

Abstract
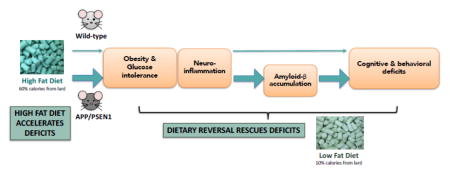
This study assessed the extent to which high fat diet (HFD)-induced β-amyloid accumulation and cognitive decline in APP/PSEN1 mice are reversible through control of fat intake. Ten months of HFD (60% calories from fat) led to significant deficits in a 2-trial Y maze task, and nest building assay, and decreased voluntary locomotor activity. The HFD induced an inflammatory response, indicated by increased expression of several inflammatory markers. Substituting a low fat diet led to pronounced weight loss and correction of glucose intolerance, decreases in the inflammatory response, and improved performance on behavioral tasks in both wild-type and APP/PSEN1 transgenic mice. Insoluble β-amyloid levels, and extent of tau phosphorylation were also lower following dietary reversal in APP/PSEN1 mice compared to high fat-fed animals, indicating that the inflammatory response may have contributed to key pathogenic pathways in the Alzheimer’s disease model. The data suggest that weight loss can be a vital strategy for cognitive protection, but also highlight potential mechanisms for intervention when sustained weight loss is not possible.
Keywords: Obesity, Cognition, Behavior, Inflammatory response, β-amyloid, High fat diet
Graphical Abstract
Introduction
Obesity and Alzheimer’s disease both have alarmingly high prevalence in Western societies (Alzheimer's-Association, 2015, Ogden, et al., 2014), and both are associated with cognitive decline and diabetes (Beydoun, et al., 2008,Gustafson, et al., 2003, Profenno, et al., 2010, Whitmer, et al., 2007). High fat diets (HFD) are thought to contribute directly to several of the key facets of Alzheimer’s disease, including increased accumulation of β-amyloid (Aβ), hyperphosphorylation of tau, and an inflammatory state in peripheral organs and brain (including astrocyte and microglial activation)(Levin-Allerhand, et al., 2002, Puig, et al., 2012, Ramos-Rodriguez, et al., 2014, Shie, et al., 2002, Zhang, et al., 2016). Altered Aβ levels are not universally found (Knight, et al., 2014, Peng, et al., 2014, Studzinski, et al., 2009), and may depend on the mouse model used, the level and type of dietary fat, and the duration of treatments. Tau phosphorylation has been studied less frequently, in part because of the weighting towards Aβ pathology in the most commonly-used mouse models of Alzheimer’s disease. Each of these pathological factors may contribute to cognitive factors individually, and also via changes to blood brain barrier (BBB) permeability. MRI imaging techniques have indicated BBB breakdown in hippocampus may be an early event in the Alzheimer’s disease pathological cascade (Montagne, et al., 2015,van de Haar, et al., 2016b). BBB leakage is also directly increased by HFD (Loffler, et al., 2016, Ramos-Rodriguez, et al., 2014, Takechi, et al., 2013), and long-term feeding with both saturated fatty acids and high cholesterol appear to exacerbate the effects of aging on BBB permeability (Takechi, et al., 2013).
Studies that included measures of cognition also demonstrate that HFD induces deficits in learning and memory, although experimental design strongly impacts the severity of deficits, and whether changes are observed in both control animals and the disease model (Knight, et al., 2014, Pistell, et al., 2010, Ramos-Rodriguez, et al., 2014, Yeh, et al., 2015). Negative effects on cognitive ability are found across several behavioral tasks, despite the challenges in testing in these populations, which vary dramatically in body size and adiposity.
Now that the links between HFD, inflammatory response and cognitive decline have been established, in both Aβ-positive and – negative populations, it is critical to investigate whether these changes are reversible, and thus whether the mechanisms may potentially be targeted by nutritional or pharmacological interventions. To that end, we designed a study in which long-term HFD feeding was initiated in young APP/PSEN1 mice and maintained for 8 to 10.5 months to induce obesity and inflammatory signaling. The obese state was then reversed by providing a low fat diet (LFD) for 2 months (Reversal condition, REV) in a subset of these animals. Our objective was to assess the effects of young to mid-life obesity and its reversal on cognitive ability, and multiple markers of disease pathogenesis, in addition to glucose homeostasis. Specifically included in our analyses were assessments of both APP/PSEN1 and their non-transgenic littermates (wild-type, WT). Obesity and cognitive decline are problems observed during otherwise normal aging, and sporadic (late onset) Alzheimer’s disease, and we therefore seek mechanisms that are not specific to populations carrying inherited mutant copies of Alzheimer’s related genes (e.g. APP, PSEN1).
Material and methods
Animals
Male and female APPSwe/PS1ΔE9 transgenic AD mice (Borchelt, et al., 1997) and wildtype littermates bred in-house were used in the present study (N=5–11/group). Founder mice were obtained from Jackson laboratories (ME. USA, #005864) and maintained on a C57Bl/6J background (#000664). Mice were weaned at 21 days old, genotyped via PCR using tail snips, and group-housed by sex (3–5 per cage) until 3 days prior to sacrifice, at which point they were moved to individual housing for nest building analysis. Food and water, including experimental diets were provided ad libitum, and mice were maintained on a 12/12 hr light/dark cycle. All procedures were approved in advance and conducted in accordance with Vanderbilt University institutional (IACUC) policies and guidelines.
Dietary Groups
At 2-months of age, standard lab chow (Purina 5001, 4% kcal/fat) was replaced with either high-fat diet (HFD; Research Diets: #D12492) in which 60% kcal are derived from lard, 20% from carbohydrate and 20% from protein, or low-fat diet control (LFD; Research Diets: #D12450J) in which 10% kcal are derived from lard and additional calories are derived from corn starch, with 70% carbohydrate and 20% from protein. Sucrose content per gram was equivalent between the two diets. Low fat diet contained approx. 3.85 kcal/gram, high fat diet contained 5.24 kcal/gram. Full details are provided in Supplementary Table 1. Mice were weighed weekly following the initiation of experimental diets. At 9.5 months of age, a subset of mice on the HFD were provided with LFD for the duration of the study (reversal group, REV). All other mice were maintained on their original diets until the end of the study, an additional 10 weeks (see Fig. 1, Experimental outline).
Figure 1. Experimental design.

Mice were randomly allocated to high fat diet (HFD) or low fat diet (LFD) at 2 months of age and remained on the same diet until sacrifice, except in the case of Reversal (REV) mice. Half of surviving high fat diet mice were switched to low fat diet (Reversal) at 9.5 months of age. Y-maze (spontaneous alternation) and fear conditioning were conducted at 6 months of age following activity and elevated zero maze. a Denotes tasks that were conducted but are not described below. Methods and Results for these tasks are given in Supplementary File 1.
Dietary intake
Average food consumption was calculated per day/ per mouse, from weight of food consumed per cage, per week. All cages contained both WT and APP/PSEN1 mice, and thus intake is not calculated according to genotype.
Behavioral Tests
Behavioral testing began at 5–6 months of age to assess potential early cognitive changes, with major testing beginning at 9 and 12 months. This represents a middle-aged state in the mouse as we sought to investigate how diet may accelerate the cognitive and amyloid pathology shown by the mice. All behavioral tests were performed in the Vanderbilt Neurobehavioral Core Facility, during the second half of the light cycle. Animals were transferred from the colony to the testing room and allowed to acclimate for 30 min. prior to the start of behavioral testing. All apparatuses were cleaned before and after each mouse with a 10% ethanol solution to reduce the influence of odor cues on behavior. Behavioral tests were conducted Monday through Friday, with no more than one task conducted on a single day, and a minimum of 20 hours between separate tests. Testing for each age-group was completed within 2 weeks. All tests conducted are shown in Figure 1. Additional information concerning methods and results not shown are included in Supplemental File 1.
2-trial Y-maze
Memory for familiar spatial locations (2 arms of the maze) is indexed by greater exploration of a novel (previously-blocked) arm of the maze during a second trial. The two 5-min. trials were given on the same day, with a 5-min. inter-trial interval. Testing was undertaken at two time points (see Fig. 1) using a different testing room and different extra maze cues at the second test point, as previously described (Kennard and Harrison, 2014). Trials were automatically tracked and analyzed using Anymaze tracking software (Stoelting, IL). Exploration data was checked to confirm that each mouse entered all open arms on a given trial.
Locomotor activity
Exploratory locomotor activity was measured during multiple 60-min. sessions in automated chambers (30cm x 30cm x 16cm; Med Associates Inc, VT).
Nest Building
Nest building is a species-specific and natural behavior of rodents, which is important for their survival (Deacon, 2006). Three days before sacrifice and just prior to the start of the dark cycle, mice were transferred from group to individual housing and given clean cages containing cotton nestlets (5g each; Ancare, NY). 14- and 28-hrs later unshredded material was weighed and nests were scored on a 1–5 scale, as described (Deacon, 2006).
Fasting blood glucose levels
Blood glucose levels were measured 20 weeks after experimental diets were initiated, using a standard glucometer (Accu-Chek, Roche) with blood obtained from the tail tip.
Glucose tolerance test (GTT)
Glucose tolerance tests were performed following an overnight fast. Body weight was confirmed and baseline blood glucose (mg/dl) was measured using a glucometer (Accu-Chek, Roche), following which mice were injected (i.p.) with a glucose bolus (2g/kg). Additional blood glucose measurements were taken from a tail snip at 30, 60, 90, and 120 minutes post-injection
Sacrifice and tissue collection
Mice were anesthetized with isofluorane, followed by cervical dislocation. Following removal and dissection of brain, tissue samples were put immediately on dry ice, and then stored at −80°C until used in biochemical assays. Serum was collected in tubes containing 2 μl 0.5M EDTA and kept at −80°C.
Amyloid-β levels
Amyloid-β levels were quantified in cortex using anti-human Aβ1-40 and Aβ1-42 sandwich ELISA kits (cat. no. KHB3441 and KHB3481; Life Technologies, CA) according to the manufacturer’s instructions for soluble and insoluble Aβ.
Western Blotting
Cortex samples were prepared using standard techniques and probed using the following primary antibodies: GFAP (MAB360, 1:2,000; Millipore, Bedford, MA)(Buckman, et al., 2013), Neprilysin/CD10 (Ab79423, 1:1,000), Receptor for Advanced Glycation End Products (AGEs), RAGE (Ab37674, 1:1,000), Tau-5 (ab80579, 1:400) and phospo-Tau (S396; ab109390, 1:5,000) all from Abcam (Cambridge, MA). TNFa (D2D4, 1:500-1:1,000), and Synaptophysin (D35E4, 1:1,000) Cell Signaling (Danvers, MA), and Actin (D35E4, 1:400-1,5,000; Santa Cruz Biotechnology, Dallas Texas). Appropriate secondary antibodies were selected from Anti-Goat IgG (A5420, 1:5,000), Anti-Guinea Pig IgG (A7289, 1:5,000) and Anti-Rabbit IgG (A0545, 1:5,000) all from Sigma-Aldrich (USA).
Tau phosphorylation
Expression of phosphorylated tau (phosphorylated at serine 395) was calculated as a percentage of expression of total tau.
Inflammatory markers in serum
TNF-α, IL-6, IL-1b and IL-10 were measured using Multiplex assays via the Luminex100 system, by the Vanderbilt Hormone Assay and Analytical Services Core.
Malondialdehyde (MDA)
MDA was measured as thiobarbituric acid reactive substances as previously described (Harrison, et al., 2009).
Statistical analyses
Data are reported in figures and text as Mean + S.E.M. unless otherwise stated. Most measures were analyzed with 2(Genotype) x 2(Diet) Univariate ANOVA (9 months), or 2(Genotype) x 3(Diet) ANOVA (12 months). Main effects from significant omnibus ANOVA were clarified using pairwise follow-up comparisons with Bonferroni corrections. Significant interactions were followed with an additional Univariate ANOVA followed by Tukey-corrected multiple comparisons to establish differences among all groups. Testing across multiple time points (e.g. locomotor activity) was analyzed using Repeated Measures ANOVA. All tests were first conducted with sex as an independent variable. Where there were significant differences between males and females we ran separate analyses for each sex (including weight, glucose tolerance, behavioral measures). Where there were no sex effects data were combined and analyzed together (biochemistry measures). Main effects from significant omnibus ANOVA were clarified using pairwise follow-up comparisons with Bonferroni corrections. Aβ measurements were performed in APP/PSEN1 mice only, and were analyzed by Univariate ANOVA followed by Fisher LSD-corrected post hoc analyses. Where assumptions of sphericity were violated, Greenhouse-Geisser corrections were used to obtain adjusted P values.
Results
Body weight, fasting blood glucose levels and glucose tolerance are all dependent on diet
Food intake
Average food intake was calculated per mouse, from total consumed per cage per week. After 20 weeks of feeding experimental diets (5 months of age), the mice fed HFD ate slightly more than LFD-fed mice, but this was only significant for females (P<0.05, Fig. 2A) and not males (P=0.11). Intake of diet decreased rapidly in REV mice in the first week that HFD was exchanged for LFD for both male and female mice (Ps<0.01, Fig. 2B).
Figure 2. High fat diet increases food consumption and leads to weight gain Food intake.

A) Food intake was assessed per cage per week, and is calculated as intake per mouse, (male mice F1, 20=2.892, P=0.105; female mice F1, 18=5.575, P=0.03), N=8–14 cages per group. B) Intake patterns remained similar in LFD and HFD mice at 9.5 months, but a dramatically decreased intake was observed in REV mice (males F1, 7=11.223, P=0.007; females F1, 8=29.911, P=0.001 ) N=3–5 cages per group. Mouse weight up to 9 months. Weights were monitored weekly, and are presented here in 4-week blocks for Ci) males, and Cii) females up to 9 months of age when the first set of mice were sacrificed. HFD weighed significantly more than LFD following instigation of the diets (week x diet interaction: males F7, 532=92.63, P<0.001, females F7, 518=100.81, P<0.001). N=15–29 per group. Mouse weight following reversal diet initiation Weights are shown in 2-week time bins following instigation of REV diets in Di) males and Dii) females. Weights varied according to diet and time in males (diet x time interaction F2, 41=229.20, P<0.001) and females (diet x time F2, 44=131.102, P<0.001, diet x genotype F2, 44=4.16, P<0.001). N=5–11 per group. A, B) Data were analyzed separately for male and female mice using Univariate ANOVA with diet as the independent variable. C, D) Data were analyzed by 2(genotype) x 2(diet) x 2(time point) RM-ANOVA . *, **, *** P<0.05, 0.01, 0.001 versus all other diet groups, +++ P<0.001 effect of genotype within HFD.
Diet induced weight change
Weights are shown for the entire experimental period, but statistics are performed on initial and final weights only. We confirmed that there were no differences in the initial weights of the four groups (Ps>0.103. Fig. 2Ci, ii). As expected the HFD led to prolonged weight gain in both males and females that was highly significant after 28 weeks of feeding (approx. 9.5 months of age) (Ps<0.001), but there was no difference according to genotype (Ps>0.209). Mice from the HFD were randomly allocated to the REV group. Following this point, female APP/PSEN1-HFD weighed significantly more than WT-HFD mice (P<0.01, Fig. 2Dii,). There were no genotype effects in LFD or REV mice (Ps>0.869). Decreased fat intake in REV mice led to rapid and substantial weight loss in all mice. In both sexes 10 weeks of LFD-feeding in REV mice led to significant decreases in weight compared to HFD mice (P<0.001, Fig. 2Di, ii). However, REV mice still weighed more than LFD mice that had never been fed the HFD (Ps<0.05).
Diet-induced alterations to glucose metabolism
Twenty weeks after initiation of diets (approx. 6.5 months of age) we confirmed that HFD had initiated a diabetic, (or pre-diabetic), state by measuring blood glucose following a 5-hour fast. Blood glucose was higher in HFD than LFD groups in both males and females (Ps<0.001, Fig. 3A, B), but APP/PSEN1 mice were not more affected than WT mice (Ps>0.24). A blood glucose level of greater than 250 mg/dl was recorded in some HFD mice, indicating that these mice had already reached a diabetic state. However, the distribution did not differ according to genotype (males: 3 WT (10.3%), 4 APP/PSEN1 (22.2%); females: 6 WT (31.5%), 5 APP/PSEN1 (20.0%)). To more fully assess glucose metabolism, we conducted a glucose tolerance test prior to sacrifice. Area under the curve (AUC) was calculated for blood glucose using baseline (pre-glucose), and 30, 60, 90 and 120-minute time points following the 2g/kg (i.p.) glucose bolus. For both males and females APP/PSEN1-HFD mice had a greater AUC than all three other groups (Ps<0.001; Fig. 3Ci, ii, Di, ii) reflecting both greater magnitude of initial response and inability to clear the glucose within the same time frame.
Figure 3. Diet-induced obesity leads to glucose intolerance to a greater extent in APP/PSEN1 mice and is rescued by reversal to LFD.

Blood glucose: HFD significantly increased fasted blood glucose levels in A) male (F1, 77=68.59, P<0.001) and B) female mice (F1, 73=114.79, P<0.001) following 20 weeks on diet. There were no effects of genotype on this measure (Fs<1.38, Ps>0.24). N=15–29 per group. Glucose tolerance test-9.5 months: In both C) males and D) females Area Under the Curve (AUC) measurements were significantly elevated in APP/PSEN1-HFD mice compared to other groups (Genotype x Diet interaction F1, 27=13.340, P=0.001 and F1, 27=16.343, P=0.001 respectively) indicating that groups responded to the glucose bolus differently. N=5–10 per group. Glucose tolerance test-12 months REV diets completely reversed glucose intolerance phenotype in both WT and APP/PSEN1 mice. E) In males AUC was larger in APP/PSEN1 mice (F1, 41 = 9.592, P=0.004), and in HFD-fed mice (F2, 41 = 37.243, P<0.001), with no interaction among the factors. F) In females the effect of HFD was even greater in APP/PSEN1 mice (F2, 42 = 7.136, P=0.002). N=5–10 per group.
Data were analyzed by 2(genotype) x 2 or 3(diet) ANOVA. *, **, *** P<0.05, 0.01, 0.001 indicates different compared to all other groups, or as marked. For Eii) ## P<0.001, indicates main effect of genotype; +++ P<0.001 indicates main effect of diet, HFD compared to LFD and REV, regardless of genotype.
Replacing HFD with LFD led to a complete reversal of glucose intolerance in REV mice of both genotypes, in both males and females (Fig. 3Ei, ii, Fi, ii). In males, AUC was greater in APP/PSEN1 than in WT mice (P<0.01), and in HFD-fed mice compared to both groups of LFD-fed mice (LFD, REV, Ps<0.001). In female mice, there was a significant interaction among the factors of diet and genotype (P<0.001) because while all APP/PSEN1 mice had an elevated AUC indicating they were less able to respond to the dietary challenge than WT mice, the effect was magnified in HFD mice. No differences were observed between LFD and REV mice indicating a recovery of glucose processing in these mice.
High fat diet decreases activity and impairs cognitive ability in both wild-type and APP/PSEN1 mice
Activity
Mice were given four 1-hour activity sessions between 5 and 6 months to ensure that they were fully habituated to the chambers. This permitted later comparisons of voluntary locomotor behavior at multiple time points without additional potential confounds of anxiety in a novel situation. By the final habituation session all mice had reached a baseline level of exploration. In males HFD led to decreased exploration compared to LFD (P<0.001) but there were no effects of genotype (Ps>0.757, Fig. 3A). In females a more modest decrease in activity in HFD mice was not significant at this time point, and there were no differences according to genotype (Ps>0.110, Fig. 3A). At the 9-month testing point the decreased activity was still evident in HFD fed mice in both males and females (Ps<0.05, Fig. 3B). In males hyperactivity was observed in some APP/PSEN1 mice, but only those on the low fat diet (P<0.05). In females there were no differences according to genotype (Ps>0.212).
We hypothesized that reversal to LFD and the resulting weight loss would also result in recovery of normal activity levels. REV mice increased activity following just 4 weeks of REV diets (Ps<0.05; Fig. 3Ci, Cii). Hyperlocomotion in APP/PSEN1 was only observed in male mice, and only in LFD condition (P<0.001).
Two trial Y-maze
At 9 months APP/PSEN1 mice showed poorer memory in the 2-trial Y maze as evidenced by less time spent in the novel arm on the test trial (Ps<0.05, Fig. 3D). Treatment with HFD also significantly decreased percent time spent in the novel arm (Ps<0.05). Greater exploration was recorded in male APP/PSEN1 mice compared to WT but only in LFD (P<0.01) and not HFD (P=0.96). The same effect was not observed in females. HFD led to decreased activity in both genotypes (Ps<0.01).
At 12 months the poorest performance on the Y-maze was seen in APP/PSEN1-HFD mice, which had poorer memory retention than all other groups (Ps<0.05, Fig. 3E). There was a striking rescue of spatial memory in APP/PSEN1-REV mice. Critically, REV mice did not differ from LFD. Distance travelled in the Y-maze on the test trial was dependent on dietary treatment for both sexes (P<0.001, data not shown) but not genotype (Ps>0.061). HFD mice travelled less than LFD groups, which was partially rescued by REV treatments in males, and fully rescued in females.
Nest building
Each mouse underwent nest building testing once, immediately prior to sacrifice. At 9 months male APP/PSEN1 mice formed poorer nests than WT mice (P<0.01, Fig. 3E), and HFD also led to poorer nest quality in both genotypes (P<0.001). In female mice, significant impairments compared to WT-LFD were only observed in the APP/PSEN1 mice on HFD (Diet X Genotype interaction, P<0.001).
In 12-month-old mice, the same pattern was observed in both male and female mice in nest building (Fig. 3H). APP/PSEN1 mice formed poorer nests than wild-type mice (Ps<0.001), but dietary reversal rescued the impairments induced by the HFD in both WT and APP/PSEN1 mice (Ps<0.001).
Lipid peroxidation is not driven by HFD treatments in brain
Malondialdehyde (MDA) was used as a marker for lipid peroxidation in brain (a mixed cortical area) and liver. In brain greater MDA was detected in APP/PSEN1 than WT mice (P<0.05, Fig. 5A), as previously observed (Dixit, et al., 2015) but only in LFD and not HFD (P<0.05). MDA levels increased by approximately 5-fold in the 2.5 months between measurements (Fig. 5B). However, this increase was lower in HFD groups, which had significantly lower MDA than either of the LFD-fed groups (P<0.01). At 12-months no difference was observed according to genotype (P=0.317). We also looked at MDA in liver as a marker of changes that may be occurring in the peripheral organs. At 9.5 months MDA levels in the liver were greater in APP/PSEN1 than wild-type mice (P<0.05, Fig. 5C) regardless of diet (Ps>0.28). At 12 months, overall levels of MDA in the liver were similar to the younger age (Fig. 5D), although there were now no differences between genotypes (P=0.73), but greater MDA was detected in the HFD mice than either LFD or REV groups (P<0.01).
Figure 5. High fat diet drives changes in oxidative stress and inflammatory response in brainand periphery.

A–D) Lipid peroxidation. A) At 9.5 months Malondialdehyde (MDA) in brain was elevated in APP/PSEN1 mice, but only those on LFD (diet X genotype interaction F1, 34=6.545, P<0.05). N=8–12 per group. B) At 12 months MDA in brain was lower in HFD mice than the other diet groups (F2, 76=8.682, P<0.001), but there was no effect of genotype (F1, 76=1.015, P=0.317). N=12–16 per group. C) At 9.5 months MDA in liver was significantly greater in APP/PSEN1 mice than WT (F1, 52=4.10, P<0.05), regardless of diet. N=11–17 per group. D) At 12 months MDA in liver was not affected by genotype (F1, 87 = 0.119, P=0.73) but was elevated in HFD mice (F2, 87 = 5.019, P<0.01). N=13–17 per group. E–L) Inflammatory response E) At 9.5 months GFAP expression was increased by both APP/PSEN1 genotype (F1, 31=28.92, P<0.001), and HFD (F1, 31=20.86, P<0.001). N=8–9 per group. A representative Western Blot is shown for GFAP and actin for both age groups under the relevant panel. F) At 12 months GFAP expression was elevated in APP/PSEN1 mice HFD-fed mice compared to every other group tested (Genotype F1, 25=43.42, P<0.001; diet F2, 25 =26.659, P<0.001; Interaction F2, 25=20.297, P<0.001). N=5–6 per group. G) At 9.5 months TNF- α expression was greater in HFD animals than LFD (F1, 30=9.31, P<0.01) regardless of genotype (F1, 30=0.42, P=0.52, interaction F1, 30=2.94, P=0.09). N=7–10 per group. H) By 12 months TNF-α expression was still elevated in HFD compared to other diet groups at 12 months (F1, 18=8.39, P<0.01) with difference according to genotype (F1, 18=2.09, P=0.17, diet x genotype interaction F1, 18=3.46, P=0.054). N=3–5 per group. Representative images from Western Blot experiments are shown in panel M. I-L) Plasma inflammatory markers. No effects of diet or genotype were observed in any of the markers assayed Il-1b, IL-6, IL-10, TNFα (Fs<2.88, Ps>0.073), with no interactions (Fs<3.0, Ps>0.066). N=5–6 per group *, ** P<0.05, 0.01 indicates differences as marked, a P<0.05 indicates main effect of genotype regardless of diet group.
Neuroinflammatory response is dependent on current dietary status
At 9.5 months both APP/PSEN1 genotype (P<0.001) and HFD diet (P <0.001, Fig. 5E, M), increased expression of GFAP with the greatest expression in APP/PSEN1-HFD mice. At 12 months the effects of diet and genotype were even more evident, with strikingly elevated GFAP expression in APP/PSEN1-HFD compared to other groups (genotype x diet interaction P<0.001, Fig. 5F, M), which was decreased in REV mice. TNF-α was measured as a further indicator of inflammatory response more reflective of microglial activation. TNF-α was elevated in HFD compared to LFD, including REV groups, at both age points (P<0.01; Fig. 5G, H, M). By 12 months, cortical TNF-α expression in REV mice did not differ from LFD mice (P=0.43).
Multiplex analyses were conducted for several markers in plasma to assess inflammatory response in the periphery. 5–6 animals were left in each group after outliers (>2 s.d. from group mean) were removed, (up to 1 per group). There were no significant main effects of diet or genotype (Ps>0.073), and no interactions (Ps>0.066) for IL-1b, IL-6, IL-10 or TNF-α (Fig. 5I–L).
High fat diet leads to greater Aβ accumulation and tau-phosphorylation in APP/PSEN1 mice
At 9.5 months there was a slight trend toward increased tau phosphorylation observed in APP/PSEN1 mice, but this difference was not significant (P=0.08, Fig. 6A, C), and there was no effect of the HFD (P=0.52). By 12.5 months, APP/PSEN1 mice that remained on the HFD showed greater levels of tau phosphorylation than WT-HFD mice (P<0.05), an effect that was not observed in either of the other two diet groups (Ps>0.9, Fig. 6B, C).
Figure 6. High fat diet leads to greater tau phosphorylation and Aβ accumulation in APP/PSEN1 mice.

Tau phosphorylation in hippocampus was calculated as p-tau/Tau, made relative to WT-LFD mice. A,C) At 9.5 months there were no significant effects of genotype of diet on tau phosphorylation (Fs1, 20<3.38, Ps>0.08). B,C) By 12.5 months, there were no overall main effects of diet or genotype (Fs<1.78, Ps>0.12), but pairwise comparisons indicated that tau phosphorylation had increased in the APP/PSEN1 compared to WT in the HFD mice only (P=0.026), but not LFD (P=0.932), or REV groups (P=0.899).
Amyloid-β levels were measured in cortex of APP/PSEN1 mice only using ELISA. D) At 9.5 months soluble Aβ1-40 levels did not differ between LFD and HFD fed mice (F1, 18=3.958, P=0.062). Insoluble Aβ1-40 and Soluble Aβ1-42 were significantly elevated in HFD mice (F1, 18=11.483, P<0.01; F1, 18=4.915, P<0.05 respectively). Insoluble Aβ1-42 was unchanged according to diet (F1, 18=0.255, P=0.619). Data are combined for males and females, N=9–11 per group. E) At 12 months female mice had greater soluble Aβ1-40 than males, but there were no effects of diet for either group (F2, 35=1.71, P=0.196). There were no effects of sex on any other measure. Data are shown collapsed across sex for all measures. Insoluble Aβ1-40, Soluble Aβ1-42, and Insoluble Aβ1-42 were all significantly altered according to dietary treatment (F2, 38=5.966, P<0.006, F2, 38=8.026, P<0.001, F2, 38=5.653, P<0.01, respectively). N=12–15 per group. F,H) At 9.5 months expression of RAGE protein was increased by APP/PSEN1 genotype (F1, 25=6.3, P<0.05), and HFD (F1, 25=19.2, P<0.001). A pairwise comparisons following the significant interaction (F1, 25=4.99, P<0.05) showed that APP/PSEN1 had greater RAGE expression than each of the other groups (Ps<0.05). N=5–9 per group. G,H) At 12 months there RAGE expression was elevated in APP/PSEN1 mice (F1, 22=17.98, P<0.001). Treatment with HFD led to elevated RAGE expression (F2, 22=12.82, P<0.001). Follow up comparisons indicated that although REV diets decreased RAGE expression in WT mice, the same was not observed in APP/PSEN1. N=3–6 per group. *, **, *** P<0.05, 0.01, 0.001 indicates differences between groups as marked based on Univariate ANOVA (Tukey) or post hoc analyses (Bonferroni) following significant omnibus ANOVA. C,H) Representative Western blots. Where only data for APP/PSEN1 mice are present, bars are in the order: D) LFD, HFD, E) LFD, HFD, REV.
We measured guanidine-HCl-soluble and – insoluble Aβ1-40 and Aβ1-42 in cortex of APP/PSEN1 mice by ELISA to determine the extent of differences in our HFD group, and how far this damage may be attenuated by REV diets. No differences were found according to sex so data were collapsed for analysis. Soluble Aβ1-40 was slightly but not significantly increased in HFD (P=0.062, Fig. 6D), whereas insoluble Aβ1-40 was significantly greater in HFD mice (P<0.01). In contrast, while soluble Aβ1-42 was also higher in HFD mice (P<0.05), insoluble Aβ1-42 was not different among the groups (P=0.619). The ratio of total Aβ1-42/1-40 did not differ between the groups (LFD 2.74 +0.32, HFD 2.09 +0.35; P =0.19). At 12 months, there was a significant effect of mouse sex only on the measure of soluble Aβ1-40 where females had greater soluble Aβ1-40 than males (P<0.05), although there was no effect of diet (Ps>0.196). (Data in Fig 6E are presented as combined males and females for simplicity.) For all other analyses, data were combined across sex. Early HFD appeared to have increased production of soluble Aβ1-42 (Ps<0.01) and this was not rescued by REV diet. Insoluble Aβ1-40 and Aβ1-42 were significantly decreased in REV compared to HFD (P<0.01). Total Aβ1-42/1-40 ratios were greatest in REV mice, but this was not significant (P=0.053; LFD 1.94 +0.14, HFD 2.29 +0.15, REV 2.46 +0.16).
Neprilysin (CD10) is involved in Aβ degradation and altered expression could explain changes in Aβ levels. We found no significant effects of diet or genotype at either age point (Fs<1.46, Ps>0.26, data not shown).
Failure to clear Aβ, or transport it from CSF into brain is one potential explanation for higher Aβ levels. We therefore checked levels of RAGE, which mediates transport of Aβ into the brain (Deane, et al., 2003). At 9.5 months, RAGE in hippocampus was elevated in HFD-APP/PSEN1 mice compared to WT and LFD mice (P<0.001, Fig. 6F, H). By 12 months, RAGE expression was still elevated in APP/PSEN1 mice compared to WT, (P<0.001, Fig 6G, H). The effect of HFD increasing RAGE expression also persisted (P<0.001). Reversal of high fat intake resulted in lower RAGE expression in WT mice, but the same rescue was not observed in APP/PSEN1 mice.
Discussion
To model a lifetime of poor diet and induce an obese state, WT and APP/PSEN1 mice were fed HFD (60% calories from lard) from 2 months of age. Mice on HFD consumed more fat and calories overall, which led to significant weight gain. Instigation of LFD feeding following 28 weeks of HFD led to profound weight loss and reversal of obesity, and diabetic phenotypes, as well as cognitive, and neuropathological changes. APP/PSEN1 mice were more affected by diets in almost all of these categories, despite lack of an overt difference in weights between genotypes except in one group. It is unclear whether the greater weights recorded in female APP/PSEN1-HFD mice after the group was divided at 9.5 months reflects a true increased susceptibility to weight gain in these mice as has been observed in some studies (Mody, et al., 2011), or whether it reflects a sampling bias created during the random allocation of mice to continued HFD or REV diets. Genotypic difference in response to HFD was not observed in several other studies with similar design (Graham, et al., 2016, Ramos-Rodriguez, et al., 2014), and was not observed in the males in our study, or prior to 9.5 months.
Despite similar weights, glucose clearance was significantly impacted in APP/PSEN1 mice. Glucose tolerance tests revealed a greater initial glucose spike in APP/PSEN1 mice on HFD at both ages, plus slower glucose clearance in those mice. Deterioration of glucose tolerance is likely due to HFD-induced insulin resistance. The decreased ability of APP/PSEN1 and other Alzheimer’s disease mouse models to clear a glucose load may be due not to differences in insulin resistance, but to failure to produce, store, and release enough new insulin to counter the elevated circulating glucose (Maesako, et al., 2012b, Mody, et al., 2011). A similar effect of impaired glucose tolerance in a different APP/PSEN1 transgenic line was linked to differences in a number of factors involved in insulin signaling in the brain (Mody, et al., 2011). Competition between amyloid and insulin or amylin for insulin degrading enzyme has also been proposed as a contributory factor to altered insulin function (Schilling, 2016). It has also been proposed that brain-derived amyloid may accumulate in the pancreas, compromising integrity and function of islets (Vandal, et al., 2015, Vandal, et al., 2014). The APP protein is also found in pancreas, however mutations in these Alzheimer’s disease mouse models are driven by brain-specific promoters and thus pancreatic amyloid is not likely to be produced in situ owing to the inserted human mutations. This additional negative role for amyloid in Alzheimer’s disease supports the argument for the bidirectional link between Alzheimer’s disease and diabetes.
The REV diet led to fast and sustained weight loss, and completely reversed abnormal glucose clearance, indicating that although APP/PSEN1 mice were more affected by the diets, this was not a permanent state. The forced weight reduction was designed to allow us to identify which neural and behavioral processes were indeed reversible following chronic high fat feeding. It is important to note that this type of dramatic weight loss of 30–40% is not readily achievable in clinical populations, particularly in such a short time frame (Batsis, et al., 2016). The only exception is in the case of bariatric surgeries where food intake is far more limited, following which improvements in cognitive ability have been described (Alosco, et al., 2014, Spitznagel, et al., 2015). Nevertheless a goal of healthy weight maintenance throughout life should be maintained given the deficits in learning and memory and cognitive function in obese populations (Elias, et al., 2003, Elias, et al., 2005, Waldstein and Katzel, 2006).
Negative effects of HFD on cognitive function have been observed within just a few weeks of starting the diets (Knight, et al., 2014). Unfortunately, the reality in human populations is that obesity is a chronic problem spanning years or decades. At both 9 and 12 months a modest deficit in Y-maze in APP/PSEN1 mice was amplified by chronic HFD. Performance was poorer in HFD mice overall indicating that cognitive deficits were not only due to pathology in APP/PSEN1 mice. Performance in the Y-maze is calculated from percent time spent exploring the novel arm permitting assessment despite differing activity levels. Although Y-maze exploration was sensitive to diet and genotype, activity differences cannot fully explain the different patterns of exploration of the novel arm given that all mice entered all three arms at least once, and locomotor activity was not lower in APP/PSEN1 mice than WT. Reversal of the obese state in the mice significantly rescued cognitive performance on this task, and this effect appeared more dramatic in APP/PSEN1 mice. The same pattern of behavioral deficits in APP/PSEN1 mice, worsened by HFD, and rescued by dietary reversal was observed in the nest building task. Nest building is analogous to a ‘Daily Living Activity’ for mice (Torres-Lista and Gimenez-Llort, 2013) and requires intact executive function. Although male and female mice undergo different evolutionary and hormonal pressures regarding nest building for the protection of offspring, all adults build nests given the opportunity and this task is sensitive to familial Alzheimer’s disease mutations, to diet and streptozotocin-induced diabetes, and in both sexes (Filali, et al., 2009, Torres-Lista and Gimenez-Llort, 2013, Wesson and Wilson, 2011, Yeh, et al., 2015).
Methodological variations among published studies often preclude the finding of consistent results, even when ostensibly the same task has been employed. Length and age-of-onset of dietary treatment, age, sex and genotypic mutations and the background of mice can all affect magnitude of deficits observed. Fat content and source (animal or vegetable-derived, (Julien, et al., 2010, Koivisto, et al., 2014)) are critical in determining physiological response, and age of initiation and length of treatment vary widely amongst studies. Experimental high fat diets range from 20–70% fat content. Some studies begin diets in young mice (2 months, e.g. (Maesako, et al., 2012a, Theriault, et al., 2016)) and others not until far later in the life-span and disease processes (Mody, et al., 2011, Theriault, et al., 2016). Nevertheless, cognitive deficits have been reported in a wide range of cognitive tasks, driven both by diet and genotype, although magnitude of deficits and compounding of diet and genotype effects vary (Knight, et al., 2014, Maesako, et al., 2012b, Peng, et al., 2014, Ramos-Rodriguez, et al., 2014, Yeh, et al., 2015). Even within our own study, the finding of cognitive deficits was not universal across tasks. We did not find significant differences among groups in commonly used tasks of Y-maze alternation and fear conditioning, both of which require intact hippocampal function. This could be due to task complexity or other methodological components age of testing and duration of diets (these tasks were begun at 6 months of age), although HFD had clearly induced significant weight gain and led to a higher fasted blood glucose level by 6 months. We report significantly decreased activity in HFD mice, which is an important finding on its own as it may reflect reversible neurological changes not assessed in this study. These issues highlight the importance of task choice and the need for inclusion of multiple measures to avoid false negatives, or overstatement of outcomes.
Oxidative changes have been observed after short-term HFD feeding (e.g. 1 month), in the absence of changes in Aβ, indicating that it may be an early initiating factor in the disease process (Studzinski, et al., 2009). In our study HFD did not increase lipid peroxidation in the brain as measured by MDA. In fact, MDA was lower in HFD mice. We do not propose that HFD is protective against oxidative stress, rather that there has been some change in the lipid metabolism of these mice that causes this result.
Chronic inflammatory response is a hallmark of Alzheimer’s disease that ultimately contributes to irreversible damage within the brain (Lue, et al., 2010). Astrocytes play a key a role in amyloid degradation, and enhanced microglial activation is typically focused around areas of Aβ deposition (Graham, et al., 2016, Ramos-Rodriguez, et al., 2014, Simard, et al., 2006). We therefore measured both GFAP and TNF-α in hippocampus by Western blot as a marker for glial activation. Both were increased significantly by HFD, and the increase in GFAP was greater in APP/PSEN1 mice. Both GFAP and TNF-α were also responsive to the reversal of high fat intake, indicating systems that may be responsive to therapeutic intervention. Similar changes in inflammatory response were seen in WT and APP/PSEN1 mice, including GFAP, Iba-1, s100b, and IL-6 (Graham, et al., 2016, Pistell, et al., 2010, Simard, et al., 2006, Yeh, et al., 2015). Increased fat intake in C57Bl6 mice caused isolated microglia to secrete more TNF-α, but the same effect was not observed in peripheral macrophages, indicating that despite the strong effect of peripheral adiposity, there are specific changes in the brain (Puig, et al., 2012). Indeed, although peripheral inflammatory markers showed a strong trend toward greater inflammatory response in the HFD-fed APP/PSEN1 mice the variability seen in the data indicates that this is likely not the primary, or only source of inflammatory response in the brain. To obtain a thorough understanding of these changes, a full panel of cytokines and chemokines and pro-inflammatory molecules should be studied in future. Identifying the changes that most strongly correlate with cognitive decline, and which are most amenable to reversal, may help identify the best areas for therapeutic targets. For example, high fat and sugar consumption increased a number of pro-inflammatory markers in prefrontal cortex of young (<5 months) C57Bl/6 mice (including IL-6, IL-Iβ, but not TNF-α)(Carlin, et al., 2016). Interestingly, voluntary exercise (running wheels) prevented this increase only in CCL2 and CXCL10.
HFD has been shown to have a negative effect on the two major pathological pathways of Alzheimer’s disease; tau phosphorylation and Aβ accumulation. Hyperphosphorylation of tau ultimately contributes to microtubule breakdown and formation of neurofibrillary tangles, both of which can then contribute to learning and memory deficits. Insulin deficiency may contribute to tau phosphorylation through modulation of activity of tau kinases (Bosco, et al., 2011, Zhang, et al., 2016). HFD increased tau phosphorylation in the APP/PSEN1 line, but not in wild-type mice, an effect that was observed in cortex but not hippocampus (Ramos-Rodriguez, et al., 2014). Here we report an increase in tau phosphorylation in the hippocampus of APP/PSEN1 mice that was driven by high fat diet, but that was prevented by dietary reversal.
Amyloid accumulation may result from increased production, failed degradation and clearance, or a combination of these factors. HFD can promote cleavage of APP by altered regulation of BACE1 trafficking, and thus ultimately resulting in greater production of Aβ (Dall'Armellina, et al., 2013, Maesako, et al., 2015). Studies that have used long-term HFD interventions, or earlier starting points tend to find greater changes in amyloid accumulation (Maesako, et al., 2012a, Maesako, et al., 2012b, Shie, et al., 2002). Aβ clearance is at least partially dependent on activity of astrocytes and microglia. Plaques are often surrounded by microglia, which may explain why HFD-fed animals exhibit a heavier microglial burden than WT or LFD mice (Ramos-Rodriguez, et al., 2014, Shi, et al., 2013). The reversible effect of HFD on both GFAP and TNF-α, supports the hypothesis that glial cells play an important role in the changes in Aβ level reported here. Nevertheless, whether the detected changes occurred in glial cells which were directly associated with plaques was not determined as part of this study.
Increased formation of AGEs and expression of RAGE are seen in both Alzheimer’s disease and type 2 diabetes (Guglielmotto, et al., 2012, Srikanth, et al., 2011, Yan, et al., 2009). We found significant upregulation of RAGE by both HFD and APP/PSEN1 genotype, with the greatest effects in the combination group. RAGE was increased in blood vessels of 6 months old APP/PSEN1 mice by 8 weeks of HFD feeding (32% fat), in combination with microglial activation. This was despite no change in plaque load (Herculano, et al., 2013) indicating that amyloid load per se may not be the main factor in cognitive decline. The increase in RAGE was prevented by treatment with carnosine, which has anti-glycation properties, indicating that there may be an area for intervention, and indeed our data suggest that dietary reversal was effective against RAGE over-expression, although the effect was mostly limited to WT mice. Wistar rats fed a 45% fat diet for 17 weeks also increased RAGE expression although this was unaffected by treatment with proposed anti-inflammatory blackberry extract in the HFD-fed mice (Meireles, et al., 2015).
The literature is mixed as to extent of increases in soluble versus insoluble Aβ, changes in plaque deposition (number and size) and even differences in cortical compared to hippocampal areas (Graham, et al., 2016, Knight, et al., 2014, Ramos-Rodriguez, et al., 2014, Takeda, et al., 2010, Yeh, et al., 2015). As with other measures, this may depend largely on type and duration of dietary treatment. Our data are in agreement with the majority of studies, which point to a clear ability of HFD to increase plaque deposition in mouse models of AD e.g. (Ettcheto, et al., 2016, Levin-Allerhand, et al., 2002, Maesako, et al., 2015). There is less consensus as to the specific cause of this accumulation with evidence for and against enzymatic activity, amyloid transport across the blood brain barrier, and glial cell response as driving forces behind the pathogenesis. HFD has also been shown to drive BBB leakage. Vascular integrity was more compromised in APPSL mice than WT following just 12 weeks of 23% HFD (Loffler, et al., 2016). 27 weeks feeding with 60% HFD in APP/PSEN1 mice increased cortical haemorrhage burden as indicated by Prussian blue staining (Ramos-Rodriguez, et al., 2014). If the BBB is compromised, the influx and efflux of AB would be less dependent on transport by LRP-1 and RAGE (Deane, et al., 2003, Zlokovic, et al., 2010). Advanced magnetic resonance imaging techniques indicate that altered neurovascular function and BBB integrity, including BBB leakage and lower cerebral blood flow, are indeed early pathological events in Alzheimer’s disease that are strongly related to cognitive decline (Montagne, et al., 2015,van de Haar, et al., 2016a,van de Haar, et al., 2016b). These findings are particularly significant because they suggest that amelioration of these neurovascular factors, such as by reversal of high fat diet intake, could aid in stabilization of the BBB and thereby directly contribute to slowing cognitive decline.
Conclusions
In this study we showed that HFD feeding led to weight gain, diabetic state (glucose intolerance), cognitive impairment, neuroinflammatory response, tau phosphorylation and Aβ accumulation in APP/PSEN1 mice. Each of these was reversed, at least partially, by substituting a LFD for HFD, even after 7 months of HFD feeding when neuropathological burden was reasonably advanced. These data support the adverse role of HFD in Alzheimer’s disease pathogenesis and suggest that early to midlife diet may be critical in triggering accelerated amyloid pathology. Although this suggests that lifestyle changes, or targeted pharmacological interventions, may be effective against pathological and cognitive changes, it also highlights the importance of early interventions and preventative measures to protect neural health in aging and disease. Extreme weight loss as observed here in our mouse model, and the instigation of such austere dietary change, is complicated in human populations, particularly in the elderly or cognitively compromised individuals. Nevertheless, continued work to clarify the mechanisms by which dietary reversal can improve cognition and neural function is critical to the development of new therapeutics to obtain the same cognitive improvement in both obese, and non-obese individuals.
Supplementary Material
Figure 4. HFD-induce cognitive deficits are reversed by switching to LFD in both APP/PSEN1 and WT mice.

Locomotor activity. A) At 6 months of age male mice on HFD travelled less in the activity chambers than LFD (F1, 77=28.031, P<0.001), but activity was not dependent on genotype, (Ps>0.757). Female mice explored similar amounts regardless of diet and genotype (Ps>0.110). N=15–29 per group. B) At 9 months of age decreased activity induced by HFD-feeding was observed in both males (F1, 68=22.23, P<0.001) and females (F1, 66=10.067, P<0.01). Hyperactivity was observed in male APP/PSEN1 mice, but only in the LFD condition (diet X genotype interaction, F1, 68=4.085, P<0.05). There were no differences according to genotype in female mice (Fs1, 66<1.585, Ps>0.212). N=15–26 per group. C) Locomotor activity. Significant recovery of HFD-induced hypolocomotion following REV diets was observed in both males (Ci; Main effects of both Genotype and Diet (Ps<0.05), and significant interactions: Session X Diet F4, 82=2.95, P<0.05, Genotype X Diet F2, 41=4.50, P<0.05) and females (Cii; Main effect of Diet, Session x Diet interaction (Ps<0.05); Genotype x Diet x Session F4, 86=2.64, P<0.05). Y maze. D) APP/PSEN1 mice spend less time in novel arm, (males F1, 77=4.47, P<0.05; females F1, 66=11.92, P<0.001). HFD feeding also decreased percent time spent in novel arm (males F1, 77=13.97, P<0.001; females F1, 66=4.23, P<0.05). There was no Diet X Genotype interaction for either sex (Fs<0.87, Ps>0.35). N=15–29 per group. E) In male mice poorest performance (least percent time spent in novel arms) was observed in HFD-APP/PSEN1 mice. There were significant main effects of both genotype (F1, 42=4.41, P<0.05) and diet (F2, 42=6.93, P<0.01) but no interaction among the factors (F2, 42= 1.68, P=0.20). A similar pattern was observed in female mice with poorest performance recorded in HFD-APP/PSEN1 mice and a recovery of function in REV mice (F2, 43=5.07, P<0.05). There was no significant effect of genotype (F1, 43=1.32, p=0.26). Nest building. F) Nest building in male mice was poorer in APP/PSEN1 mice than WT (F1,27=9.06, P<0.01) and in HFD compared to LFD mice (F1,27=21.93, P<0.001). In female mice significant impairments in nest building behavior were only observed in the APP/PSEN1 mice on HFD (Diet X Genotype interaction, F1,21=15.08, P<0.001). G) Both male and female APP/PSEN1 mice built poorer quality nests than WT counterparts regardless of diet (male F1,40=25.66, P<0.001, female F1,42=23.78, P<0.001). HFD also significantly impaired nest quality, and this deficit was reversed in REV mice (males F2,40 =11.73, P<0.001, females F2,42=22.65, P<0.001). *, **, *** P<0.05, 0.01, 0.001 indicates difference compared to all other diet groups, or between two diet groups as marked; a, b, c P<0.05, 0.01, 0.001 according to genotype (WT vs. APP/PSEN1 regardless of diet).
Highlights.
High fat diet-induced cognitive deficits were reversible by low fat diet treatment
High fat diet-induced inflammatory response was reversible by low fat diet treatment
High fat diet-induced acceleration of β-amyloid accumulation was partially reversible
APP/PSEN1 mice on high fat diet showed greater glucose intolerance than wild-type mice
Acknowledgments
The authors would like to acknowledge funding provided by the National Institute on Aging R01 AG038739 to FEH. JMW was also supported by T32 Training Grant DK7061, and DK050435 to JMM. All behavioral experimentation was performed in the Vanderbilt Neurobehavioral Core Facility, which receives support from the Vanderbilt Kennedy Center through the National Institute for Child Health and Human Development, U54HD083211. The Vanderbilt Hormone Assay Core is supported by the Vanderbilt Diabetes Research Training Center and grants DK059637 and DK020593. We would like to acknowledge Navila Sharif, Casey Freeman and Robert Zhang for their technical assistance in conducting these experiments.
Abbreviations
- HFD
High fat diet
- LFD
Low fat diet
- HFD to LFD; REV
Reversal diet
- Aβ
β-amyloid
- APP
Amyloid Precursor Protein
- PSEN1
Presenilin 1
- RAGE
Receptor for Advanced Glycation End Products
- AGE
Advanced Glycation End Products
- EZM
Elevated Zero Maze
- GTT
Glucose Tolerance Test
- TNF-α
Tumor Necrosis Factor-alpha
- IL:IL-1b, IL-6, IL-10
Inter-Leukin
- ELISA
Enzyme-linked Immunosorbent Assay
- GFAP
Glial Fibrillary Acidic Protein
- MDA
Malondialdehyde
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alosco ML, Spitznagel MB, Strain G, Devlin M, Cohen R, Paul R, Crosby RD, Mitchell JE, Gunstad J. Improved memory function two years after bariatric surgery. Obesity. 2014;22(1):32–8. doi: 10.1002/oby.20494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's-Association. 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 2015;11(3):332–84. doi: 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Batsis JA, Gill LE, Masutani RK, Adachi-Mejia AM, Blunt HB, Bagley PJ, Lopez-Jimenez F, Bartels SJ. Weight Loss Interventions in Older Adults with Obesity: A Systematic Review of Randomized Controlled Trials Since 2005. Journal of the American Geriatrics Society. 2016 doi: 10.1111/jgs.14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beydoun MA, Beydoun HA, Wang Y. Obesity and central obesity as risk factors for incident dementia and its subtypes: a systematic review and meta-analysis. Obesity reviews : an official journal of the International Association for the Study of Obesity. 2008;9(3):204–18. doi: 10.1111/j.1467-789X.2008.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19(4):939–45. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. Journal of cellular and molecular medicine. 2011;15(9):1807–21. doi: 10.1111/j.1582-4934.2011.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman LB, Thompson MM, Moreno HN, Ellacott KL. Regional astrogliosis in the mouse hypothalamus in response to obesity. The Journal of comparative neurology. 2013;521(6):1322–33. doi: 10.1002/cne.23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin JL, Grissom N, Ying Z, Gomez-Pinilla F, Reyes TM. Voluntary exercise blocks Western diet-induced gene expression of the chemokines CXCL10 and CCL2 in the prefrontal cortex. Brain, behavior, and immunity. 2016 doi: 10.1016/j.bbi.2016.07.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Armellina E, Ferreira VM, Kharbanda RK, Prendergast B, Piechnik SK, Robson MD, Jones M, Francis JM, Choudhury RP, Neubauer S. Diagnostic value of pre-contrast T1 mapping in acute and chronic myocardial infarction. JACC Cardiovascular imaging. 2013;6(6):739–42. doi: 10.1016/j.jcmg.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Deacon RM. Assessing nest building in mice. Nature protocols. 2006;1(3):1117–9. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nature medicine. 2003;9(7):907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Dixit S, Bernardo A, Walker JM, Kennard JA, Kim GY, Kessler ES, Harrison FE. Vitamin C Deficiency in the Brain Impairs Cognition, Increases Amyloid Accumulation and Deposition, and Oxidative Stress in APP/PSEN1 and Normally Aging Mice. ACS chemical neuroscience. 2015;6(4):570–81. doi: 10.1021/cn500308h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D'Agostino RB. Lower cognitive function in the presence of obesity and hypertension: the Framingham heart study. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity. 2003;27(2):260–8. doi: 10.1038/sj.ijo.802225. [DOI] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D'Agostino RB. Obesity, diabetes and cognitive deficit: The Framingham Heart Study. Neurobiology of aging. 2005;26(Suppl 1):11–6. doi: 10.1016/j.neurobiolaging.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Ettcheto M, Petrov D, Pedros I, Alva N, Carbonell T, Beas-Zarate C, Pallas M, Auladell C, Folch J, Camins A. Evaluation of Neuropathological Effects of a High-Fat Diet in a Presymptomatic Alzheimer's Disease Stage in APP/PS1 Mice. J Alzheimers Dis. 2016;54(1):233–51. doi: 10.3233/JAD-160150. [DOI] [PubMed] [Google Scholar]
- Filali M, Lalonde R, Rivest S. Cognitive and non-cognitive behaviors in an APPswe/PS1 bigenic model of Alzheimer's disease. Genes, brain, and behavior. 2009;8(2):143–8. doi: 10.1111/j.1601-183X.2008.00453.x. [DOI] [PubMed] [Google Scholar]
- Graham LC, Harder JM, Soto I, de Vries WN, John SW, Howell GR. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer's disease. Scientific reports. 2016;6:21568. doi: 10.1038/srep21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, Catalano MG, Smith MA, Perry G, Danni O, Boccuzzi G, Tabaton M. AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation. Neurobiology of aging. 2012;33(1):196.e13–27. doi: 10.1016/j.neurobiolaging.2010.05.026. [DOI] [PubMed] [Google Scholar]
- Gustafson D, Rothenberg E, Blennow K, Steen B, Skoog I. An 18-year follow-up of overweight and risk of Alzheimer disease. Archives of internal medicine. 2003;163(13):1524–8. doi: 10.1001/archinte.163.13.1524. [DOI] [PubMed] [Google Scholar]
- Harrison FE, Hosseini AH, Dawes SM, Weaver S, May JM. Ascorbic acid attenuates scopolamine-induced spatial learning deficits in the water maze. Behavioural brain research. 2009;205(2):550–8. doi: 10.1016/j.bbr.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano B, Tamura M, Ohba A, Shimatani M, Kutsuna N, Hisatsune T. beta-alanyl-L-histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. 2013;33(4):983–97. doi: 10.3233/JAD-2012-121324. [DOI] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiology of aging. 2010;31(9):1516–31. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Kennard JA, Harrison FE. Intravenous ascorbate improves spatial memory in middle-aged APP/PSEN1 and wild type mice. Behavioural brain research. 2014;264:34–42. doi: 10.1016/j.bbr.2014.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight EM, Martins IV, Gumusgoz S, Allan SM, Lawrence CB. High-fat diet-induced memory impairment in triple-transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiology of aging. 2014;35(8):1821–32. doi: 10.1016/j.neurobiolaging.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivisto H, Grimm MO, Rothhaar TL, Berkecz R, Lutjohann DD, Giniatullina R, Takalo M, Miettinen PO, Lahtinen HM, Giniatullin R, Penke B, Janaky T, Broersen LM, Hartmann T, Tanila H. Special lipid-based diets alleviate cognitive deficits in the APPswe/PS1dE9 transgenic mouse model of Alzheimer's disease independent of brain amyloid deposition. The Journal of nutritional biochemistry. 2014;25(2):157–69. doi: 10.1016/j.jnutbio.2013.09.015. [DOI] [PubMed] [Google Scholar]
- Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. The journal of nutrition, health & aging. 2002;6(5):315–9. [PubMed] [Google Scholar]
- Loffler T, Flunkert S, Temmel M, Hutter-Paier B. Decreased Plasma Abeta in Hyperlipidemic APPSL Transgenic Mice Is Associated with BBB Dysfunction. Frontiers in neuroscience. 2016;10:232. doi: 10.3389/fnins.2016.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Beach T, Walker DG. Microglia activation and anti-inflammatory regulation in Alzheimer's disease. Molecular neurobiology. 2010;41(2–3):115–28. doi: 10.1007/s12035-010-8106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Asada M, Watanabe K, Hayashida N, Ihara M, Ito H, Shimohama S, Kihara T, Kinoshita A. Environmental enrichment ameliorated high-fat diet-induced Abeta deposition and memory deficit in APP transgenic mice. Neurobiology of aging. 2012a;33(5):1011.e11–23. doi: 10.1016/j.neurobiolaging.2011.10.028. [DOI] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Hayashida N, Asada-Utsugi M, Watanabe K, Uemura M, Kihara T, Takahashi R, Shimohama S, Kinoshita A. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. The Journal of biological chemistry. 2012b;287(27):23024–33. doi: 10.1074/jbc.M112.367011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesako M, Uemura M, Tashiro Y, Sasaki K, Watanabe K, Noda Y, Ueda K, Asada-Utsugi M, Kubota M, Okawa K, Ihara M, Shimohama S, Uemura K, Kinoshita A. High Fat Diet Enhances beta-Site Cleavage of Amyloid Precursor Protein (APP) via Promoting beta-Site APP Cleaving Enzyme 1/Adaptor Protein 2/Clathrin Complex Formation. PloS one. 2015;10(9):e0131199. doi: 10.1371/journal.pone.0131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meireles M, Marques C, Norberto S, Fernandes I, Mateus N, Rendeiro C, Spencer JP, Faria A, Calhau C. The impact of chronic blackberry intake on the neuroinflammatory status of rats fed a standard or high-fat diet. The Journal of nutritional biochemistry. 2015;26(11):1166–73. doi: 10.1016/j.jnutbio.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Mody N, Agouni A, McIlroy GD, Platt B, Delibegovic M. Susceptibility to diet-induced obesity and glucose intolerance in the APP (SWE)/PSEN1 (A246E) mouse model of Alzheimer's disease is associated with increased brain levels of protein tyrosine phosphatase 1B (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia. 2011;54(8):2143–51. doi: 10.1007/s00125-011-2160-2. [DOI] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311(8):806–14. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Liu J, Tang Y, Liu J, Han T, Han S, Li H, Hou C, Liu J, Long J. High-Fat-Diet-Induced Weight Gain Ameliorates Bone Loss without Exacerbating AbetaPP Processing and Cognition in Female APP/PS1 Mice. Front Cell Neurosci. 2014;8:225. doi: 10.3389/fncel.2014.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010;219(1–2):25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders. Biological psychiatry. 2010;67(6):505–12. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Puig KL, Floden AM, Adhikari R, Golovko MY, Combs CK. Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PloS one. 2012;7(1):e30378. doi: 10.1371/journal.pone.0030378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Rodriguez JJ, Ortiz-Barajas O, Gamero-Carrasco C, de la Rosa PR, Infante-Garcia C, Zopeque-Garcia N, Lechuga-Sancho AM, Garcia-Alloza M. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology. 2014;48:123–35. doi: 10.1016/j.psyneuen.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Schilling MA. Unraveling Alzheimer's: Making Sense of the Relationship between Diabetes and Alzheimer's Disease1. J Alzheimers Dis. 2016;51(4):961–77. doi: 10.3233/JAD-150980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi JQ, Wang BR, Tian YY, Xu J, Gao L, Zhao SL, Jiang T, Xie HG, Zhang YD. Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci Ther. 2013;19(11):871–81. doi: 10.1111/cns.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Jin LW, Cook DG, Leverenz JB, LeBoeuf RC. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport. 2002;13(4):455–9. doi: 10.1097/00001756-200203250-00019. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Spitznagel MB, Hawkins M, Alosco M, Galioto R, Garcia S, Miller L, Gunstad J. Neurocognitive Effects of Obesity and Bariatric Surgery. European eating disorders review : the journal of the Eating Disorders Association. 2015;23(6):488–95. doi: 10.1002/erv.2393. [DOI] [PubMed] [Google Scholar]
- Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Munch G. Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiology of aging. 2011;32(5):763–77. doi: 10.1016/j.neurobiolaging.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Studzinski CM, Li F, Bruce-Keller AJ, Fernandez-Kim SO, Zhang L, Weidner AM, Markesbery WR, Murphy MP, Keller JN. Effects of short-term Western diet on cerebral oxidative stress and diabetes related factors in APP x PS1 knock-in mice. Journal of neurochemistry. 2009;108(4):860–6. doi: 10.1111/j.1471-4159.2008.05798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takechi R, Pallebage-Gamarallage MM, Lam V, Giles C, Mamo JC. Aging-related changes in blood-brain barrier integrity and the effect of dietary fat. Neuro-degenerative diseases. 2013;12(3):125–35. doi: 10.1159/000343211. [DOI] [PubMed] [Google Scholar]
- Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(15):7036–41. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault P, ElAli A, Rivest S. High fat diet exacerbates Alzheimer's disease-related pathology in APPswe/PS1 mice. Oncotarget. 2016 doi: 10.18632/oncotarget.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Lista V, Gimenez-Llort L. Impairment of nesting behaviour in 3xTg-AD mice. Behavioural brain research. 2013;247:153–7. doi: 10.1016/j.bbr.2013.03.021. [DOI] [PubMed] [Google Scholar]
- van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, Hofman PA, Verhey FR, Backes WH. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology. 2016a:152244. doi: 10.1148/radiol.2016152244. [DOI] [PubMed] [Google Scholar]
- van de Haar HJ, Jansen JF, van Osch MJ, van Buchem MA, Muller M, Wong SM, Hofman PA, Burgmans S, Verhey FR, Backes WH. Neurovascular unit impairment in early Alzheimer's disease measured with magnetic resonance imaging. Neurobiology of aging. 2016b;45:190–6. doi: 10.1016/j.neurobiolaging.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Vandal M, White PJ, Chevrier G, Tremblay C, St-Amour I, Planel E, Marette A, Calon F. Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer's disease. FASEB J. 2015;29(10):4273–84. doi: 10.1096/fj.14-268482. [DOI] [PubMed] [Google Scholar]
- Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, Lefrancois D, Virgili J, Planel E, Giguere Y, Marette A, Calon F. Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes. 2014;63(12):4291–301. doi: 10.2337/db14-0375. [DOI] [PubMed] [Google Scholar]
- Waldstein SR, Katzel LI. Interactive relations of central versus total obesity and blood pressure to cognitive function. International journal of obesity. 2006;30(1):201–7. doi: 10.1038/sj.ijo.0803114. [DOI] [PubMed] [Google Scholar]
- Wesson DW, Wilson DA. Age and gene overexpression interact to abolish nesting behavior in Tg2576 amyloid precursor protein (APP) mice. Behavioural brain research. 2011;216(1):408–13. doi: 10.1016/j.bbr.2010.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmer RA, Gunderson EP, Quesenberry CP, Jr, Zhou J, Yaffe K. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Current Alzheimer research. 2007;4(2):103–9. doi: 10.2174/156720507780362047. [DOI] [PubMed] [Google Scholar]
- Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med (Berl) 2009;87(3):235–47. doi: 10.1007/s00109-009-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CW, Yeh SH, Shie FS, Lai WS, Liu HK, Tzeng TT, Tsay HJ, Shiao YJ. Impaired cognition and cerebral glucose regulation are associated with astrocyte activation in the parenchyma of metabolically stressed APPswe/PS1dE9 mice. Neurobiology of aging. 2015;36(11):2984–94. doi: 10.1016/j.neurobiolaging.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yin F, Liu J, Liu Z. Geniposide Attenuates the Phosphorylation of Tau Protein in Cellular and Insulin-deficient APP/PS1 Transgenic Mouse Model of Alzheimer's Disease. Chem Biol Drug Des. 2016;87(3):409–18. doi: 10.1111/cbdd.12673. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid beta-peptide elimination from the brain. Journal of neurochemistry. 2010;115(5):1077–89. doi: 10.1111/j.1471-4159.2010.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.