

Abstract
Background/Aims
Genetic defects in Polycystins -1 or -2 (PC1 or PC2) cause polycystic liver disease associated with ADPKD (PLD-ADPKD). Progressive cyst growth is sustained by a cAMP-dependent Ras/ERK/HIFα pathway leading to increased autocrine/paracrine VEGF-A signalling. In PC2-defective cholangiocytes, store-operated Ca2+ entry (SOCE), intracellular and endoplasmic reticulum [Ca2+]ER levels are reduced, while cAMP production in response to [Ca2+]ER depletion is increased. We hypothesized that in PC2-defective cells, in response to [Ca2+]ER depletion, the Ca2+-inhibitable adenylyl-cyclases AC5 or AC6 are activated by the ER chaperon STIM1 resulting in cAMP/PKA-dependent Ras/ERK/HIFα pathway activation.
Methods/Results
PC2/AC6 conditional double-KO mice were generated (Pkd2/AC6-KO) and compared to Pkd2-KO mice, however no decrease in liver cyst was found and cellular cAMP generated by [Ca2+]ER depletion decreased only by 12%. Conversely, in PC2-defective cells, inhibition of AC5 with siRNA or SQ22,536 and NKY80 significantly reduced [Ca2+]ER depletion-stimulated cAMP production, and pERK1/2 expression and VEGF-A secretion. AC5 inhibitors significantly reduced also growth of biliary organoids derived from Pkd2-KO and Pkd2/AC6-KO mice. Consistent with these data, in vivo treatment with SQ22,536 significantly reduced liver cystic area and cell proliferation in PC2-defective mice. Confocal imaging and proximity ligation assay demonstrated that in PC2-defective cells, after [Ca2+]ER depletion, STIM1 interacts with AC5 but not with Orai1, the Ca2+ channel that mediates SOCE.
Conclusion
in PC2-defective cells, in response to [Ca2+]ER depletion, activation of AC5 results in stimulation of cAMP/ERK1–2 signalling, VEGF production and cyst growth. As shown by in vivo experiments this mechanism is of pathophysiological relevance and may represent a novel therapeutic target.
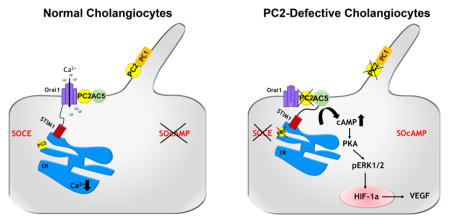
Graphical Abstract
INTRODUCTION
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is characterized by formation of multiple cysts in the kidney, liver and pancreas[1]. In the liver (PLD-ADPKD), multiple fluid-filled cysts progressively dilate and grow until their complications mandate surgery or liver transplantation [2, 3]. PLD-ADPKD is associated with mutations in one of two genes: PKD1, and PKD2, that encode, respectively, for polycystin-1 (PC1) and polycystin-2 (PC2). These two proteins are involved in intracellular signalling, cell differentiation and epithelial morphogenesis[4]. PC1, is mostly localized in the primary cilium and functions as a mechanoceptor and a signaling molecule. PC2 (or TRPP2) also localizes in the cilium, where it physically interacts with PC1, however, the largest pool of PC2 is located in the ER and in the plasma membrane[5].
PC2, a member of the transient receptor potential channels (TRP) family, has the ability to multimerize with other proteins that determine its function[6]. In fact, PC2 can function as a mechano-chemo-osmo-sensor, or a receptor operated Ca2+ channel, depending on its interaction with PC1, TRPV4, ryanodine receptors, etc. In the ER, PC2/TRPP2 interacts with ryanodine[5, 7] and Insp3 receptors[8] and participates to the regulation of agonist-induced Ca2+ release from the ER. ER calcium is lower in PC-2 defective cells[5].
Using PC2 conditional knock-out mice, we have previously demonstrated that VEGF/VEGFR2 stimulate liver cyst growth through paracrine effects on pericystic vascular cells, and autocrine stimulation of cystic epithelium proliferation[9, 10]. We showed that in PC2-defective cholangiocytes, cellular Ca2+ homeostasis is altered while intracellular cAMP is increased, leading to PKA/Ras/Raf/ERK1/2-mediated stimulation of mTOR and HIF−1α−regulated VEGF production[9–13]. We have also found that in PC2-defective cholangiocytes, store-operated calcium entry (SOCE)[12], a mechanism that uses extracellular Ca2+ to replenish intracellular Ca2+ stores[14], is inhibited and cells respond to an acute reduction in ER Ca2+ concentration with increased cAMP production and a PKA-dependent ERK1/2 phosphorylation[12]. Stromal interacting molecule 1 (STIM1) is the molecular sensor that couples reduction in intraluminal [Ca2+]ER and the activation of Ca2+ entry from the plasma membrane[15]. When [Ca2+ ]ER decreases, STIM1 dimerizes and redistributes within the ER close to the plasma membrane. This mechanism activates store-operated Ca2+ entry channels, recently described as members of the Orai channels family[14, 16, 17]. Our previous studies suggest that PC2-defective cells are very sensitive to conditions that further decrease ER Ca2+ stores and trigger oligomerization and membrane translocation of STIM1; this leads to derepression of Ca2+-inhibitable adenylyl cyclases (ACs), and inappropriate production of cAMP (SOcAMP)[18].
Cyclic-AMP is synthesized from ATP by enzymes of the adenylate cyclase (AC) family, which consist of 9 membrane bound (AC1–9) and 1 cytosolic member (soluble AC, sAC)[19]. Among the different AC isoforms, two Ca2+-inhibited ACs (AC6 and AC5) are expressed in cholangiocytes and have similar properties, however the molecular identity of the AC responsible for inappropriate cAMP production in PLD is still unknown. Thus, the aim of this study is to investigate the hypothesis that in PC2 defective cholangiocytes, in response to [Ca2+]ER depletion, STIM1-dependent AC6 or AC5 activation is responsible for PKA/ERK/VEGF-mediated cyst growth.
MATERIAL AND METHODS
Materials and reagents
Mouse models
Pkd2flox/−:pCxCreERTM mice (a kind gift from Dr. S. Somlo, Yale University) is an ADPKD mouse model characterized in prior papers[9, 10, 13]. This conditional knock-out mice, abbreviated as Pkd2-KO is generated by an inducible defect in polycystin 2 (Pkd2flox/−:pCxCreERTM), targeted through a Cre system, fused to the ligand-binding domain of a mutated estrogen receptor, as previously described[9].The deletion of floxed PC2 alleles is achieved 28 days after birth by exposing the mice to tamoxifen (0.2 mg/g/day) for five days. Pkd2-KO mice developed a liver phenotype resembling human ADPKD[9]. AC6 knock-out mice (a kind gift from Dr. M.H. Nathanson, Yale University) were generated by disruption of the AC6 gene by homologous recombination with a targeting vector containing exon1 replaced by the PGKneo-bpA cassette. AC6 −/− mice were fertile, and no physical abnormalities were obvious[20]. Pkd2-KO mice were double crossed with AC6−/− mouse to generate Pkd2fl/fl/AC6−/− mice. Mice with this genotype were treated with tamoxifen (0.2 mg/g/day) for five days in order to obtain Pkd2−/−/AC6−/− mice. Successful deletion of AC6 was assessed by RT/qPCR. Moreover, the levels of expression of AC5 and AC6 was assessed by RT-PCR in liver tissues as well as in cholangiocytes isolated from WT, Pkd2-KO and Pkd2/AC6-KO mice (Supplemenatry figure 1). Animals were treated with AC5 inhibitor SQ22,536(300μg/Kg/day)[21] or with the vehicle (Phosphate Buffered Saline) intraperitoneally (i.p.) every day for 8 weeks, starting 1 week after induction with tamoxifen. All animal experimental protocol were approved by the Yale Animal Care and Use Committee (IACUC) and the Office of Animal Research Support (OARS). At the end of the treatment, mice were sacrificed and liver tissue (two main lobes) were harvested and fixed in formalin and then embedded in paraffin for histochemical analysis; the small liver lobes were snap frozen in liquid nitrogen. Liver slides 4 μm thick were processed and stained with hematoxylin/eosin or other specific markers.
Cell Isolation
Cholangiocytes were isolated as already described[9, 10]. Methods for cell isolation, culture and their full phenotypic characterization have been previously described[22–24]; see also supplementary materials.
Generation of biliary organoids form WT and conditional KO mice
Mouse cholangiocytes isolated from WT, Pkd2-KO and Pkd2/AC6-KO mice were cultured in Matrigel (BD Biosciences) in a non-attaching 8 well chamber. Culture medium was based on DMEM/F12 (GIBCO) supplemented with 1% N2 and 1% B27 (GIBCO) and the growth factors: 50 ng/ml EGF (SIGMA),100 ng/ml FGF4 (R&D), 25 ng/ml HGF (R&D), 10 mM Nicotinamide (Sigma) and 10 uM FSK (SIGMA). Culture medium was supplemented during the first 3 days with 30% Wnt3a (R&D)[25]. When cultured in this conditions, cells grew as organoids and maintained differentiated cholangiocytes such as expression of K19, SOX9, the presence of primary cilium and responsiveness to forskolin (Supplementary figure 2). Images of WT, Pkd2-KO and Pkd2/AC6-KO organoids were taken using 10× magnification for ten days using Axio Observer.Z1 Zeiss Microscopy. The AC5 inhibitors, SQ22,536 (1uM) and NKY80 (1uM) were added at day 5 of culture. To assess the volume of organoids, three diameters were measured for eight random organoids every day for the following 5 days.
RNA Interference Silencing and Real-Time PCR
Gene silencing and Real-Time PCR were performed as previously described[12]; see also supplementary materials.
Intracellular cAMP Assay
Intracellular cAMP Assay was performed as described[12] and see also supplementary material.
Immunohistochemical studies and Morphometric quantization of Keratin 19 and pERK positive structures
Immunohistochemical studies and morphometric quantization of Keratin 19, pERK pERK and PCNA positive structures were performed as decribed[9] and see also supplementary material.
Measurement of VEGF Secretion in Cultured Cells
Measurement of VEGF Secretion in cultured cells were performed as previously described[9] and see supplementary material.
MTS Cell Proliferation Assay
Measurement of cell proliferation (MTS Assay) in cultured cells were performed as previously described[9] and see supplementary material.
Immunofluorescence and confocal microscopy
Polarized WT, Pkd2-KO and Pkd2/AC6-KO cholangiocytes were grown on transwell insert until confluence and stained using antibody against Orai1, STIM1, AC5 and AC6, see supplementary material for details.
Transmission Electron Microscopy
Organoids samples used for transmission electron microscopy (TEM) were processed using standard techniques, see also supplementary material for details.
Proximity Ligation Assay (PLA)
PLA experiments were done using Duolink In Situ reagents (Olink Bioscience)[26], see supplementay material for details.
Statistical Analysis
Results are shown as mean ± standard deviation. Statistical comparisons were made using Student’s t tests, or one-way analysis of variance (ANOVA), where appropriate. Statistical analysis was performed using SAS software (SAS Institute, Onc. Cary, NC). P values <.05 were considered significant.
RESULTS
Blockage of AC5, rather than AC6, inhibits the cAMP/pERK/VEGF pathway in C2-defective cholangiocytes and liver cystic area in Pkd2-KO mice
We previously reported that siRNA silencing AC6 in PC2-defective cholangiocytes in vitro, reduced cAMP production after intracellular Ca2+ store depletion [12], suggesting that AC6 was responsible for increased levels of cAMP and stimulation of cyst growth in Pkd2-KO mice. To confirm this hypothesis in vivo, we generated a double Pkd2/AC6-KO mouse line and evaluated liver cyst area, 8 weeks after induction of Cre-mediated PC2 excision. Contrary to our expectation, we found that the cystic area and the liver/body ratio in Pkd2/AC6-KO mice were not significantly different from Pkd2-KO mice (Figure 1A–B and Table 1). Furthermore, in cultured Pkd2/AC6-KO cystic cholangiocytes, exposed to TPEN (1mM) (to induce intracellular Ca2+ store depletion)[12, 18], cAMP was still clearly increased with respect to WT cells, being reduced only by 12% with respect to Pkd2-KO cells. These data suggest that an additional Ca2+-inhibitable ACs isoform (AC5) is involved in liver cysts growth and that the cAMP increase generated by this second AC is enough to stimulate cyst growth in vivo (Figure 1C and Table 1). By repeating our previously published AC6 silencing experiment [12], we found that the siRNA used to silence AC6, actually decreased also gene expression of AC5 (Supplementary figure 3), which, in hindsight was not surprising, given the high molecular homology between AC5 and AC6[27]. These findings clearly indicate that the cAMP reduction observed in our previous study was likely AC5-dependent. Consistent with this hypothesis, silencing AC5 with specific siRNA (Figure 1C) or treatment with different AC5 inhibitors (SQ22,536 or NKY80 1uM))[28, 29] significantly reduced cAMP in Pkd2-KO and Pkd2/AC6-KO cholangiocytes both at baseline and after treatment with TPEN, to levels similar to those measured in WT cells (Figure 2A–B and Table 1).
Figure 1. Cystic area and [cAMP]i levels are not reduced in Pkd2/AC6-KO mice.

Representative micrographs of liver specimens, labeled with K19 antibody, obtained from Pkd2-KO (n=9) and Pkd2/AC6-KO (n=5) (A). No reduction in cystic area was observed in Pkd2/AC6-KO mice, with respect to WT, as well as in the liver body weight ratio (B). Bar scale=100 μm. (C) cAMP levels induced by TPEN (1mM) were not reduced in Pkd2-KO and Pkd2/AC6-KO cholangiocytes, compared with WT cells (**p< .01 vs unstimulated cells; n=4). On the contrary, in WT, Pkd2-KO and Pkd2/AC6-KO cells treated with AC5 siRNA (50 nM) or scramble RNA (50 nM) the production of cAMP induced by TPEN was significantly inhibited (B) (●p < .05 versus unstimulated cells; ○p < .01 versus TPEN-treated cells; n= 5).
Table 1.
| Cystic Area | |||
|---|---|---|---|
| Sample | Mean±SD (% Cystic Area) | p Value | n |
| Pkd2-KO Ctrl | 5.29±0.85 | 9 | |
| Pkd2-KO + SQ22,536 | 2.95±0.46 | ***p<.0001 | 6 |
| Pkd2/AC6-KO Ctrl | 6.52±0.47 | 5 | |
| Pkd2/AC6-KO + SQ22,536 | 3.56±1.19 | ***p<.0001 | 4 |
| Liver/Body Weight | |||
|---|---|---|---|
| Sample | Mean±SD (g) | p Value | n |
| Pkd2-KO Ctrl | 0.064±0.004 | 9 | |
| Pkd2-KO + SQ22,536 | 0.045±0.003 | ***p<.0001 | 6 |
| Pkd2/AC6-KO Ctrl | 0.073±0.007 | n.s. | 5 |
| Pkd2/AC6-KO + SQ22,536 | 0.042±0.004 | ***p<.0001 | 4 |
| cAMP | |||
|---|---|---|---|
| Sample | Mean±SD (pmo/mg protein) | p Value | n |
| WT Ctrl | 0.15±0.10 | 9 | |
| WT + TPEN | 0.39±0.17 | *p<.05 | 9 |
| WT + TPEN + SQ22,536 | 0.089±0.008 | *p<.05 | 3 |
| WT + SQ22,536 | 0.093±0.003 | 3 | |
| WT + TPEN + NKY80 | 0.11±0.014 | *p<.05 | 3 |
| WT + NKY80 | 0.10±0.008 | 3 | |
| Pkd2-KO Ctrl | 0.44±0.21 | 9 | |
| Pkd2-KO + TPEN | 1.19±0.45 | ***p<.0001 | 9 |
| Pkd2-KO + TPEN +SQ22,536 | 0.28±0.033 | ***p<.0001 | 3 |
| Pkd2-KO + SQ22,536 | 0.18±0.005 | 3 | |
| Pkd2-KO + TPEN + NKY80 | 0.29±0.016 | ***p<.0001 | 3 |
| Pkd2-KO + NKY80 | 0.20±0.010 | 3 | |
| Pkd2/AC6-KO Ctrl | 0.39±0.12 | 9 | |
| Pkd2/AC6-KO + TPEN | 1.04±0.18 | ***p<.0001 | 9 |
| Pkd2/AC6-KO + TPEN+ SQ22,536 | 0.24±0.039 | ***p<.0001 | 3 |
| Pkd2/AC6-KO + SQ22,536 | 0.15±0.015 | 3 | |
| Pkd2/AC6-KO + TPEN+ NKY80 | 0.41±0.073 | **p<.001 | 3 |
| Pkd2/AC6-KO + NKY80 | 0.20±0.005 | 3 | |
| VEGF | |||
|---|---|---|---|
| Sample | Mean±SD (ng/mg protein) | p Value | n |
| WT Ctrl | 211±50 | 3 | |
| WT + Thapsi | 291±80 | *p<.05 | 3 |
| WT + Thapsi + SQ22,536 | 229±43 | n.s. | 3 |
| WT + SQ22,536 | 204±69 | 3 | |
| Pkd2-KO Ctrl | 666±112 | 3 | |
| Pkd2-KO + Thapsi | 1469±247 | 3 | |
| Pkd2-KO + Thapsi +SQ22,536 | 453±85 | **p<.001 | 3 |
| Pkd2-KO + SQ22,536 | 267±65 | **p<.001 | 3 |
| Pkd2/AC6-KO Ctrl | 575±98 | 3 | |
| Pkd2/AC6-KO + Thapsi | 1449±184 | 3 | |
| Pkd2/AC6-KO + Thapsi+SQ22,536 | 492±58 | ***p<.0001 | 3 |
| Pkd2/AC6-KO + SQ22,536 | 252±100 | *p<.05 | 3 |
| Cysts Organoids Volume | |||
|---|---|---|---|
| Sample | Mean±SD (μm3) | p Value | n |
| WT Ctrl | 1.78×107±3.81×106 | 8 | |
| WT + SQ22,536 | 1.62×107±5.12×106 | n.s. | 5 |
| WT + NKY80 | 2.09×107±1.94×106 | n.s. | 4 |
| Pkd2-KO Ctrl | 3.33×107±4.07×106 | *p<.05 | 8 |
| Pkd2-KO + SQ22,536 | 7.68×106±2.17×107 | **p<.001 | 5 |
| Pkd2-KO + NKY80 | 6.48×106±6.98×106 | **p<.001 | 4 |
| Pkd2/AC6-KO Ctrl | 2.73×107±6.95×106 | *p<.05 | 8 |
| Pkd2/AC6-KO + SQ22,536 | 5.83×106±1.75×106 | *p<.05 | 5 |
| Pkd2/AC6-KO + NKY80 | 6.58×106±5.31×106 | *p<.05 | 4 |
Figure 2. Inhibition of AC5 decrease [c-AMP]i, pERK1–2 and VEGF secretion induced by ER- Ca2+ depletion.

Treatment with SQ22,536 (1uM) (A) and NKY80 (1uM) (B) significantly reduced the TPEN(1mM)-induced cAMP levels (n= 3, *** p<.0001 vs TPEN- treated cells, **p<.001 vs TPEN, *p<.05 vs Pkd2-KO cell treated with TPEN. (C) SQ22,536-reduced ERK phosphorylation induced by Thapsigargin (2uM) in Pkd2-KO and Pkd2/AC6-KO cystic cholangiocytes. (**p<.01 vs Pkd2-KO+Thapsigargin and **p<.01 vs Pkd2/AC6-KO+Thapsigargin). (D) Treatment with SQ22,536 (1uM) significantly reduced the VEGF secretions in Pkd2-KO and Pkd2/AC6-KO (** p< .01, ***p<.001 vs CTRL; n=3. SQ=SQ22,536 and NKY=NKY80).
Furthermore, both in Pkd2-KO and in Pkd2/AC6-KO cells, inhibition of AC5 (SQ22,536) blocked the Thaspsigargin (2μM) induced increase in ERK1/2 phosphorylation and VEGF secretion (Figure 2C–D and table 1) as well as cell proliferation (Supplementary figure 5). Further indicating that AC5 is responsible for the cAMP production that activates the ERK1/2-VEGF pathway.
Inhibition of AC5 reduces cyst growth in vitro and in vivo
Biliary organoids currently represent the most advanced cellular model to study biliary physiology and pathophysiology [30, 31]. We generated biliary organoids from WT, Pkd2-KO and Pkd2/AC6-KO mice and grew them, as described in the methods section. Under these conditions, WT, as well as PC2 defective mice formed small spheroids that progressively enlarged. As shown in Figure 3A–B and Table 1 the size of the Pkd2-KO organoids was significantly higher with respect to WT, Pkd2/AC6-KO being intermediate between the two. When organoids were grown in the presence of SQ22,536 or of NKY80 their size, measured at day 5 was significantly lower with respect to that organoids cultured in the absence of AC5 inhibitors (Figure 3A–C and table 1). These data are consistent with a major role for AC5 in mediating liver cyst growth. Contrary to what found in vivo, cultured Pkd2/AC6-KO organoids showed a growth rate that was intermediate between WT and Pkd2-KO (Figure 3C). This difference in growth rate is in line with the small decrease in intracellular cAMP levels shown in Figure 1C, and suggest that there is a small role for AC6 in cyst growth in vitro.
Figure 3. Inhibition of AC5 (SQ,22536) reduces the size of organoids derived from Pkd2-KO and Pkd2/AC6-KO cholangiocytes.

(A) Representative micrographs of organoids derived from WT, Pkd2-KO and Pkd2/AC6-KO mouse cholangiocytes untreated (Control), treated with SQ22,536 (1uM) or with NKY80 (1uM) for 5 days. Bar scale,100 μm. (B) Bar graphs show that the volume is significant reduced in Pkd2-KO and Pkd2/AC6-KO organoids treated with SQ22,536 (1uM) and NKY80 (1uM). (C) Organoids were cultured for 10 days and pictures were taken every day and volume was measured as described in materials and methods. The graph show the rate of different growth rate in WT, Pkd2-KO and Pkd2/AC6-KO (*p<.05 vs WT and ● p<.05 vs Pkd2/AC6-KO; n=4).
Finally, to firmly establish the pathophysiological relevance of these mechanisms, we studied the effects of AC5 inhibition in vivo. To this aim, we treated Pkd2-KO and Pkd2/AC6-KO mice with SQ22,536 (300 μg/Kg/day for 8 weeks, i.p.)[21]. The dosage was well tolerated, without mortality or toxicity. At the end of the treatment, mice were sacrificed, liver tissues were harvested and analyzed. Cystic area was significantly reduced in both Pkd2-KO and Pkd2/AC6-KO mice treated with SQ22,536 as compared to untreated mice (Figure 4A–B). Furthermore, treatment with SQ22,536 decreased also the liver weight/body weight ratio (Figure 4C), the amount of pERK (figure 5A–B) and of PCNA (figure 5C–D) in Pkd2-KO and Pkd2/AC6-KO mice. These results suggest that targeting AC5 activity significantly reduces intracellular levels of cAMP activation of the pERK1/2-VEGF pathway and consequently liver cysts growth. It is worth noting that the effects of AC5 inhibition in PC2-defective mice were similar irrespective to the expression of AC6, further suggesting that AC6 has a negligible role in cyst growth in vivo.
Figure 4. Inhibition of AC5 (SQ,22536) reduces cystic area and liver body weight ratio in Pkd2-KO and Pkd2/AC6-KO mice.

(A) Representative micrographs of K19 stained liver tissues from Pkd2-KO (n=9) and Pkd2/AC6-KO (n=5) mice untreated (CTRL) or treated with SQ22,536, (300 ug/Kg/day i.p.) for 8 weeks (n=6) and (n=4) respectively. Computer-assisted morphometric analysis of K19 positive areas showed a significant reduction in the cystic area as well as in the liver body weight (g) ratio (B), as well as the liver body weight (g) ratio (C), in Pkd2/AC6-KO mice (**p<.01 in Pkd2-KO+SQ22,536 vs. CTRL; ***p<.001 in Pkd2/AC6-KO+SQ22,536 vs. CTRL). Bar scale, 100 μm.
Figure 5. pERK1/2 and PCNA are reduced after SQ22,536 treatments in Pkd2-KO and Pkd2/AC6-KO cholangiocytes.

(A) Rapresentative micrographs showing that treatment with SQ22,536 (300 ug/Kg/day for 8 weeks, i.p.) reduces pERK1/2 in Pkd2-KO and Pkd2/AC6-KO cystic cholangiocytes. (B) Bar graphs showing a computer-assisted morphometric analysis of pERK1/2 expression. (Pkd2-KO CTRL (n=9), Pkd2/AC6-KO CTRL (n=5), Pkd2-KO+SQ22,536 (n=6) and Pkd2/AC6-KO+SQ22,536 (n=4); ***p<.001 in Pkd2-KO SQ22,536 vs. CTRL; **p<.01 in Pkd2/AC6-KO SQ22,536 vs. CTRL). Bar scale, 50 μm. (C) Representative micrographs showing that treatment with SQ22,536 (300 ug/Kg/day for 8 weeks, i.p.) reduces PCNA proliferation marker in Pkd2-KO and Pkd2/AC6-KO cystic cholangiocytes (Pkd2-KO CTRL (n=9), Pkd2/AC6-KO CTRL (n=5), Pkd2-KO+SQ22,536 (n=6) and Pkd2/AC6-KO+SQ22,536 (n=4); ***p<.001 in Pkd2-KO SQ22,536 vs. CTRL; ***p<.001 in Pkd2/AC6-KO SQ22,536 vs. CTRL).
STIM1 and AC5 co-localize in Pkd2/AC6-KO cholangiocytes after ER-Ca2+ depletion
We recently showed that inhibition of STIM1, reduced [Ca2+]ER-stimulated cAMP production in PC2-defective cholangiocytes[12]. Here, we treated WT, Pkd2-KO and Pkd2/AC6-KO cells grown on transwell inserts as polarized monolayers, with thapsigargin to induce [Ca2+]ER depletion, and analyzed the co-localization of STIM1, Orai1 and AC5 or AC6, using confocal microscopy and specific antibodies. We found that in WT cells STIM1 and Orai1 colocalized as expected at the plasma membrane after intracellular calcium store depletion, whereas, in Pkd2-KO and Pkd2/AC6-KO cells the co-localization of STIM1 and Orai1 was missing (Figure 6A). On the contrary, in Pkd2-KO and Pkd2/AC6-KO cholangiocytes, under the same conditions, we clearly observed the co-localization of AC5 but not of AC6 with STIM1 (Figure 6B and supplementary figure 6A).
Figure 6. STIM1 and AC5 co-localize in membrane of Pkd2-KO and Pkd2/AC6-KO but not in WT cholangiocytes.

Confocal images of polarized WT, Pkd2-KO and Pkd2/AC6-KO cholangiocytes co-stained for STIM1/Orai1 (A) or STIM1/AC5 (B). Serial optical sections (0.5 μm thick) were collected for orthorgonal view(x,y,z). STIM1 and Orai1 co-localize in WT cholangiocytes after treatment Thapsigargin (2uM) but not in Pkd2-KO and Pkd2/AC6-KO, while STIM1 co-localize with AC5 but not with Orai1 in Pkd2-KO and Pkd2/AC6-KO cells (B).
To further demonstrate the physical interaction between STIM1 and Orai1 channels or between STIM1 and AC5 or AC6, we used an in situ proximity ligation assay (PLA), based on the formation of fluorescent spots when two proteins of interest in their native status are located within a distance of 40 nm. As shown in Figure 7A, signals indicating STIM1/Orai1 was induced by thaspsigargin in WT cholangiocytes, but not in Pkd2-KO and Pkd2/AC6-KO cells. On the other hand, also in this case in Pkd2-KO and Pkd2/AC6-KO cells but not in WT cells, STIM1 was interacting with AC5 (Figure 7B) while there was no interaction between STIM1 and AC6 (Supplementary Figure 6B). As negative control, we performed similar experiments omitting one on the two antibodies and no signal was detected as expected (not shown).
Figure 7. STIM1 co-localize with AC5 in Pkd2-KO and Pkd2/AC6-KO cells but not in WT cells.

Fluorescence imaging of in-situ proximity ligation assay (PLA), showed interaction of STIM1 and Orai1 (green dots) in WT cells but not in Pkd2-KO and Pkd2/AC6-KO cells after Thapsigargin (2uM) (A) while STIM1 interact with AC5 (green dots) in Pkd2-KO and Pkd2/AC6-KO cells but not in WT cholangiocytes (B).
DISCUSSION
Defective function of PC2, a member of the transient receptor potential channel family is associated with PLD-ADPKD[2, 3]. In this condition multiple large cysts develop in the liver causing a massive increase in liver weight with compression of vital districts, or complications such as rupture or infection of a cyst[2]. The pathophysiology of polycystic and fibropolycystic liver diseases is being actively investigated in the hope to find better treatments for these conditions and also for acquired cholangiopathies, as some pathophysiologic mechanisms are actually similar[2, 11].
It is well established that growth of liver cysts depends on increased cAMP production by PC2 defective cells[9, 10, 13], leading to PKA-dependent activation of Ras/ERK1/2/mTOR/HIF-1α– dependent production of VEGF-A. In turn, VEGF-A stimulates cyst growth and disease progression[9, 10, 13]. The pathophysiological relevance of this pathway is demonstrated by the reduction of cysts volume achieved in vivo in rodents by somatostatin[32], VEFGR2 inhibitors[13] and rapamycin [33], and, in human, by somatostatin analogs[34].
A known feature of cystic cells in ADPKD is also their lower cytoplasmic Ca2+ [5] levels AND impaired intracellular Ca2+ homeostasis. We have shown that in PC2-defective cells ER Ca2+ stores are reduced and store-operated calcium entry (SOCE) is significantly inhibited[12]. We have also shown that altered Ca2+ homeostasis and increased cAMP signaling are linked, as [Ca2+]ER-depletion stimulates cAMP production. It is known that stimulation of SOCE activates AC8, but inhibits AC5/6[17]. Thus, we hypothesized that cAMP production in PC2-defective cells in response to store depletion was due to stimulation of AC5/6 activity.
Cholangiocytes express 7 out of the 9 known ACs isoform, each with distinct subcellular localization[19]. The Ca2+−Calmodulin-stimulated AC8 is activated by SOCE and in response to secretin receptor stimulation and is linked to increased Cl−/HCO3− secretion. On the other hand, the Ca2+-inhibitable AC6 and AC5 isoforms are inhibited by SOCE and inactive at the normal resting [Ca2+]i[17], but can be activated at the [Ca2+]i reported in PC2-defective cholangiocytes[12]. AC6 is expressed in the cilia of cholangiocytes, and is believed to be involved in shear stress-induced signaling[35]. Stimulation of cAMP/PKA is known to have a proliferative effects in cholangiocytes, but, on the other hand, sAC was recently shown to promote bile acid-induced apoptosis[36]. The explanation of the protean effects of this common intracellular mediator, lies in the compartmentalization (microdomains) of ACs and cAMP production[37]. Compartmentalization is achieved through a number of mechanisms, including the presence of specific ACs in multiprotein complex into raft domain, and their link with different A-kinase anchoring proteins (AKAPs)[38].
Previous studies suggested a role for AC5/6 in the pathogenesis of ADPKD. Indeed, Masyuk et al, observed that the primary cilium of a rat cholangiocytes cell line expresses a protein complex including AKAP150 and ACs5/6 [35]. In renal epithelial cells, Choi and colleagues showed that AC5/6 is present in a proteins complex with PDE4C and PC2[38]. These authors hypothesized that PC2 functions as a Ca2+ entry channel able to inhibit the activity of the Ca2+ -sensitive AC5/6. In these studies the authors did not distinguish among the two isoforms. Indeed, AC5 and AC6 share several functional and molecular features and most studies, refer to ACs5/6, without attempt to separate the two isoforms.
We have reported that cAMP production after ER Ca2+ depletion in Pkd2-KO cholangiocytes was reduced by silencing AC6 [12], a result apparently at odds with the present findings. However, we later found that silencing AC6 with siRNA resulted also in a significant silencing of AC5 expression, an observation that reconciliates the past and present findings.
It is also interesting to note that while in Pkd2/AC6-KO double knock-out mice, the total liver cysts area was not different from Pkd2-KO mice, in Pkd2/AC6-KO cells [Ca2+]ER-depletion stimulated cAMP production was 12 % lower than in single KO mice, and the growth of biliary organoids was reduced suggesting that part of cAMP is produced by AC6, likely in cilia, but these reduced amounts are not enough to prevent cyst formation.
Thus, we investigated the role of AC5 by silencing AC5 with specific siRNA and by testing the effects SQ22,536 and NKY80, two known inhibitors of AC5[28, 29], on cAMP production after store depletion. After AC5 silencing and with both inhibitors, cAMP was reduced almost to WT levels. AC5 inhibitors also inhibited ERK1/2 phosphorylation, VEGF secretion and cell proliferation in PC2-defective cells, suggesting a major role for AC5. Studies on cholangiocytes monolayers may not be fully representative of the cellular physiology, as this preparation lacks of a three-dimensional architecture. Epithelial organoids represents a novel technical development that overcomes part of these limitations. WT and mutant cholangiocyte monolayers, dispersed into Matrigel and exposed to a short stimulus with Wnt3a [25] grow as organoids that progressively enlarge. We found that organoids deriving form Pkd2-KO mice enlarged significantly faster than those deriving from WT mice. Those deriving from the double Pkd2/AC6-KO grew at an intermediate rate. Using this experimental approach, we found that inhibition of AC5 significantly reduced the size of the organoids in single and double mutant, confirming that that AC5 has a central role in the signaling mechanisms leading to cyst enlargement. An AC5-KO mouse to generate a triple PC2/AC6/AC5 mutant was not available and most likely, even if available, it would not be viable given the functions of these two isoforms in heart, brain and kidney[39, 40]. Therefore, to investigate the role of AC5 in vivo, we exposed Pkd2/AC6-KO and Pkd2-KO mice to SQ22,536, at a dose that has been reported to inhibit only AC5 (300mg/Kg/day)[21]. Our results clearly show a significant decrease of cysts volume in treated mutant organoids suggesting a key pathogenetic role for AC5/6 in PLD-ADPKD. These data and our previous findings establish that PC2 is a necessary component of the SOCE mechanisms, and that in its absence, AC5-dependedent cAMP signaling is instead activated, leading to ERK1/2 phosphorylation, VEGF production and cyst growth.
Activation of AC5 could be an effect of decreased inhibition by cytoplasmic calcium in the absence of SOCE. However, based on our previous observation that cAMP production stimulated by ER store depletion was inhibited by exposure to STIM1 inhibitors (store-operated cAMP signaling), we decided to investigate this mechanism further. It is known that STIM1 at the ER membrane, senses the reduction in ER Ca2+ concentration, dimerizes and binds to the Orai1 Ca2+ channel at the plasma membrane. Since physical interaction of these proteins is necessary to induce SOCE to replenish the intracellular stores and in particular the ER, a tightly controlled assembly mechanism is required. When the ER is loaded with Ca2+ STIMs distribute homogeneously at the ER-membrane and are inactive. When the ER store is depleted STIM1 dimerizes following the Ca2+ dissociation from the EF-hand domain. This leads to unfolding and destabilization of the EF-hand-SAM complex which triggers activation and oligomerization of STIM1[41]. Cluster of STIM1 at the junction ER/plasma membrane junction are then able to interact in lipid rafts with Orai, and/or TRPC1 (a channel also mediating Ca2+ influx); Orai also binds AC8 through a sequence in the N-terminal of AC8[42]. There is evidence for direct protein–protein interaction between STIM1 and Orai1, but there is also evidence for the involvement of other, as yet unidentified, proteins. An unidentified ‘calcium influx factor’ has been proposed to mediate the actions of STIM1 on Orai, as well as on ACs [18, 43]. Indeed, to add complexity to this model we have found that in the absence of PC2, SOCE is inhibited, and by confocal microscopy and PLA, STIM1 is unable to colocalize with Orai, after store depletion. These data suggest that in cholangiocytes, PC2, another member of the TPR family, is also a required member of the SOCE complex. In its absence, cAMP is produced in response to calcium store depletion.
Lefkimmiatis et al showed that Ca2+ store depletion can facilitate a STIM1-dependent activation of ACs [18]. Consistent with this hypothesis, using confocal imaging and PLA we were able to show also a co-localization between STIM1 and AC5, but not between STIM1 and AC6 in Pkd2-KO and Pkd2/AC6-KO cells after Ca2+ store depletion but not in WT cells. The data suggest that levels of PC2 determine the nature of the response to Ca2+ store depletion, directing it towards the cAMP.
In conclusion, we provide strong evidence that AC5 activation plays a pathogenic role in ADPKD. Our study unveils a signaling pathway that, in the absence of PC2, directly link, via STIM1, Ca2+ homeostasis with the production of cAMP. Increased cAMP/PKA stimulates the activity of the ERK pathway, which actually mediates both the increased secretion of VEGF and the increased response to VEGF in the cysts epithelium (Supplementary Figure 7). These studies improve our understanding of the mechanism leading to the progressive growth of cysts in ADPKD and indicate a possible target for its treatment. As the levels of PC2 can be down-regulated in cholestasis via a post-translational mechanism [11], pharmacologic modulation of AC5 activity may be of relevance also in acquired cholangiopathies.
Supplementary Material
Lay Summary.
Polycystic liver diseases are charcterized by progressive cyst growth cysts until their complications mandate surgery or liver transplantation. In this manuscript we demonstrate that inhibiting cell proliferation induced by increased levels of cAMP may represent a novel therapeutic target to slow the progression of the disease.
Acknowledgments
Financial Support: Supported by NIH Grant DK079005 to MS, by a NIH Grant DK34989: Silvio O. Conte Digestive Diseases Research Core Centers to MS and CS.
The authors are indebted with Dr S. Somlo (Yale University) for providing the Pkd2-KO mice and with Dr. M. Nathanson (Yale University) for providing the AC6-KO mice.
List of Abbreviations
- PC1
Polycystin-1
- PC2
Polycystin-2
- ADPKD
Autosomal Dominat Polycystic Kidney Disease
- PLD
Polycystic Liver Disease
- cAMP
3',5'-cyclic adenosine monophosphate
- ERK1/2
extracellular signal–regulated kinases type 1/2
- HIF1a
hypoxia-inducible factor type 1a
- VEGF-A
vascular endothelial growth factor type A
- SOCE
store-operated calcium entry
- SOcAMP
store-operated cyclic AMP
- STIM1
stromal interaction molecule 1
- Orai1
ORAI Calcium Release-Activated Calcium Modulator 1
- TRPP2
Transient Receptor Potential Polycystic-2
- VEGFR2
VEGFR, vascular endothelial growth factor receptor type 2
- PKA
protein kinase A
- Raf
proto-oncogene serine/threonine-protein kinase
- mTOR
mammalian target of rapamycin
- ACs
adenylyl cyclase
- sAC
solube adenylyl cyclase
- EDTA
Ethylenediaminetetraacetic acid
- TPEN
N’,N’,N’,N’-tetrakis-(2-pyridylmethyl)-ethylenediamide
- PFA
Paraformaldehyde
- DMEM/F12
Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12
- EGF
Epidermal Growth Factor
- FGF4
Fibroblast growth factor 4
- HGF
Hepatocyte growth factor
- FSK
forskolin
- Wnt3a
Wingless-type MMTV integration site 3a
- K19
cytokeratin 19
- DAPI
4',6-diamidino-2-phenylindole
- TEM
transmission electron microscope
- PLA
proximity ligation assay
- siRNAs
short interfering RNAs
- AKAPs
A-kinase anchor proteins
- PCNA
Proliferating cells nuclear antigen..
- PDE4C
cAMP-specific 3',5'-cyclic phosphodiesterase 4C
Footnotes
Conflict of Interest: All the authors have no conflict of interest to disclose.
Authors Contribution: C.S. was involved in the study design, acquisition, analysis and interpratation of data and drafting the manuscript; V.M. was involved acquisition, analysis and interpratation of data, A.V. and R.F. were involved in data acquisition; L.F. was involved in study design; M.S. designed the study and supervised the acquisition, analysis and interpretation of data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Author names in bold designate shared co-first authorship
- 1.Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015;88:17–27. doi: 10.1038/ki.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strazzabosco M, Somlo S. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology. 2011;140:1855–1859. 1859 e1851. doi: 10.1053/j.gastro.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strazzabosco M, Fabris L, Spirli C. Pathophysiology of cholangiopathies. J Clin Gastroenterol. 2005;39:S90–S102. doi: 10.1097/01.mcg.0000155549.29643.ad. [DOI] [PubMed] [Google Scholar]
- 4.Nigro EA, Castelli M, Boletta A. Role of the Polycystins in Cell Migration, Polarity, and Tissue Morphogenesis. Cells. 2015;4:687–705. doi: 10.3390/cells4040687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banner KH, Igney F, Poll C. TRP channels: emerging targets for respiratory disease. Pharmacol Ther. 2011;130:371–384. doi: 10.1016/j.pharmthera.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Geng L, Boehmerle W, Maeda Y, Okuhara DY, Tian X, Yu Z, et al. Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc Natl Acad Sci U S A. 2008;105:15920–15925. doi: 10.1073/pnas.0805062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sammels E, Devogelaere B, Mekahli D, Bultynck G, Missiaen L, Parys JB, et al. Unraveling the role of polycystin-2/inositol 1,4,5-trisphosphate receptor interaction in Ca signaling. Commun Integr Biol. 2010;3:530–532. doi: 10.4161/cib.3.6.12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, et al. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin- 2 defective mice. Gastroenterology. 2010;138:360–371. e367. doi: 10.1053/j.gastro.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, et al. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology. 2010;51:1778–1788. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spirli C, Villani A, Mariotti V, Fabris L, Fiorotto R, Strazzabosco M. Posttranslational regulation of polycystin-2 protein expression as a novel mechanism of cholangiocyte reaction and repair from biliary damage. Hepatology. 2015;62:1828–1839. doi: 10.1002/hep.28138. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Spirli C, Locatelli L, Fiorotto R, Morell CM, Fabris L, Pozzan T, et al. Altered store operated calcium entry increases cyclic 3',5'-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes. Hepatology. 2012;55:856–868. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 13.Spirli C, Morell CM, Locatelli L, Okolicsanyi S, Ferrero C, Kim AK, et al. Cyclic AMP/PKA-dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin-2 defective mice treated with sorafenib. Hepatology. 2012;56:2363–2374. doi: 10.1002/hep.25872. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 14.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 15.Putney JW., Jr New molecular players in capacitative Ca2+ entry. J Cell Sci. 2007;120:1959–1965. doi: 10.1242/jcs.03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Deng X, Gill DL. Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci Signal. 2010;3:pe42. doi: 10.1126/scisignal.3148pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper DM. Store-operated Ca(2)(+)-entry and adenylyl cyclase. Cell Calcium. 2015;58:368–375. doi: 10.1016/j.ceca.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 18.Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11:433–442. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 19.Strazzabosco M, Fiorotto R, Melero S, Glaser S, Francis H, Spirli C, et al. Differentially expressed adenylyl cyclase isoforms mediate secretory functions in cholangiocyte subpopulation. Hepatology. 2009;50:244–252. doi: 10.1002/hep.22926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, et al. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008;117:61–69. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 21.Jimenez-Castro MB, Casillas-Ramirez A, Massip-Salcedo M, Elias-Miro M, Serafin A, Rimola A, et al. Cyclic adenosine 3',5'-monophosphate in rat steatotic liver transplantation. Liver Transpl. 2011;17:1099–1110. doi: 10.1002/lt.22359. [DOI] [PubMed] [Google Scholar]
- 22.Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology. 2011;141:1498–1508. 1508 e1491–1495. doi: 10.1053/j.gastro.2011.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fabris L, Cadamuro M, Fiorotto R, Roskams T, Spirli C, Melero S, et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 24.Scirpo R, Fiorotto R, Villani A, Amenduni M, Spirli C, Strazzabosco M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-gamma limits NF-kappaB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology. 2015;62:1551–1562. doi: 10.1002/hep.28000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albarran L, Lopez JJ, Amor NB, Martin-Cano FE, Berna-Erro A, Smani T, et al. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci Rep. 2016;6:24452. doi: 10.1038/srep24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- 28.Klotz KN, Kachler S. Inhibitors of membranous adenylyl cyclases with affinity for adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:349–352. doi: 10.1007/s00210-015-1197-z. [DOI] [PubMed] [Google Scholar]
- 29.Mou TC, Masada N, Cooper DM, Sprang SR. Structural basis for inhibition of mammalian adenylyl cyclase by calcium. Biochemistry. 2009;48:3387–3397. doi: 10.1021/bi802122k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dianat N, Dubois-Pot-Schneider H, Steichen C, Desterke C, Leclerc P, Raveux A, et al. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology. 2014;60:700–714. doi: 10.1002/hep.27165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampaziotis F, Cardoso de Brito M, Madrigal P, Bertero A, Saeb-Parsy K, Soares FA, et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol. 2015;33:845–852. doi: 10.1038/nbt.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 33.Perico N, Antiga L, Caroli A, Ruggenenti P, Fasolini G, Cafaro M, et al. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21:1031–1040. doi: 10.1681/ASN.2009121302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Keimpema L, de Man RA, Drenth JP. Somatostatin analogues reduce liver volume in polycystic liver disease. Gut. 2008;57:1338–1339. doi: 10.1136/gut.2008.155721. [DOI] [PubMed] [Google Scholar]
- 35.Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang JC, Go S, de Waart DR, Munoz-Garrido P, Beuers U, Paulusma CC, et al. Soluble adenylyl cyclase regulates bile salt-induced apoptosis in human cholangiocytes. Hepatology. 2016 doi: 10.1002/hep.28550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin AC, Cooper DM. Layers of organization of cAMP microdomains in a simple cell. Biochem Soc Trans. 2006;34:480–483. doi: 10.1042/BST0340480. [DOI] [PubMed] [Google Scholar]
- 38.Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, et al. Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc Natl Acad Sci U S A. 2011;108:10679–10684. doi: 10.1073/pnas.1016214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vatner SF, Park M, Yan L, Lee GJ, Lai L, Iwatsubo K, et al. Adenylyl cyclase type 5 in cardiac disease, metabolism, and aging. Am J Physiol Heart Circ Physiol. 2013;305:H1–8. doi: 10.1152/ajpheart.00080.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Defer N, Best-Belpomme M, Hanoune J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am J Physiol Renal Physiol. 2000;279:F400–416. doi: 10.1152/ajprenal.2000.279.3.F400. [DOI] [PubMed] [Google Scholar]
- 41.Hooper R, Soboloff J. STIMATE reveals a STIM1 transitional state. Nat Cell Biol. 2015;17:1232–1234. doi: 10.1038/ncb3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willoughby D, Everett KL, Halls ML, Pacheco J, Skroblin P, Vaca L, et al. Direct binding between Orai1 and AC8 mediates dynamic interplay between Ca2+ and cAMP signaling. Sci Signal. 2012;5:ra29. doi: 10.1126/scisignal.2002299. [DOI] [PubMed] [Google Scholar]
- 43.Putney JW., Jr SOC: now also store-operated cyclase. Nat Cell Biol. 2009;11:381–382. doi: 10.1038/ncb0409-381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.