplantation), which blocks early T cell response, prolonged graft survival to > 50 days (Fig. 1A) but histologically, the heart allografts demonstrated prominent vascular occlusion (Fig. 1B, D) and fibrosis (Fig. 1E, G), which are characteristic for chronic rejection, impaired heart muscle integrity (Fig. 1H), and heavy total and inflammatory macrophages infiltration (Fig. 2A, C, E). In contrast, cardiac allografts from Lyz2Cre+/− RhoAflox/flox recipients treated with CTLA4-Ig showed not only prolonged survival (Fig. 1A) but also the abrogation of chronic rejection with low (statistically significant) vascular occlusion (Fig. 1C, D) and markedly improved heart muscle integrity (Fig. 1I). Histological examination showed that these allografts had reduced, although statistically nonsignificant, collagen deposition (Fig. 1F, G). The immunostaining with total (F4/80) and activated macrophage (Mac2) markers showed that allografts from Lyz2Cre+/− RhoAflox/flox recipients had statistically significant reduction of infiltration with total and activated (inflammatory) macrophages (Fig. 2B, D, E).
Figure 1. Macrophage specific RhoA deletion inhibits chronic rejection.

(A) Comparison of cardiac allograft survival in RhoAflox/flox (no Cre, labeled wild type, WT) and Lyz2Cre+/−RhoAflox/flox (labeled RhoA KO) recipients. Recipients received 0.25 mg of CTLA4-Ig (administered i.p.) on day 2 and day 4 post- transplantation. (B, C) Sections of transplanted hearts at 50 days post-transplantation stained with VVG. (B) Fully occluded vessel from the RhoAflox/flox (no Cre) recipient and (C) un-occluded vessel from the Lyz2Cre+/− RhoAflox/flox recipient. (D). Graph showing the difference in neointimal inedx between hearts from RhoAflox/flox (no Cre) and Lyz2Cre+/−RhoAflox/flox recipients. Y-axis indicates percentage of occluded area. The difference in vessel occlusion (neointima formation) is statistically significant with P value <0.05. The VVG stained sections were used to calculate neointimal index (vessel occlusion). For comparison of vessel occlusion we inspected 5–8 vessels in each mouse, and 20 vessels were used to calculate neointimal indexes. 3 mice from each group were used. (E, F) Sections of transplanted hearts at 50 days post-transplantation stained with trichrome Masson’s stain. Blue color shows collagen deposition. Heart from RhoAflox/flox (no Cre) recipient (E) shows much more collagen deposition than heart from Lyz2Cre+/−RhoAflox/flox recipient (F). (G) Comparison of collagen deposition between hearts from RhoAflox/flox (no Cre) and Lyz2Cre+/−RhoAflox/flox recipients. Although there is a visible difference in the collagen deposition this difference is statistically insignificant with P value > 0.05. 3 mice from each experimental group were used. (H, I) High magnification of tissue fragments from heart sections shown in E and F illustrates dramatic difference in cardiac muscle tissue integrity. (B, C, H, I) Bar is equal to 50 μm. (E, F) Bar is equal to 100 μm.
Figure 2. Macrophage specific RhoA deletion inhibits macrophage infiltration of the allograft.
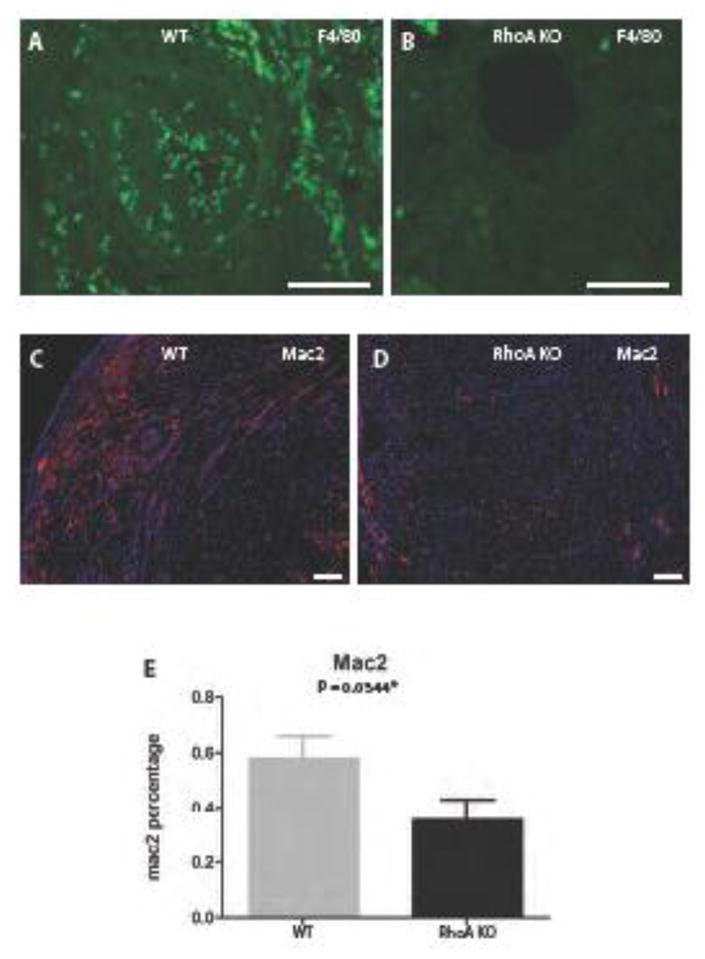
(A, B) Sections of transplanted hearts at 50 days post-transplantation immunostained with antibody against macrophage marker F4/80 conjugated with FITC. (A) High macrophage (green spots) infiltration is present in the heart from the RhoAflox/flox (no Cre; labeled WT) recipient and (B) negligible macrophage infiltration in the heart from Lyz2Cre+/−RhoAflox/flox (labeled RhoA KO) recipient. (C, D) Sections of transplanted hearts at 50 days post-transplantation immunostained with antibody against activated macrophage marker Mac-2 and secondary antibody conjugated with red fluorescein protein. (C) High macrophage (red spots) infiltration is present in the heart from the RhoAflox/flox (no Cre) recipient and (D) low macrophage infiltration in the heart from Lyz2Cre+/−RhoAflox/flox recipient. (E) Graph showing the difference in the Mac-2 positive macrophages between hearts from RhoAflox/flox (no Cre) and Lyz2Cre+/−RhoAflox/flox recipients. The difference is statistically significant with the P value <0.05. 3 mice from each experimental group were used. (A, B) Bar is equal to 50 μm; (C, D) Bar is equal to 100μm.
2. Cardiac allografts show negligible apoptosis and RhoA deleted macrophages are not more prone to apoptosis induction than control macrophages
It is known that RhoA pathway regulates apoptosis (27). Thus, one of the explanations for low macrophage infiltration of the allografts from RhoA-deleted (Lyz2Cre+/−RhoAflox/flox) recipients is the possibility of heightened apoptosis of macrophages within the graft. To address this issue we performed apoptosis Tunel assay on transplanted hearts procured 50 days post-transplantation. Figure 3A–C clearly shows that in contrast to high level of apoptosis in the acutely rejecting allografts from the untreated recipients (positive control for apoptosis), the allografts from control (RhoAflox/flox (no Cre) and RhoA-deleted (Lyz2Cre+/−RhoAflox/flox) recipients show negligible level of apoptosis. To see if the RhoA-deleted macrophages were not more prone to apoptosis induction than control macrophages we checked in vitro how they respond to TNFα (apoptosis inducer) treatment. Flow cytometry analysis showed (Fig. 3D) that apoptosis in untreated and TNFα treated control peritoneal macrophages was comparable to apoptosis in RhoA-deleted peritoneal macrophages. This indicates that RhoA-deleted macrophages are not more prone to apoptosis induction than control macrophages.
Figure 3. Apoptosis in cardiac allografts and peritoneal macrophages.
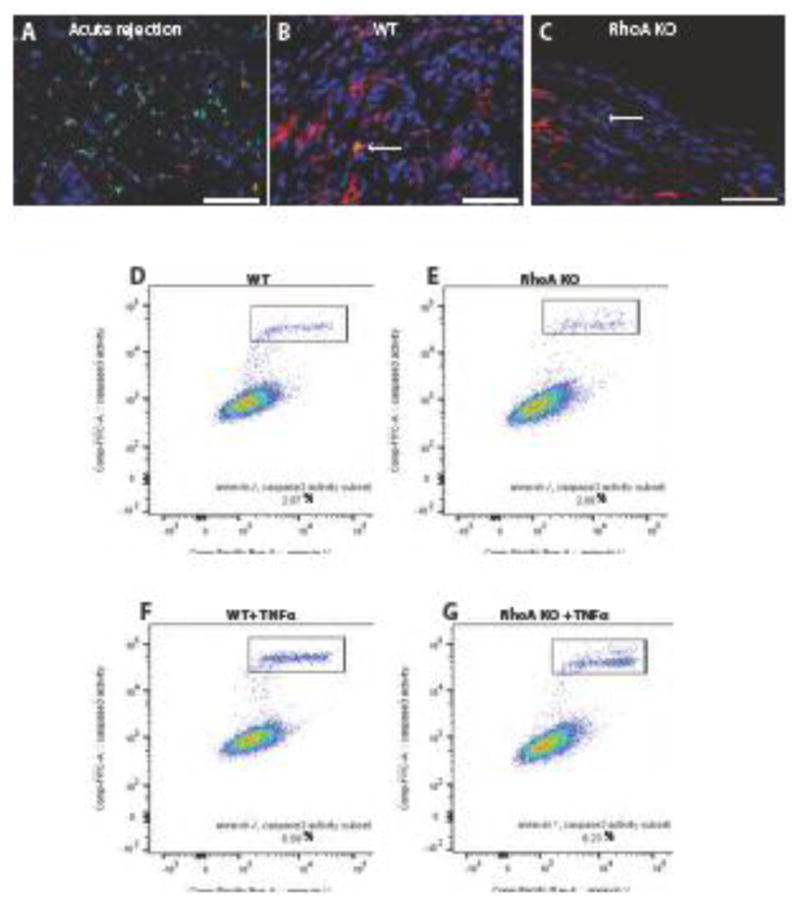
(A–C). Sections of hearts fixed at 7 days post-transplantation (acutely rejecting positive control) and at 50 days post-transplantation were stained using Tunel Assay and immunostained with macrophage marker Mac-2 antibody. Nuclei were counterstained with Hoechst. Apoptotic cells are green, macrophages are red and nuclei are blue. (A) Section of the acute rejecting heart at 7 days post-transplantation (positive control) shows high macrophage infiltration (red) and high number of apoptotic cells (green). Hearts from RhoAflox/flox (no Cre; labeled WT) recipient (B) and Lyz2Cre+/−RhoAflox/flox (labeled RhoA KO) recipient (C) show very low number of Tunel positive apoptotic cells. Arrows point to Tunel positive cells. Bar is equal to 50 μm. (D–G) Flow cytometry of apoptosis assay (gating on F4/80+/CD11b+ cells) performed on peritoneal macrophages isolated from RhoAflox/flox (no Cre) (Control) and Lyz2Cre+/−RhoAflox/flox (RhoA KO) mice. (D, E) There is no difference in apoptotic cell percentage between control and RhoA deleted-macrophages. (E, F) RhoA-deleted macrophages are not more vulnerable to the apoptosis-inducing factor (TNFα) than control macrophages. 3 mice from each experimental group were used.
3. RhoA deletion does not impair bone marrow proliferation and frequency of blood monocytes
Knowing that RhoA besides apoptosis also regulates cell proliferation (28) we considered another possible explanation for low macrophage infiltration of the allografts from RhoA-deleted recipients. The possibility was that already in naïve mice the RhoA deletion caused reduction of macrophage precursors in the bone marrow, which would resulted in the lower number of monocytes released to the blood. The BRDU proliferation assay and flow cytometry of bone marrow of naïve (pre-transplantation) mice showed that there was no statistically significant difference in the bone marrow proliferation (Fig. 4A, B), and in the frequency of CD45+/CD115+/CD11b+ bone marrow monocytes, which are potentially able to migrate into the blood i.e. are CCR2 positive (Fig. 4C, D) between control and RhoA-deleted naïve mice. Moreover, there was also no significant difference between the number of CD115+/CD11b+monocytes in the blood of control and RhoA-deleted mice (Fig. 4E, F). These results strongly suggest that similar to lack of apoptosis, the reduction in macrophage precursors (monocytes) number is not a likely cause of low macrophage infiltration observed in the allografts of RhoA-deleted mice.
Figure 4. RhoA deletion does not impair bone marrow and blood monocytes.
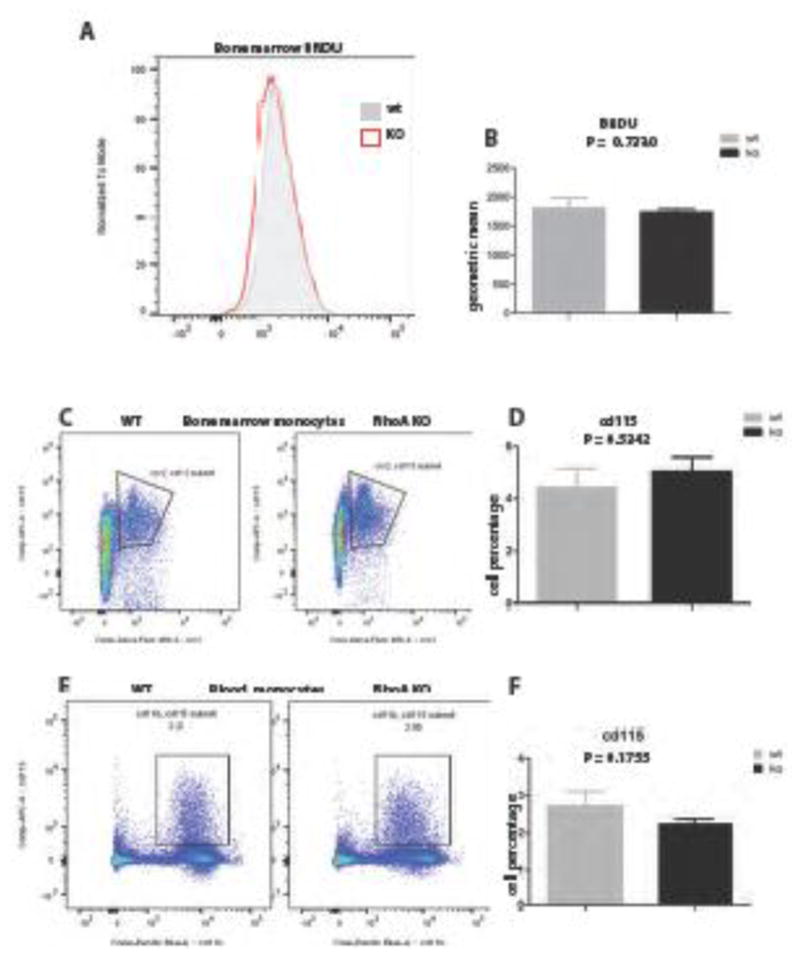
(A–B) The BRDU proliferation assay of bone marrow of CD11b+ monocytes shows no statistically significant difference in proliferation between wild type and RhoA-deleted mice. Flow cytometry analysis of CCR2+/CD115+ /CD11b+cells (monocytes) in bone marrow (C, D) and CD11b+/CD115+/CD45+ monocytes in blood (E, F) shows no significant difference between wild type and RhoA-deleted mice. Y-axis in graph D represents percentage of CD115+ cells among all bone marrow cells and in graph F the percentage of CD115+ monocytes among all blood cells. 3 mice from each experimental group were used.
4. RhoA deletion down-regulates fractalkine receptor CX3CR1 in the monocytes and macrophages
Another possible explanation for low macrophage infiltration of the allografts is that the RhoA-deleted mice were impaired in the chemotactic recruitment of monocytes/macrophages to the allograft. The most important pathways involved in the recruitment of monocytes/macrophages to the inflammation/injury site are CX3CL1/CX3CR1 and CCL2/CCR2 chemotactic pathways. The fractalkine (CX3CL1) - a chemokine abundantly expressed by endothelial cells of vasculature, chemoattracts monocytes/macrophages expressing CX3CR1 receptor, to the blood vessel walls. Thus, we checked what was the level of CX3CR1 expression in blood and bone marrow monocytes in control and RhoA-deleted naïve (pre-transplantation) mice. Flow cytometry analysis clearly showed that there was a statistically highly significant reduction of CX3CR1 expression in bone marrow and blood monocytes from RhoA-deleted mice (Fig. 5 A–D). For comparison we also checked the expression of CCR2 - the receptor of CCL2 chemokine and found that the frequency of CCR2 expressing blood monocytes was slightly lower (at the border of being statistically significant) in RhoA deleted mice (Fig. 5E, F). We also checked the level of CX3CR1 protein and mRNA expression in peritoneal macrophages. Western blot and RTPCR analyses showed that deletion of RhoA down regulated CX3CR1 protein expression in peritoneal macrophages while the expression of CCR2 receptor remained unchanged (Fig. 5). We also found that upon RhoA deletion there is up-regulation of CX3CR1 mRNA (Fig. 5 I). We believe that the up-regulation of CX3CR1 mRNA is a compensatory response to the insufficient level of receptor protein and faulty receptor recycling (see below and Discussion). Taken together all these results strongly suggest that the impairment of fractalkine (CX3CL1/CX3CR1) chemotactic pathway is the main reason for the low abundance of macrophages in the allografts transplanted into RhoA-deleted recipients.
Figure 5. RhoA deletion down-regulates expression of CX3CR1 receptor in blood and bone marrow monocytes and peritoneal macrophages.

(A–D) Flow cytometry analysis of monocytes in blood and bone marrow shows statistically highly significant decrease in CXC3R1 expression level in RhoA-deleted mice. (E, F) Flow cytometry analysis of CCR2 expressing monocytes in blood shows slight difference (at the border of being significant) between wild type and RhoA- deleted mice. (G, H) Flow cytometry analysis of peritoneal macrophages shows statistically highly significant decrease in CX3CR1 expression level in RhoA- deleted macrophages. (I) The level of CX3CR1 mRNA, as measured by RTPCR, is highly and statistically significantly upregulated in RhoA-deleted macrophages. (J, K) Flow cytometry analysis of peritoneal macrophages shows no significant difference between expression levels of CCR2 in wild type and RhoA- deleted macrophages. 3 mice from each experimental group were used.
5. RhoA deletion changes actin cytoskeleton and impairs CX3CR1 association with endosomes
Actin staining of RhoA-deleted macrophages showed profound changes in actin cytoskeleton organization caused by inability to disassembly focal adhesions and retract the tail (described in detail in Liu et al. 25 and Fig. 6). Because it is known that receptor recycling i.e. translocation to the membrane and its post-activation internalization and degradation depend on endosome pathway, which in turn, is regulated by actin cytoskeleton (29, 30) we wanted to see if the RhoA deletion influenced CX3CR1 association with endosomes. The co-immunostaining with antibodies against CX3CR1 and endosomal marker transferrin receptor (CD71) showed (Fig. 6) that while in control macrophages the CX3CR1 co-localizesd with endosomes, in RhoA-deleted macrophages the CX3CR1 association with endosomes was disrupted and it aggregated at macrophage membrane instead (Fig. 6 D, arrows, and E).
Figure 6. RhoA deletion affects actin cytoskeleton and inhibits association of CX3CR1 with the endososmes.

(A, B) Actin and (A1, B1) anti-vinculin antibody staining of wild type and RhoA deleted bone marrow derived M0 macrophages. RhoA-deleted macrophages show extreme elongation of the tail with visible aggregations of focal adhesions (arrows). (C, D) Wild type and RhoA-deleted bone marrow derived M0 macrophages stained with anti-CX3CR1 antibody and (C1, D1) anti-transferrin receptor (CD71; endosome marker) antibody. (C2, D2) Merged images of CX3CR1 (red), transferrin receptor (green) and nuclear Hoechst staining (blue). In wild type macrophages CX3CR1 co-localized with endosomes, while in RhoA-deleted macrophages the co-localization with endosome is disrupted and CX3CR1 localizes at the macrophage plasma membrane (arrows). Bar is equal to 50 μm.
6. RhoA deletion decreases frequency of CX3CR1+ macrophages in the allograft
Because the deletion of RhoA down-regulated expression of CX3CR1 in blood and bone marrow monocytes we wanted to see if this had a bearing on identity of macrophages present in the allograft. Flow cytometry analysis showed that the allografts from RhoA-deleted mice had significantly less F4/80+/CD11b+ macrophages than allografts from control mice (0. 67% versus 2.11%; Fig. 7 AC), while the macrophage proliferation remained unchanged (Fig. 7 H, I). The analysis of the CX3CR1 and CCR2 expression in F4/80+/CD11b+ macrophages showed that both in control and RhoA deleted mice all these macrophages expressed high level of CX3CR1 and CCR2 receptors (Fig. 7 D–G). This indicates that only the monocytes/macrophages expressing high level of CX3CR1/ CCR2 were efficiently recruited to the allografts. Thus, the reduction of CX3CR1 expression in monocytes/macrophages in RhoA-deleted mice resulted in a very low abundance of CX3CR1+ monocytes/macrophages recruited into the allograft, which in turn drastically reduced the CX3CR1-dependent immune response.
Figure 7. RhoA deletion reduces number of CX3CR1+ macrophages in the allograft.

(A–C) Flow cytometry analysis of F4/80+/CD11b+ macrophages showed their reduction in the allografts of RhoA-deleted mice at 30 days post-transplantation. (D–G) Flow cytometry analysis of F4/80+/CD11b+ macrophages showed that in the allografts from wild type and RhoA-deleted mice at 30 days post-transplantation these macrophages expressed high levels of CX3CR1 and CCR2 receptors. (H, I) The BRDU proliferation assay of allograft macrophages at 30 days post-transplantation shows no statistically significant difference in proliferation between wild type and RhoA-deleted mice. 3 mice from each experimental group were used.
Discussion
We showed here that the monocyte/ macrophage lineage- targeted deletion of RhoA down-regulates fractalkine receptor CX3CR1 in monocytes and macrophages, decreases macrophage infiltration and abrogates arterial neointimal hyperplasia and chronic rejection of cardiac allografts in transgenic mouse model system.
RhoA is a member of small GTPase protein family and regulates actin dependent cell functions including motility of monocytes and macrophages (18- 20). During inflammatory response, the infiltration of the inflamed tissue/organ, depend on the subpopulation of chemokine receptors belonging to the G protein-coupled proteins family. These receptors (including fractalkine receptor CX3CR1), upon binding their chemokine ligands undergo conformational changes and become the GEFs (17), which activate RhoA and regulate actin-dependent cell functions. The fractalkine, which is expressed by endothelial cells of the blood vessel walls, recruits macrophage precursors (monocytes) and macrophages in the vicinity of blood vessels where they mount inflammatory responses, collagen deposition (fibrosis) and neointimal hyperplasia (11, 12, 31–33). Recent studies indicate that CX3CR1/CX3CL1 pathway is responsible for the monocyte/macrophage directed migration into artherosclerotic plaque (8) and for smooth muscle differentiation and neiontima formation in vessel wall after the injury (11–13). It has been also shown that CX3CR1 knockout prolongs survival of cardiac allografts in mouse model system (14) Recently, we showed that RhoA pathway interference affected actin organization and macrophage phenotype (24,25). However, we showed that in in vitro assay, the RhoA-deleted peritoneal macrophages were able to degrade extracellular matrix (a prerequisite for intra- and inter-tissue migration) and migrate normally (25). Although this observation does not prove that RhoA-deleted macrophages can migrate normally in vivo, it strongly suggests that low macrophage infiltration in the allografts in RhoA-deleted mice is not caused by the impairment of macrophage migration but rather a defect in their recruitment. In addition, although we have not directly proven that macrophages actually cause chronic rejection, our findings indicate that macrophages are crucial for development of chronic rejection.
Considering all these data we propose the following scenario of events (Fig. 8, 9). Under naïve conditions (untransplanted mice) majority of bone marrow derived and blood monocytes and peritoneal macrophages express CX3CR1 receptors (8). The organ injury such as transplantation results in increased production and release of inflammatory chemokine (CX3CL1, fractalkine) and high recruitment of monocytes/macrophages to the allograft. The interaction of CX3CR1 receptor (which is the G coupled protein) with CX3CL1 causes conformational changes of the receptor into GEF (Fig. 8). The GEF activates RhoA, which in turn, changes actin polymerization/dynamics, which promotes monocyte/macrophage migration to the blood vessel wall (main source of CX3CL1), differentiation of smooth muscle cells in the wall and hyperplasia of neointima. This, over time, occludes blood vessels and leads to chronic rejection/organ failure (Fig. 8, 9). The deletion of RhoA causes faulty RhoA-dependent actin polymerization/dynamics, which interferes with normal recycling (and transcription/translation) of CX3CR1 receptors and leads to decreased expression of CX3CR1 in monocytes and macrophages (Fig. 8), which in turn reduces CX3CR1/CX3CL1- dependent recruitment to the allografts. Our study showed that deletion of RhoA caused decrease in CX3CR1 protein expression and increase in CX3CR1 mRNA level. It is known that the recycling/turnover of G protein coupled receptors, such as CX3CR1 occurs through endocytotic/exocytotic pathway and is actin- and RhoA- dependent (29, 30–34). Proper receptor recycling (turnover), in turn, determines receptor’s expression at transcriptional (mRNA) and translational (protein) level. We showed that RhoA deletion disrupted CX3CR1 association with endosomes. Thus, we believe that the increase in the expression of CX3CR1 mRNA, which we had observed in RhoA–deleted macrophages, is a compensatory response to the low expression of its protein resulting from a faulty CX3CR1 turnover (Fig. 8). All these defects, upon transplantation, lead to a dramatic decrease in the abundance of CX3CR1 positive macrophages in the allograft and its vasculature, decrease in CX3CR1/CX3CL1- dependent neointimal hyperplasia and resulted in abrogation of chronic rejection (Fig. 8, 9).
Figure 9. Hypothetical model of the effect of monocyte/macrophage-targeted RhoA deletion on neointima hyperplasia and chronic rejection.

(A) Normal (undeleted) monocytes and macrophages have normal actin organization/dynamics and express high level of CX3CR1 receptor. In response to transplantation, the allograft and the endothelium of its vasculature produce fractalkine chemokine (CX3CL1), which is a ligand of CX3CR1 receptor. Binding of fractalkine to its receptor induces chemotactic response and recruits monocytes/macrophages toward the allograft. While in the allograft, the monocytes differentiate into macrophages and together with co-recruited macrophages mount inflammatory response and cause over-proliferation of smooth muscle cells in the vascular walls. This in turn leads to the neointimal hyperplasia, occlusion of arteries and chronic rejection of the allograft. (B) RhoA deleted monocytes and macrophages have abnormal actin organization/dynamics, which lowers expression of CX3CR1 receptors. The CX3CR1-deficient monocytes/macrophages are unable to respond to fractalkine signaling to be recruited to the allograft. Thus, only very low number of monocytes/macrophages still expressing high level of CX3CR1 is recruited to the allograft. The very low number of these monocytes/macrophages results in lower inflammatory response, lower neointimal hyperplasia and diminished chronic rejection of the allograft.
It is known that the monocyte/macrophage inflammatory response does not depend exclusively on CX3CR1/CX3CL1 signaling but also, among multitude of other pathways, on the CCR2/CCL2 pathway (8, 35). The CCR2 receptor belongs to CC chemokine receptors family, which similar to CX3CR1 are the G protein-coupled proteins involved in regulation of, and regulated by, the RhoA pathway. Our study showed that while RhoA-deletion had impaired CX3CR1 it had only slight effect on CCR2 expression in blood monocytes. The question is why RhoA-deletion has different effect on two different receptors from the (broadly) same family of G protein-coupled receptors. Although answering this question requires further study, one of the possible explanations is that the changes caused by RhoA deletion are detrimental to the receptors with a specific structure only. Another explanation is that because the CCL2/CCR2 mediates RhoA activity through Smad3/ MAPK (36) and/or PKCα signaling (37) while the CX3CR1/CX3CL1 interactions with RhoA pathway are more direct (12) the CX3CR1/CX3CL1 pathway is more “sensitive” to RhoA deletion.
Another fascinating possibility, which would explain the difference between the involvement and importance of CX3CR1 versus CCR2 pathways in chronic rejection may be related to selective utilization of CX3CR1 pathway, which was described in development of atherosclerotic plaque (8) or the phenomenon of CCR2 to CX3CR1 receptor switching, which was described by Barlic et al. (38). These authors showed that human macrophages present in human coronary artery have ability to switch CCR2 off and CX3CR1 on, which causes inhibition of CCR2-dependent migration in favor of CX3CR1-dependent macrophage accumulation in the vessel wall. If such switch also occurs during anti-allograft immune response than it would be quite understandable why the inhibition of CX3CR1 but not the CCR2 pathway had such profound inhibitory effect on neointimal hyperplasia and chronic rejection.
Recent study from our laboratory (25) also showed that RhoA deletion up-regulates the activity of its downstream effector ROCK1 in peritoneal macrophages, and that this effect depends on non-apoptotic Caspase-3. Thus, in the future, it will be important to study if and how Caspase-3 regulates monocyte/macrophage CX3CR1/CX3CL1 pathway during chronic rejection. Further studies are needed to eliminate a very remote possibility that the differences between the macrophage number in the grafts of RhoA-deficient and wild type recipients might in part reflect differences in severity of ischemia reperfusion injury of the grafts or in the properties of acute alloimmune responses. If these were true than the low number of macrophages in RhoA-deficient mice could then have reflected the lesser degree of the initial damage of allograft tissue. Although our findings can potentially lead to the development of novel anti-chronic rejection therapies in human transplantation, one has to remember, when extrapolating mouse model results to human situation, that human allograft vasculopathy (based on fibroblast functions and smooth muscle cells proliferation) differ from that in the mouse (based on smooth muscle cells proliferation only).
Further studies are also necessary to establish what is the indirect effect of monocyte/macrophage specific RhoA-deletion on other types of immune cells participating in immune response toward the allograft.
Conclusions
Our finding that monocyte/macrophage specific deletion of RhoA inhibits cardiac allograft chronic rejection via down-regulation of CX3CR1/CX3CL1 pathway may lead to development of novel anti-chronic rejection therapies.
Acknowledgments
Funding
We are extremely grateful for the support from William Stamps Farish Fund and Donald D. Hammill Foundation.
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mannon RB. Macrophages: Contributors to allograft dysfunction, repair or innocent bystanders? Curr Opin Organ Transplant. 2012;17:20–25. doi: 10.1097/MOT.0b013e32834ee5b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salehi S, Reed EF. The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant. 2015;20:446–53. doi: 10.1097/MOT.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. 2015;144:541–548. doi: 10.1111/imm.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epelman S, Lavine KJ2, Randolph GJ3. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pilmore HL, Painter DM, Bishop GA, et al. Early up-regulation of macrophages and myofibroblasts: a new marker for development of chronic renal allograft rejection. Transplantation. 2000;69:2658–62. doi: 10.1097/00007890-200006270-00028. [DOI] [PubMed] [Google Scholar]
- 10.Skaro AI, Liwski RS, Johnson P, Legare J-F, Lee TDG, Hirsch GM. Donor versus recipient: Neointimal cell origin in allograft vascular disease. Graft. 2002;5:390–398. [Google Scholar]
- 11.Kumar AH, Metharom P, Schmeckpeper J, Weiss S, Martin K, Caplice NM. Bone marrow-derived CX3CR1 progenitors contribute to neointimal smooth muscle cells via fractalkine CX3CR1 interaction. FASEB J. 2010;24:81–92. doi: 10.1096/fj.09-132225. [DOI] [PubMed] [Google Scholar]
- 12.Kumar AH, Martin K, Turner EC, Buneker CK, Dorgham K, Deterre P, Caplice NM. Role of CX3CR1 receptor in monocyte/macrophage driven neovascularization. PLoS One. 2013;8(2):e57230. doi: 10.1371/journal.pone.0057230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Feng X, Cai W, Liu T, Liang Z, Sun Y, Yan C, Han Y. Chemokine CX3CL1 and its receptor CX3CR1 are associated with human atherosclerotic lesion vulnerability. Thromb Res. 2015;135:1147–1153. doi: 10.1016/j.thromres.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 14.Haskell CA, Hancock WW, Salant DJ, Gao W, Csizmadia V, Peters W, Faia K, Fituri O, Rottman JB, Charo IF. Targeted deletion of CX(3)CR1 reveals a role for fractalkine in cardiac allograft rejection. J Clin Invest. 2001;108:679–688. doi: 10.1172/JCI12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dichmann S, Herouy Y, Purlis D, Rheinen H, Gebicke-Härter P, Norgauer J. Fractalkine induces chemotaxis and actin polymerization in human dendritic cells. Inflamm Res. 2001;50:529–533. doi: 10.1007/PL00000230. [DOI] [PubMed] [Google Scholar]
- 16.Gevrey JC, Isaac BM, Cox D. Syk is required for monocyte/macrophage chemotaxis to CX3CL1 (Fractalkine) J Immunol. 2005;175:3737–3745. doi: 10.4049/jimmunol.175.6.3737. [DOI] [PubMed] [Google Scholar]
- 17.Gilman AG. G proteins: transducers of receptor-generated signals. Ann Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 18.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiological reviews. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 19.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Allen WE, Jones GE, Pollard JW, Ridley AJ. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110:707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 21.Kloc M, Li XC, Ghobrial RM. RhoA Cytoskeletal Pathway to Transplantation. J Immunol Clin Transpl. 2014;2:1012–1017. [Google Scholar]
- 22.Ridley AJ, Allen WE, Peppelenbosch M, Jones GE. Rho family proteins and cell migration. Biochem Soc Symp. 1999;65:111–123. [PubMed] [Google Scholar]
- 23.Vicente-Manzanares M, Webb DJ, Horwitz AR. Cell migration at a glance. J Cell Sci. 2005;118:4917–4919. doi: 10.1242/jcs.02662. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Tejpal N, You J, Li XC, Ghobrial RM, Kloc M. ROCK inhibition impedes macrophage polarity and functions. Cell Immunol. 2016;300:54–62. doi: 10.1016/j.cellimm.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Minze LJ, Mumma L, Li XC, Ghobrial RM, Kloc M. Mouse macrophage polarity and ROCK1 activity depend on RhoA and non-apoptotic Caspase 3. Exp Cell Res. 2016;341:225–236. doi: 10.1016/j.yexcr.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Clausen B, Burkhardt C, Reith, Renkawitz W, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 27.Tsai NP, Wei LN. RhoA/ROCK1 signaling regulates stress granules formation and apoptosis. Cell Signal. 2010;22:668–75. doi: 10.1016/j.cellsig.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Zheng F, Zhang S, Lu J. Loss of RhoA expression prevents proliferation and metastasis of SPCA1 lung cancer cells in vitro. Biomed Pharmacother. 2015;69:361–6. doi: 10.1016/j.biopha.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Leung SM, Rojas R, Maples C, Flynn C, Ruiz WG, Jou TS, Apodaca G. Modulation of Endocytic Traffic in Polarized Madin-Darby Canine Kidney Cells by the Small GTPase RhoA. Mol Biol Cell. 1999;10:4369–4384. doi: 10.1091/mbc.10.12.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garnacho C, Shuvaev V, Thomas A, McKenna L, Sun J, Koval M, Albelda S, Muzykantov V, Muro S. RhoA activation and actin reorganization involved in endothelial CAM-mediated endocytosis of anti-PECAM carriers: critical role for tyrosine 686 in the cytoplasmic tail of PECAM-1. Blood. 2008;111:3024–33. doi: 10.1182/blood-2007-06-098657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apostolakis S, Krambovitis E, Vlata Z, Kochiadakis GE, Baritaki S, Spandidos DA. CX3CR1 receptor is up-regulated in monocytes of coronary artery diseased patients: impact of pre-inflammatory stimuli and renin-angiotensin system modulators. Thromb Res. 2007;121:387–395. doi: 10.1016/j.thromres.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Apostolakis S, Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin. 2013;34:1251–1256. doi: 10.1038/aps.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, Jiang D. Fractalkine/CX3CR1 and atherosclerosis. Clin Chim Acta. 2011;412:1180–1186. doi: 10.1016/j.cca.2011.03.036. [DOI] [PubMed] [Google Scholar]
- 34.Puthenveedu MA, Lauffer B, Temkin P, Vistein R, Carlton P, Thorn K, Taunton J, Weiner OD, Parton RG, von Zastrow M. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell. 2010;143:761–773. doi: 10.1016/j.cell.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu C, Yu P, Tao M, Gupta T, Moldawer LL, Berceli SA, Jiang Z. Monocyte chemoattractant protein-1/CCR2 axis promotes vein graft neointimal hyperplasia through its signaling in graft-extrinsic cell populations. Arterioscler Thromb Vasc Biol. 2012;32:2418–2426. doi: 10.1161/ATVBAHA.112.255786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem. 2012;287:36593–36608. doi: 10.1074/jbc.M112.365999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stamatovic SM, Dimitrijevic OB, Keep RF, Andjelkovic AV. Protein kinase Calpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J Biol Chem. 2006;281:8379–8388. doi: 10.1074/jbc.M513122200. [DOI] [PubMed] [Google Scholar]
- 38.Barlic J, Zhang Y, Foley JF, Murphy PM. Oxidized lipid-driven chemokine receptor switch, CCR2 to CX3CR1, mediates adhesion of human macrophages to coronary artery smooth muscle cells through a peroxisome proliferator-activated receptor gamma-dependent pathway. Circulation. 2006;114:807–819. doi: 10.1161/CIRCULATIONAHA.105.602359. [DOI] [PubMed] [Google Scholar]