

Abstract
Background and Aims
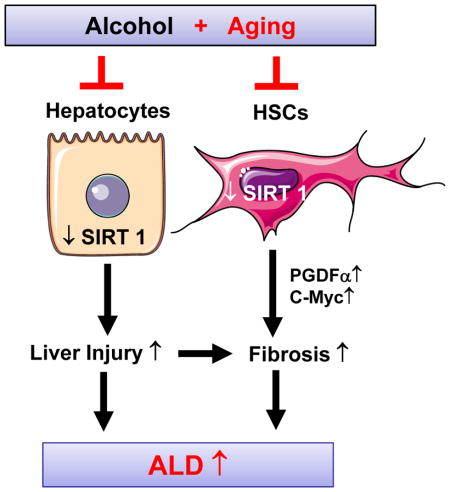
Aging is known to exacerbate the progression of alcoholic liver disease (ALD), but the underlying mechanisms remain obscure.
Methods
C57BL/6 mice were subjected to short-term (10-days) ethanol-plus-one binge or long- term (8-weeks) ethanol-plus-multiple binges of ethanol. Liver injury and fibrosis were determined. Hepatic stellate cells (HSCs) were isolated and used in in vitro studies.
Results
Compared to young (8–12 weeks) mice, middle-aged (12–14 months) and old (>16 months) mice were more susceptible to liver injury, inflammation, and oxidative stress induced by short-term-plus-one binge or long-term-plus-multiple binges of ethanol feeding. Long-term-plus- multiple binges of ethanol feeding induced greater liver fibrosis in middle-aged mice than that in young mice. Hepatic expression of Sirtuin 1 (SIRT1) protein was downregulated in the middle-aged mice compared to young mice. Restoration of SIRT1 expression via the administration of adenovirus-SIRT1 vector ameliorated short-term-plus-binge ethanol-induced liver injury and fibrosis in middle-aged mice. HSCs isolated from middle-aged mice expressed lower levels of SIRT1 protein and were more susceptible to spontaneous activation in in vitro culture than those from young mice. Overexpression of SIRT1 reduced activation of HSCs from middle-aged mice in vitro with downregulation of PDGFR-α and c-Myc, while deletion of SIRT1 activated HSCs isolated from young mice in vitro. Finally, HSC-specific SIRT1 knockout mice were more susceptible to short-term-plus-binge ethanol-induced liver fibrosis with upregulation of PDGFR-α expression.
Conclusions
Aging exacerbates ALD in mice through the downregulation of SIRT1 in hepatocytes and HSCs. Activation of SIRT1 may serve as a novel target for the treatment of ALD.
Keywords: Fatty liver, steatohepatitis, ethanol, PDGFR-α, middle-age, hepatic stellate cells (HSCs)
Graphical Abstract
INTRODUCTION
Alcoholic liver disease (ALD) is considered to be a major cause of morbidity and mortality nationwide [1–5]. It has an array of liver pathology that ranges from simple fatty liver to more severe forms of liver injury such as steatohepatitis, cirrhosis, and hepatocellular carcinoma. Many factors have been shown to affect the development and progression of ALD, including age, gender, ethnicity, genetic factors, drinking pattern, type of alcohol consumed, dose, duration, obesity, viral hepatitis infection, and environment [1–5]. Aging has been demonstrated to be associated with a progressive and widespread impairment of cellular functions. Therefore, resulting in an increasing risk of diseases in humans including diabetes, inflammatory diseases, and cancers [6]. In addition, liver function is declined during aging which involves in alterations to the hepatic structure, hepatic sinusoid and function, thereby leading to the development of age-related liver diseases [7–9]. It has been reported that aged mice are more susceptible to hepatocellular injury, inflammation, and liver fibrosis after high-fat diet feeding. This, is partly due to the sensitization to the Fas death pathway, increased M1 macrophage polarization, and increased innate immune responses [10, 11]. Moreover, accumulating evidence suggests that aged liver is more susceptible to injury due to alcohol abuse [12], which may be due to many mechanisms such as alterations to ethanol metabolism, microsomal ethanol oxidation, CYP2E1, and microsomal activity, and reduction of hepatic mitochondrial functions in the aged liver [13–16].
Although several mechanisms have been identified to be responsible for the increased susceptibility of aged livers to ALD, the exact underlying mechanisms are still unclear. Recent studies suggest that sirtuins (SIRT) play critical roles in regulating many biological and cellular processes during aging [17]. SIRTs are classified as a class III histone deacetylases, in which seven mammalian sirtuin (SIRT1–7) isoforms have been identified [17]. They are part of a family of nicotinamide adenine dinucleotide (NAD+) dependent protein deacetylases and ADP-ribosyltransferases. SIRTs are able to catalyze at specific lysine substrate deacetylation/deacylation, playing important roles in the pathogenesis of metabolic stress and control, caloric restriction, and cancer [17]. They are also associated with aging and are found at diverse subcellular locations like in the nucleus, cytosol and mitochondria [17]. For instance, SIRT1 was named after the Saccharomyces cerevisiae Sir2 (silent information regulator 2) protein and it has been proposed to serve as an anti-aging protein [18]. SIRT1 is predominately expressed in the nucleus and cytoplasm, whereas SIRT-2, 3 and 4 are expressed in the mitochondria [17]. Previous studies have reported that hepatic SIRT1 plays a major role in ameliorating steatosis and inhibiting inflammation in alcoholic and non-alcoholic fatty liver diseases (NAFLD) by modifying the acetylation status of the different target molecules [19–21]. In addition, it has also been reported that a variant of histone H2A, macroH2A1.1 is highly involved in lipid metabolism in NAFLD and binds SIRT1, which helps protect hepatocytes from lipid accumulation [22]. In the present study, we used the chronic-plus-binge ethanol feeding model to evaluate the effects of aging on alcohol induced liver injury. Chronic-plus-binge ethanol-feeding mimics the drinking pattern of alcoholic hepatitis patients who have had a history of heavy drinking for many years (chronic) and with recent excessive alcohol consumption (binge). Chronic-plus-binge ethanol feeding may also induce steatosis, liver injury and inflammation in mice [23]. Our results revealed that aged and middle-aged mice are more susceptible to chronic-plus-binge ethanol feeding-induced liver injury and fibrosis by downregulating SIRT1 in both hepatocytes and HSCs.
MATERIAL AND METHODS
Chronic-plus-binge ethanol feeding model
Wild-type (WT) and C57BL/6N mice were used. In most of experiments, female C57BL/6N mice were used. In some experiments, male C57BL/6N mice were used. HSC-specific SIRT1 knockout (SIRT1HSC−/−) mice were obtained via the several steps of crossing with SIRT1flox/flox mice [24] and LratCre+ mice (a kind gift from Dr. Robert Schwabe, Columbia University, NY) [25]. Since Cre in LratCre+SIRT1flox/flox mice do not efficiently delete floxed gene in the male mice (a personal communication with Dr. Schwabe), only female SIRT1HSC−/− and their wild-type littermate controls were used in our study.
For the short term feeding, mice were subjected to short-term (10 days)-plus-one binge ethanol feeding as previously published [23]. For the long term feeding, the same protocol was followed with the exception that mice were fed for 8 weeks with multiple binges (twice a week) of ethanol (5g/kg [b.w.]) feeding as described previously [26]. Animal studies were approved by the NIAAA Animal Care and Use Committee.
Statistical Analysis
For statistical analysis, which are expressed as the means±SEM for each group, GraphPad Prism software (v. 5.0a; GraphPad Software, La Jolla, CA) were used. To compare values obtained from three or more groups, a one-factor ANOVA was used, followed by Tukey post-hoc test. To compare values obtained from two groups, the Student t test was performed. P values of <0.05 were considered significant.
Other methods are described in supporting materials.
RESULTS
Middle-aged and aged mice are more susceptible to short-term (10 days)-plus-binge ethanol-induced liver injury and inflammation
Previous studies reported that chronic ethanol feeding induced greater liver damage and steatosis in aged rats compared to young rats [14]. In order to test whether aging also exacerbates the chronic-plus-binge ethanol feeding-induced liver injury in mice, we first tested the short term 10-day ethanol feeding plus-one binge (E10d+1B) model. After the E10d+1B ethanol feeding, female middle-aged mice (12–14 M) and aged mice (>16 M) showed significantly higher levels of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), liver to body weight ratio, and hepatic triglyceride contents when compared to young mice (8–12 weeks) (Fig. 1A, supporting Fig. 1A). Both middle-aged and aged mice exhibited greater liver damage when compared to young mice. A recent NIH publication entitled, “Consideration of Sex as a Biological Variable in NIH-funded Research” encouraged to include studies that involve both male and female vertebrate animals. Therefore, we have included studies of both sexes because it will help us gain a better understanding on the “health and disease in both men and women.” Thus, our results correlate with previous studies conducted in old rats where chronic ethanol feeding increased hepatic fat accumulation [14]. Due to the limitations of cost to house aged mice, only middle-aged mice were used in the rest of the paper in comparison to young mice.
Fig. 1. Aging exacerbates short-term chronic-plus-one binge ethanol-induced (E10d+1B) liver injury and inflammation.

Young (8–12 W) and middle-aged (12–14 M) mice were pair-fed or E10d+1B fed. (A) Serum ALT, AST and triglycerides levels are shown. (B) Representative images of H&E and Oil Red O staining (magnification of 200x). (C) Quantification of TUNEL positive cells. (D) Quantitative real-time PCR analysis of Ly6g and F4/80. Values represent means ± SEM (n= 4–12). *P<0.5, **P<0.01.
Hematoxylin and Eosin (H&E) and Oil Red O staining analyses revealed that E10d+1B treated middle-aged mice exhibited a higher degree of steatosis when compared to young mice that underwent the same ethanol treatment (Fig. 1B). TUNEL analyses demonstrated that the number of TUNEL positive apoptotic hepatocytes were higher in middle-aged mice post E10d+1B treatment (Fig. 1C). Furthermore, real-time PCR analyses of neutrophil marker (Ly6g) demonstrated that E10d+1B treated middle-aged mice had higher levels of hepatic neutrophil infiltration when compared with young mice, while hepatic expression of the macrophage marker (F4/80) was comparable between these two groups (Fig. 1D).
Aging exacerbates long-term (8wks)-plus-multiple binges-induced liver injury and inflammation but attenuates liver regeneration
The above data shows that short-term plus-binge ethanol feeding induces liver injury and inflammation. Next, we utilized a long-term 8-week ethanol feeding (E8w+nB) model with multiples binges that caused severe liver injury than that seen in the E10d+1B ethanol feeding [26]. As illustrated in Figs. 2A–C, E8w+nB feeding induced increased levels of serum ALT, AST, hepatic triglycerides, and TUNEL positive apoptotic hepatocytes in middle-aged mice when compared to young mice. Similarly, to chronic (10-d)-plus-binge ethanol feeding model (E8w+nB) feeding also induced greater levels of liver inflammation in middle-aged mice than in WT mice (supporting Fig. 2). Moreover, liver regeneration was measured by using bromodeoxyuridine (BrDU) immunohistochemistry. As illustrated in Fig. 2D, the number of BrdU+ hepatocytes were lower in E8w+nB-treated middle-aged mice compared to young mice.
Fig. 2. Aging exacerbates long-term plus-multiple binges-induced (E8w+nB) liver injury but attenuates liver regeneration.

Young and middle-aged mice were pair-fed or ethanol-fed with Lieber-DeCarli liquid diets for 8 weeks, plus two binges of ethanol weekly during 8-week chronic feeding. Mice were sacrificed 9h post the last binge. (A) Serum ALT, AST and percentage of liver to body weight [LW/BW] ratio in pair-fed and EtOH-fed mice. (B) Representative H&E staining are shown and hepatic triglyceride levels were measured. (C) Immunohistochemistry images and quantification of positive TUNEL staining shown by the red arrows (200x). (D) Brdu was injected 2h before collecting the liver tissues. BrdU incorporation was analyzed by immunohistochemistry, representative positive staining images are shown by the red arrows (100x) and quantification. Values represent means ± SEM (n= 5–10). *P< 0.05, **P<0.01.
Middle-aged mice exhibited stronger oxidative stress post chronic-plus-binge ethanol feeding when compared to young mice
Oxidative stress was determined by measuring 4-hydroxynonenal (4-HNE) adducts and protein nitration levels in livers from mice that underwent E8w+nB feeding. As illustrated in Fig. 3A, E8w+nB ethanol feeding increased hepatic levels of HNE adducts and protein nitration in middle-aged mice when compared to young mice. To further confirm these observations, we performed immunohistochemistry of 4-HNE staining, which is a stable by-product of lipid peroxidation due to oxidative stress. Immunohistochemical analysis showed positive staining in both young and middle-aged mice exposed to E10d+1B, with the most prominent 4-HNE positive staining in middle-aged mice (Fig. 3B).
Fig. 3. Aging exacerbates chronic-plus-binge-induced oxidative stress.

(A) Young and middle-aged mice were treated with chronic (8wks)-plus-multiple binges as described in Fig. 2. Hepatic levels of HNE adducts and protein nitration were measured by using an ELISA kit. (B) Young and middle-aged mice were treated with chronic (10d)-plus-one binge as described in Fig. 1. Immunohistochemistry for 4-HNE was performed, magnification at 200x. Values represent means ± SEM (n= 5). *P< 0.05, **P<0.01.
The above data indicated that middle aged mice are more susceptible to chronic-plus-binge ethanol-induced liver injury and inflammation. Next, we examined whether ethanol metabolism and CYP2E1 (which has been implicated in the increased susceptibility of age rats to ALD) [14], may also contribute to the pathogenesis of ALD in middle-aged mice. As shown in supporting Fig. 3A, ethanol concentrations were comparable in young and middle-aged mice post ethanol gavage. Real-time PCR analysis showed that hepatic expression of several ethanol metabolizing enzymes were comparable between WT and middle-aged mice in pair-fed groups. Expression of Adh1 Adh2, Aldh2, catalase, and Cyp2E1 were slightly lowered in ethanol-fed middle-aged mice than in WT mice but it did not reach statistical differences (supporting Fig. 3B). Moreover, western blotting analysis showed that ethanol feeding increased hepatic CYP2E1 levels, however these levels were comparable between young and middle-aged mice under alcohol feeding (supporting Fig. 4).
The hepatic CYP2E1 expression was upregulated post ethanol feeding. CYP2E1-mediated ethanol metabolism and it plays an important role in induction of oxidative stress [27], therefore, the elevation of oxidative stress (4-hydroxynonenal and protein nitration) in ethanol-fed mice was partly due to CYP2E1 induction. However, hepatic expression of CYP2E1 was comparable between young and middle-aged mice, thus, the higher levels of oxidative stress in middle-aged mice were probably due to other mechanisms such as decreased mitochondrial function when compared with young mice.
Increased ethanol-induced liver injury in middle-aged mice is not due to alteration of NKT cells
Several reports have recently indicated that the activation of NKT cells are involved in chronic-plus-binge ethanol-induced liver injury and inflammation [25, 28, 29]. A previous study reported that aging is associated with increased NKT cell function as demonstrated by markedly increased liver injury in middle-aged mice after α-Galcer injection [30], suggesting that hepatic NKT cells are more active in middle-aged mice than in young mice. We therefore analyzed hepatic NKT cells in E10d+1B fed young and middle-aged mice. Flow cytometry analysis showed that the percentage of NKT cells in liver lymphocytes was lower in middle-aged mice, with an average of ~17.8% when compared to young mice that had an average of ~27.4% (Fig. 4A). Furthermore, the total number of NKT cells from middle-aged livers was also lower than in young mice (Fig. 4A). Activation of NKT cells, which was determined by using the activation marker CD69, was comparable between middle-aged and young mice (Fig. 4B). Also, injection of α-Galcer only caused higher levels of liver injury in middle-aged mice when compared to young mice (Fig. 4C). In addition, aged mice (>16 months) had comparable levels of NKT cells and slightly higher levels of liver injury after injection of α-Galcer when compared to young mice (data not shown). Collectively, our results suggest that NKT cells do not account for the increased susceptibility of middle-aged mice to ethanol-induced liver injury.
Fig. 4. Increased ethanol-induced liver injury in middle-aged mice is not due to the alternation of NKT cells.

(A, B) Young and middle-aged mice were subjected to E10d+1B feeding. Liver lymphocytes were isolated and used for flow cytometry analysis of NKT number and percentage (A) and flow cytometry analysis of CD69 expression level in young and middle-aged mice (B). (C) Serum ALT levels after α-Galcer i.v. injection at 9 hrs and 24 hrs. Values represent means ± SEM (n=3–6). *P<0.05, **P<0.01.
Aging exacerbates long-term (8wks)-plus-multiple binges-induced liver fibrosis
Liver fibrosis was examined in two different models by using short-term plus one ethanol binge (E10d+1B) or long-term ethanol feeding plus multiple ethanol binges (E8w+nB). Supporting Fig. 5, shows results from E10d+1B that expression of some fibrotic genes were upregulated after E10d+1B in both young and middle-aged mice, but there were no differences between these two groups (supporting Fig. 5A). Sirius Red staining showed no significant fibrosis in young mice, but interestingly middle-aged mice demonstrated slight fibrosis after E10d+1B (supporting Fig. 5B).
Interestingly, Sirius Red staining in Fig. 5A displayed chicken wire-like fibrosis in E8w+nB-fed middle-aged mice, which is about a 4- to 5-fold greater than that observed in E8w+nB-fed young mice (Fig. 5A). Hepatic expression of α-SMA protein was increased in E8w+nB-fed middle-aged mice (Fig. 5B). At the mRNA levels, α-Sma, collagen I and III expression were also higher in E8w+nB-fed middle-aged mice when compared to young mice (Fig. 5C).
Fig. 5. Aging exacerbates long-term (8wks)-plus-multiple binges-induced liver fibrosis.

Young and middle-aged mice were treated with chronic (8wks)-plus-multiple binges as described in Fig. 2. (A) Representative images of Sirius Red staining at a magnification of 200x. Sirius Red positive area was quantified. (B) Western blot analysis of hepatic expression levels of α-SMA. (C) Quantitative real-time PCR analysis of fibrosis related genes. Values represent means ± SEM (n= 4–8). *P< 0.05, **P<0.01.
Suppression of SIRT1 contributes to enhanced alcohol induced liver injury and fibrosis in middle-aged mice
To further understand the mechanisms by which aging exacerbates ethanol-induced liver injury, we measured the protein levels of hepatic SIRT1 in mice that were exposed to E10d+1B. As illustrated in Fig. 6A, hepatic SIRT1 protein expression was significantly decreased in middle-aged mice compared to young mice. Interestingly, hepatic SIRT1 mRNA expression was not altered (data no shown). To identify the function of SIRT1 in alcoholic liver injury and fibrosis, we determined whether restoration of hepatic SIRT1 protein expression via the injection of adenovirus SIRT1 decreased susceptibility of middle-aged mice to ethanol-induced liver injury. As illustrated in Fig. 6B, adenovirus-mediated overexpression of SIRT1 ameliorated the overall morphology of the livers and reduced liver fibrosis in E10d+1B treated middle aged mice. Serum levels of ALT were also decreased by overexpression of SIRT1 in middle-aged mice when compared to Ad-GFP injected mice (Fig. 6C). In addition, real-time PCR analysis showed that overexpression of SIRT1 led to a significant decrease in expression of fibrosis related genes such as α-Sma, Tgf-β, and in Col1α1, 2α1, and 5α1 in middle-aged mice (Fig. 6D).
Fig. 6. Down-regulation of SIRT1 contributes to the enhanced alcohol induced liver injury and fibrosis in middle-aged mice.

(A) Young and middle-aged mice were treated with chronic (10d)-plus-one binge as described in Fig. 1. Western blot analysis of hepatic SIRT1 protein levels and quantification graph. (B, C, D) Middle-aged mice were injected with either control adenovirus-GFP or adenovirus-SIRT1, followed by chronic (10d)-plus-one binge feeding. Representative images of H&E and Sirius Red staining of livers (Panel B). Serum ALT and total blood neutrophils were measured (Panel C). Quantitative real-time PCR analysis of fibrosis related genes in liver tissues (Panel D). Values represent means ± SEM (n= 4–6). *P< 0.05, **P<0.01.
Downregulation of SIRT1 expression in HSCs from middle-aged mice, and SIRT1 inhibits HSC activation in vitro
The above data suggest that SIRT1 expression is downregulated in hepatocytes and contributes to the increased ethanol-induced liver injury and fibrosis in middle-aged mice when compared to young mice. Next, we examined the effects of SIRT1 in hepatic HSCs, one of the main cell types responsible for liver fibrogenesis [31]. As illustrated in Fig. 7A–B, isolated HSCs from middle-aged mice became activated faster when cultured in vitro than those from young mice, as expressions of α-Sma and collagen I mRNAs were higher in HSCs derived from middle-aged mice compared to those cells from young mice. The western blot analysis revealed decreased SIRT1 protein expression in freshly isolated HSCs (day 0) from middle-aged mice than those from young mice (upper panel in Fig. 7C). In addition, protein levels of SIRT1 were downregulated in activated HSCs (day 5) than those in freshly isolated HSCs (day 0) (lower panel in Fig. 7C).
Fig. 7. Effects of SIRT1 on HSC activation in vitro.

(A–C) HSCs were isolated from non-treated young and middle-aged mice (without ethanol feeding), and cultured in vitro. Representative morphological images of cultured primary HSCs captured by using a phase contrast microscope of cultured primary HSCs (Panel A). Quantitative real-time PCR analysis of fibrosis related genes (Panel B). Western blot analysis of SIRT1 proteins (Panel C) (D0: freshly isolated HSCs; D5: 5-day cultured HSCs). (D and E) HSCs were isolated from middle-aged mice and infected with adenovirus-GFP (Ad-GFP) or adenovirus-SIRT1 for 5 days. Quantitative real-time PCR analysis of fibrosis related genes (Panel D), and Western blot analysis (Panel E). (F) HSCs were isolated from non-treated SIRT1flox/flox mice, and infected with Ad-GFP or Ad-CRE, and cultured for 5 days. (F) Western blot analysis confirms SIRT1 deletion in Ad-Cre+SIRT1f/f HSCs. (G) Quantitative real-time PCR analysis of fibrosis related genes. (H) HSCs from non-treated WT mice were infected with adenovirus-SIRT1 or adenovirus-GFP control for 3 days. Protein extracts were prepared and used for western blotting analysis, using c-Myc protein. Values represent means ± SEM (n= 4–6). *P< 0.05, **P<0.01
Next, we determined whether the restoration of SIRT1 protein expression via the Ad-SIRT1 treatment prevented HSC activation in vitro. Our results showed that hepatic expression of fibrosis related genes including α-Sma, Pdgfr-α, Col1a1, and Timp-1 were lowered in Ad-SIRT1-infected HSCs than those in Ad-GFP-infected HSCs, (Fig. 7D). Decreased protein levels of α-SMA and PDGFR-α were also evident in the Ad-SIRT1 infected HSCs (Fig. 7E).
In addition, we examined the effects of endogenous SIRT1 on HSC activation in vitro. The SIRT1 gene deletion in HSCs was achieved via the infection of SIRT1fl/fl HSCs with Ad-CRE. As illustrated in Fig. 7F, SIRT1 expression was diminished in Ad-CRE-treated SIRT1 fl/fl HSCs, confirming that SIRT1 protein was efficiently deleted. Hepatic expressions of several fibrosis related genes were increased in Ad-CRE-infected SIRT1fl/fl HSCs compared to those in Ad-GFP infected HSCs (Fig. 7G), indicating that overexpression of SIRT1 attenuates HSC activation.
To further examine the mechanism by which SIRT1 inhibits HSC activation, we examined the expression of c-Myc that is known as an important onco-protein to promote HSC activation [32]. As illustrated in Fig. 7H, overexpression of SIRT1 via the incubation with Ad-SIRT1 markedly downregulated expression of c-Myc in cultured HSCs.
HSC-specific SIRT1 knockout (LratCre+SIRT1fl/−) mice are more susceptible to long-term chronic-plus-multiple binges ethanol-induced liver fibrosis
To examine the role of HSC-specific SIRT1 in liver fibrogenesis in vivo, we generated HSC specific HSC knockout (LratCre+SIRT1f/f) mice. As illustrated in Fig. 8A, Sirius Red staining revealed that LratCre+SIRT1f/f mice had greater degree of fibrosis than the control (LratCre−SIRT1f/f) mice after E10d+1B ethanol feeding. Serum levels of ALT and the total number of peripheral neutrophils in the blood were comparable between ethanol-fed LratCre+SIRT1f/f and WT mice (Fig. 8B). Hepatic expressions of several fibrosis related genes including α-Sma, Pdgfr, Col5a1 were higher in ethanol-fed LratCre+SIRT1f/f mice than those in ethanol-fed WT mice, (Fig. 8C). While hepatic expressions of Timp1, Col1a1 and Col3a1 had a trend that showed an increase level in ethanol-fed LratCre+SIRT1f/f mice compared to ethanol-fed WT mice, but with no statistical differences (Fig. 8C).
Fig. 8. HSC-specific SIRT1 KO mice are more susceptible to long-term-plus-multiple binges (E8w+nB) alcohol feeding-induced liver fibrosis.

Eight-week old WT (LratCre−SIRT1fl/fl) and HSC-specific SIRT1 KO (LratCre+SIRT1fl/fl) mice were subjected to long-term (8wks)-plus-multiple binges of ethanol feeding. (A) Representative images of H&E and Sirius Red staining of livers, magnification 100x. (B) ALT levels and total blood neutrophils were measured in WT and SIRT1 KO mice. (C) Quantitative real-time PCR analysis of fibrosis related genes. Values represent means ± SEM (n= 6–8). *P< 0.05, **P<0.01
DISCUSSION
Previous studies have identified several mechanisms underlying the increased susceptibility of aged rats to ethanol-induced liver injury as described in the introduction. In this current paper we used a, a mouse model of chronic-plus-binge ethanol feeding. We demonstrated that downregulation of SIRT1 in hepatocytes and HSCs contributes to the increased susceptibility of middle-aged mice to chronic-plus-binge ethanol-induced liver injury, and that SIRT1 attenuates HSC activation via the downregulation of PDGFα and c-Myc. In contrast, NKT cells did not contribute to the increased sensitivity of middle-aged mice to ethanol-induced liver injury. We have integrated all of these findings in a model that depict potential mechanisms underlying the role of SIRT1 in alcohol and aging-induced liver injury (Supporting Fig. 5).
It is known that iNKT cells are enriched in liver lymphocytes, which play an important role in host defenses against tumor transformation and pathogen infection [28, 33]. NKT cells are also involved in the pathogenesis of liver injury, inflammation and fibrosis [28, 33]. Recent studies from several laboratories including ours, demonstrated that iNKT cells were activated after undergoing chronic-plus binge ethanol feeding, which contributed to hepatic neutrophil recruitment and liver injury [25, 28, 29]. Moreover, a previous study reported that aged mice are more susceptible to the NKT activator α-Galcer-induced liver injury with a peak serum ALT levels of 5000 IU/L [30]. However, we were unable to reproduce these findings, and our results showed that middle-aged and aged mice had slightly increased liver injury after α-Galcer injection with peak serum ALT levels of 500 IU/L compared to young mice. The reasons for this discrepancy are not clear, but it may be related to different environment and gut microbiota that are known to affect NKT cell development [34]. Moreover, the number and activation of hepatic NKT cells were comparable in ethanol-fed middle-aged and young mice. Collectively, our findings suggest that NKT cells do not contribute to the increased susceptibility to chronic-plus-binge ethanol-induced liver injury and inflammation.
Previous studies have well documented that hepatocyte SIRT1 plays a critical role in protecting against alcoholic liver injury via the activation of several signaling pathways [19, 20]. Here, our results demonstrated that hepatic SIRT1 protein expression was downregulated in middle-aged mice when compared to that in young mice, and that restoration of SIRT1 protein expression in the liver ameliorated chronic-plus-binge ethanol-induced liver injury in middle-aged mice. These results suggest that the downregulation of SIRT1 contributes to the amplified susceptibility of middle-aged mice to ethanol-induced liver injury.
In addition to decreased SIRT1 protein expression in aged liver, SIRT1 activity is also downregulated via several mechanisms. First, SIRT1 is an NAD+ dependent protein deacetylase, and aging is known to decrease the NAD+ availability from various metabolic pathways including alcohol oxidation [13], causing a decrease in SIRT1 activity in aged liver. Second, a recent study reported that menin, a protein is encoded by multiple endocrine neoplasia 1 (Men1) gene, recruits SIRT1 to inhibit hepatic CD36 expression and triglyceride accumulation via histone deacetylation [35]. Expression of menin is downregulated in the liver from aged mice, which could reduce SIRT1 activity in aged liver [35].
Downregulation of SIRT1 protein expression is seen in various organs including the liver during aging [36]. However, the underlying mechanisms still remain unclear. A variety of factors have been shown to regulate SIRT1 expression at both transcriptional and posttranslational levels, including hypoxia, nutrient deprivation, DNA damage, C/EBPβ-HDAC1, and oxidative stress [36, 37]. Many of these factors likely contribute to the downregulated SIRT1 protein expression in aged liver. For example, in the present study, we found that hepatic oxidative stress was greater in chronic-plus-binge ethanol-fed middle-aged mice than in young mice. Moreover, treatment of hepatocytes with oxidative stress inducers markedly downregulated SIRT1 protein expression (data not shown). Thus, the increased oxidative stress in middle-aged mice likely contributes to the downregulation of SIRT1 protein expression in the liver. However, further studies are required to identify the mechanisms underlying oxidative stress-mediated downregulation of SIRT1 in hepatocytes. In addition, ethanol feeding also downregulated hepatic SIRT1 protein expression, which is at least in part due to severe disruption of carotenoid metabolism and subsequent reduction of its metabolite lycopenic acid, a factor known to increase hepatic SIRT1 expression and ameliorate fatty liver in mice [38].
Another interesting finding from the current study was that middle-aged mice were more susceptible to long-term chronic-plus-multiple binges-induced liver fibrosis. This increased susceptibility was partly because ethanol metabolism and age synergistically enhances the redox potential, liver injury, and inflammation, and subsequently increase HSC activation [13–16]. In addition, because deletion of the SIRT1 gene in hepatocytes exacerbated alcoholic liver fibrosis [20], the downregulation of hepatic SIRT1 protein expression in aged mice is likely an important mechanism that is responsible for enhancing alcoholic liver fibrosis in these aged mice. Moreover, our studies identified a novel mechanism that may contribute to the exacerbated alcoholic liver fibrosis in aged mice. First, SIRT1 protein expression is downregulated in HSCs from aged liver. Second, restoration of SIRT1 protein expression attenuated aged HSC activation in vitro. Third, deletion of the SIRT1 gene accelerated the activation of HSCs isolated from young mice when they were cultured in cell culture plates. This is in agreement with a previous study showing that knockdown of SIRT1 using siRNA enhanced HSC activation in vitro [39]. Finally, HSC-specific SIRT1 knockout mice were more susceptible to chronic-plus-binge ethanol-induced liver fibrosis. Collectively, our findings suggest that SIRT1 reduces liver fibrosis. Furthermore, we identified two novel mechanisms by which SIRT1 attenuates HSC activation. First, expression of PDGFR-α in cultured HSCs is inhibited by overexpression of SIRT1 and upregulated by knockdown of SIRT1. Since PDGFR-α is one of the most important factors that promotes HSC activation via the augmentation of TGF-β signaling, HSC survival and proliferation [40, 41], inhibition of PDGFR-α is probably an important mechanism by which SIRT1 attenuates HSC activation and liver fibrosis. Second, c-Myc is known to promote HSC activation and proliferation [32]. Our data showed that overexpression of SIRT1 markedly decreased expression of c-Myc in cultured HSCs, suggesting that SIRT1 inhibits HSC activation via the downregulation of c-Myc.
In summary, aging exacerbates alcoholic liver injury and fibrosis via the downregulation of SIRT1 in hepatocytes and HSCs. Activation of SIRT1 may be a potential therapeutic strategy for the treatment of ALD in elderly patients.
Supplementary Material
Lay summary.
Aged mice are more susceptible to alcohol-induced liver injury and fibrosis, which is, at least in part, due to the downregulation of sirtuin 1 in hepatocytes and hepatic stellate cells. Our findings suggest that sirtuin 1 agonists may have beneficial effects for the treatment of alcoholic liver disease in aged patients.
Acknowledgments
Financial support
This work was supported by the intramural program of NIAAA, NIH (B.G.), National Natural Science Foundation of China (No. 81522009/H0317 to H.W., 81470879/H0318 to S.Y.), the National Institutes of Health Grants (R01 DK100603 and R21AA021181 to M.Z.). No conflicts of interest exist for any of the authors.
ABBREVIATIONS
- ALT
alanine aminotransferase
- ALD
alcoholic liver disease
- AST
aspartate aminotransferase
- BrDU
bromodeoxyuridine
- Ccr2
C-C motif chemokine receptor 2
- H&E
hematoxylin and eosin
- HSCs
Hepatic stellate cells
- 4-HNE
4-Hydroxy-2-nonenal
- IL-1β
interleukin 1β
- MPO
myeloperoxidase
- NAD+
nicotinamide adenine dinucleotide
- 3-NT
3-nitrotyrosine
- PDGFR-α
platelet derived growth factor receptor alpha
- Sir2
silent information regulator 2
- SIRT1
sirtuin 1
- α-SMA
smooth muscle alpha actin
- TNFα
tumor necrosis factor α
- WT
wild-type
Footnotes
Author contributions: TR, YML, and SY performed experiments, analyzed data and wrote the paper. MJX, DF, ZZ, MZ, PM, ZV performed experiments and analyzed data. PP analyzed data and edited the paper. BG and HW designed experiments, analyzed data and wrote the paper.
Conflict of interest: All authors declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bergheim I, McClain CJ, Arteel GE. Treatment of alcoholic liver disease. Digestive diseases. 2005;23:275–284. doi: 10.1159/000090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nature reviews Gastroenterology & hepatology. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 3.Bataller R, Gao B. Liver fibrosis in alcoholic liver disease. Semin Liver Dis. 2015;35:146–156. doi: 10.1055/s-0035-1550054. [DOI] [PubMed] [Google Scholar]
- 4.Kirpich IA, Miller ME, Cave MC, Joshi-Barve S, McClain CJ. Alcoholic Liver Disease: Update on the Role of Dietary Fat. Biomolecules. 2016;6:1. doi: 10.3390/biom6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Shea RS, Dasarathy S, McCullough AJ Practice Guideline Committee of the American Association for the Study of Liver D, Practice Parameters Committee of the American College of G. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 6.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 7.Kim IH, Kisseleva T, Brenner DA. Aging and liver disease. Curr Opin Gastroenterol. 2015;31:184–191. doi: 10.1097/MOG.0000000000000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cogger VC, Svistounov D, Warren A, Zykova S, Melvin RG, Solon-Biet SM, et al. Liver aging and pseudocapillarization in a Werner syndrome mouse model. J Gerontol A Biol Sci Med Sci. 2014;69:1076–1086. doi: 10.1093/gerona/glt169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frith J, Jones D, Newton JL. Chronic liver disease in an ageing population. Age Ageing. 2009;38:11–18. doi: 10.1093/ageing/afn242. [DOI] [PubMed] [Google Scholar]
- 10.Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology. 2013;57:995–1004. doi: 10.1002/hep.26099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim IH, Xu J, Liu X, Koyama Y, Ma HY, Diggle K, et al. Aging increases the susceptibility of hepatic inflammation, liver fibrosis and aging in response to high-fat diet in mice. Age (Dordr) 2016 doi: 10.1007/s11357-016-9938-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seitz HK, Stickel F. Alcoholic liver disease in the elderly. Clin Geriatr Med. 2007;23:905–921. viii. doi: 10.1016/j.cger.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Seitz HK, Xu Y, Simanowski UA, Osswald B. Effect of age and gender on in vivo ethanol elimination, hepatic alcohol dehydrogenase activity, and NAD+ availability in F344 rats. Res Exp Med (Berl) 1992;192:205–212. doi: 10.1007/BF02576276. [DOI] [PubMed] [Google Scholar]
- 14.Seitz HK, Meydani M, Ferschke I, Simanowski UA, Boesche J, Bogusz M, et al. Effect of aging on in vivo and in vitro ethanol metabolism and its toxicity in F344 rats. Gastroenterology. 1989;97:446–456. doi: 10.1016/0016-5085(89)90082-6. [DOI] [PubMed] [Google Scholar]
- 15.Choudhury M, Jonscher KR, Friedman JE. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging (Albany NY) 2011;3:175–178. doi: 10.18632/aging.100289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meier P, Seitz HK. Age, alcohol metabolism and liver disease. Curr Opin Clin Nutr Metab Care. 2008;11:21–26. doi: 10.1097/MCO.0b013e3282f30564. [DOI] [PubMed] [Google Scholar]
- 17.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nature reviews Drug discovery. 2012;11:443–461. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You M, Jogasuria A, Taylor C, Wu J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2015;4:88–100. doi: 10.3978/j.issn.2304-3881.2014.12.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Gao Y, Li M, Geng C, Xu H, Yang Y, et al. Sirt1 mediates the effect of the heme oxygenase inducer, cobalt protoporphyrin, on ameliorating liver metabolic damage caused by a high-fat diet. J Hepatol. 2015;63:713–721. doi: 10.1016/j.jhep.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 22.Pazienza V, Borghesan M, Mazza T, Sheedfar F, Panebianco C, Williams R, et al. SIRT1-metabolite binding histone macroH2A1. 1 protects hepatocytes against lipid accumulation. Aging. 2014;6:35–47. doi: 10.18632/aging.100632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146:539–549. e537. doi: 10.1053/j.gastro.2013.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maricic I, Sheng H, Marrero I, Seki E, Kisseleva T, Chaturvedi S, et al. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology. 2015;61:1357–1369. doi: 10.1002/hep.27632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu MJ, Cai Y, Wang H, Altamirano J, Chang B, Bertola A, et al. Fat-Specific Protein 27/CIDEC Promotes Development of Alcoholic Steatohepatitis in Mice and Humans. Gastroenterology. 2015;149:1030–1041. e1036. doi: 10.1053/j.gastro.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdelmegeed MA, Choi Y, Ha SK, Song BJ. Cytochrome P450-2E1 promotes aging-related hepatic steatosis, apoptosis and fibrosis through increased nitroxidative stress. Free Radic Biol Med. 2016;91:188–202. doi: 10.1016/j.freeradbiomed.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cellular & molecular immunology. 2016;13:206–216. doi: 10.1038/cmi.2015.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1beta in mice. J Hepatol. 2015;62:1311–1318. doi: 10.1016/j.jhep.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 30.Inui T, Nakagawa R, Ohkura S, Habu Y, Koike Y, Motoki K, et al. Age-associated augmentation of the synthetic ligand- mediated function of mouse NK1. 1 ag(+) T cells: their cytokine production and hepatotoxicity in vivo and in vitro. J Immunol. 2002;169:6127–6132. doi: 10.4049/jimmunol.169.11.6127. [DOI] [PubMed] [Google Scholar]
- 31.Warren A, Cogger VC, Fraser R, Deleve LD, McCuskey RS, Le Couteur DG. The effects of old age on hepatic stellate cells. Current gerontology and geriatrics research. 2011;2011:439835. doi: 10.1155/2011/439835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arteaga M, Shang N, Ding X, Yong S, Cotler SJ, Denning MF, et al. Inhibition of SIRT2 suppresses hepatic fibrosis. Am J Physiol Gastrointest Liver Physiol. 2016;310:G1155–1168. doi: 10.1152/ajpgi.00271.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–528. doi: 10.1189/jlb.0309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dowds CM, Blumberg RS, Zeissig S. Control of intestinal homeostasis through crosstalk between natural killer T cells and the intestinal microbiota. Clinical immunology. 2015;159:128–133. doi: 10.1016/j.clim.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao Y, Xue Y, Xue L, Jiang X, Wang X, Zhang Z, et al. Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J Hepatol. 2013;59:1299–1306. doi: 10.1016/j.jhep.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Yuan Y, Cruzat VF, Newsholme P, Cheng J, Chen Y, Lu Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mechanisms of ageing and development. 2016;155:10–21. doi: 10.1016/j.mad.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Jin J, Iakova P, Jiang Y, Medrano EE, Timchenko NA. The reduction of SIRT1 in livers of old mice leads to impaired body homeostasis and to inhibition of liver proliferation. Hepatology. 2011;54:989–998. doi: 10.1002/hep.24471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chung J, Koo K, Lian F, Hu KQ, Ernst H, Wang XD. Apo-10′-lycopenoic acid, a lycopene metabolite, increases sirtuin 1 mRNA and protein levels and decreases hepatic fat accumulation in ob/ob mice. J Nutr. 2012;142:405–410. doi: 10.3945/jn.111.150052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Liu X, Zhou Q, Huang C, Meng X, Xu F, et al. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicology and applied pharmacology. 2015;289:163–176. doi: 10.1016/j.taap.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 40.Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. Journal of hepatology. 2006;45:419–428. doi: 10.1016/j.jhep.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 41.Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, et al. PDGF receptor-alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-beta receptors. American journal of physiology Gastrointestinal and liver physiology. 2014;307:G749–759. doi: 10.1152/ajpgi.00138.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.