

Summary
Lupins are important grain legume crops that form a critical part of sustainable farming systems, reducing fertilizer use and providing disease breaks. It has a basal phylogenetic position relative to other crop and model legumes and a high speciation rate. Narrow‐leafed lupin (NLL; Lupinus angustifolius L.) is gaining popularity as a health food, which is high in protein and dietary fibre but low in starch and gluten‐free. We report the draft genome assembly (609 Mb) of NLL cultivar Tanjil, which has captured >98% of the gene content, sequences of additional lines and a dense genetic map. Lupins are unique among legumes and differ from most other land plants in that they do not form mycorrhizal associations. Remarkably, we find that NLL has lost all mycorrhiza‐specific genes, but has retained genes commonly required for mycorrhization and nodulation. In addition, the genome also provided candidate genes for key disease resistance and domestication traits. We also find evidence of a whole‐genome triplication at around 25 million years ago in the genistoid lineage leading to Lupinus. Our results will support detailed studies of legume evolution and accelerate lupin breeding programmes.
Keywords: Legume comparative genomics, synteny, whole‐genome assembly, flowering time genes, polyploidy, Genistoids
Introduction
Lupins are grain legumes that form an integral part of sustainable farming systems and have been an important part of the human diet for thousands of years (Gladstones, 1970). Planted in rotation with cereal crops, lupins reduce the need for nitrogenous fertilizer, provide valuable disease breaks and boost cereal yields (Gladstones, 1970). Lupins thrive on low‐nutrient soils due to their ability to fix atmospheric nitrogen in symbiosis with beneficial bacteria and efficiently take up phosphorus from soils (Gladstones, 1970). Consequently, they are effective ecological pioneers and able to colonize extremely impoverished soils such as coastal sand dunes and new lava soils set down by recently erupted volcanoes (Lambers et al., 2013).
Lupins have emerged as both a human health food and food‐additive. Lupin seeds are rich in protein, ranging from 30% to 40% of whole seeds (Williams, 1979), with very little starch compared to other major grain legumes, for example chickpea and soya bean. The narrow‐leafed lupin kernel contains 40%–45% protein and 25%–30% dietary fibre, and low fat and carbohydrate content (Lee et al., 2006). An important property of lupin kernel flour is as a food‐additive (e.g. in bread); it increases satiety (thus reducing energy intake) and reduces insulin resistance, which are valuable properties in the context of the rising incidence of obesity and diabetes (Lee et al., 2006). Furthermore, lupin flour is increasingly used as a nongenetically modified alternative to soya bean products and is used to produce gluten‐free foods such as pasta. Studies on lupin seed proteins have provided valuable information on their number and RNA/protein expression patterns (Foley et al., 2011, 2015) as well as demonstrated that specific members are able to reduce glycaemia to comparable levels as achieved with metformin, a widely used hypoglycaemic drug (Magni et al., 2004).
Lupins belong to a single genus, Lupinus, in a legume clade known as the genistoids, which are believed to have diverged early in the evolution of papilionoid legumes (Lavin et al., 2005). There are an estimated 267 species of lupin distributed around the Mediterranean region (‘Old World’ lupins) and North and South America (‘New World’ lupins) (Drummond et al., 2012). Andean Lupinus species in particular show a rate of speciation unparalleled in the plant kingdom with broad morphological diversity ranging from small prostrate herbs to tall trees (Drummond et al., 2012; Hughes and Eastwood, 2006). Both annual and perennial species have found their niches in a vast array of ecological habitats across 100 degrees of latitude (Drummond et al., 2012). Together, these properties make this genus exceptionally useful for testing hypotheses relating to genome evolution, adaptation and speciation.
While wild lupin species were cultivated as far back as 2000 BC in the Mediterranean and Andean regions, domestication of lupin species was completed only in the 20th century (Gladstones, 1970). The most widely grown domesticated species today is narrow‐leafed lupin (L. angustifolius; NLL) (Lee et al., 2006). Its domestication was initiated in Germany in the early 20th century and completed in the 1960s in Australia with the development of the first fully domesticated cultivar with low alkaloid content, nonshattering pods, permeable seeds and early flowering. Since then, NLL cultivation has grown to span more than 600 000 hectares in over 20 countries (FAO, 2013).
Over the last decade, various legume genomes that utilized reference genetic maps to order and orient scaffolds for pseudomolecule assembly have been published, and these include those of Medicago truncatula (Young et al., 2011), chickpea (Cicer arietinum; Varshney et al., 2013), pigeon pea (Cajanus cajan; Varshney et al., 2012); common bean (Phaseolus vulgaris; Schmutz et al., 2014) and soya bean (Glycine max; Schmutz et al., 2010). In the latter two assemblies, synteny‐based refinement methods were used in addition to the dense genetic maps to order and orient assembled scaffolds into pseudomolecules (Schmutz et al., 2010, 2014). A range of genomic resources have also been produced in recent years for the study of lupins, particularly NLL. These include genetic maps for NLL and white lupin (Croxford et al., 2008; Kamphuis et al., 2015; Kroc et al., 2014; Nelson et al., 2010; Yang et al., 2013b) and large genomic insert libraries for NLL (Gao et al., 2011; Kasprzak et al., 2006). Transcriptomic resources have been developed for all four cultivated lupin species (Foley et al., 2015; Kamphuis et al., 2015; O'Rourke et al., 2013; Parra‐González et al., 2012; Secco et al., 2014; Wang et al., 2014). Preliminary draft genome data had been generated for NLL and were used to assist molecular marker design (Gao et al., 2011; Kamphuis et al., 2015; Yang et al., 2013a). In this study, we present the first high‐quality draft genome for a genistoid legume, narrow‐leafed lupin (2n = 40), report on a survey of its gene content and provide insights into its genome evolution, symbiotic relationships and host–pathogen interactions. Lupin, as a genus in the early‐diverging genistoid lineage in the papilionoid subfamily, serves as an outgroup for the many crop and model species in this subfamily—an outgroup that shares many characteristics of other papilionoid legumes (such as symbiotic nitrogen fixation), but with sufficient evolutionary distance to make inferences about the timing and histories of important molecular evolutionary events.
Results and discussion
The NLL genome assembly and gene features
The haploid genome size for NLL was previously estimated by flow cytometry to be 924 Mb (Kasprzak et al., 2006; Naganowska et al., 2003). K‐mer‐based estimation of genome size predicted a similar value of 951 Mb (Figure S1). Initial assembly of the Tanjil genome using only paired‐end Illumina data produced 191 701 scaffolds in 521 Mb, with an N50 of 10 137 and N50 length of 13.8 kb. The assembly was improved via scaffolding with additional paired‐end, mate‐pairs and BAC‐end data totalling an average coverage of 162.8 X (Table S1). This resulted in a contig assembly with 1 068 669 contigs, totalling 810 Mb or 85% of the K‐mer‐based estimated genome size. The final scaffold assembly after removing scaffolds less than 200 bp comprised 14 379 scaffolds totalling 609 Mb with a contig N50 length of 45 646 bp and scaffold N50 of 232 and scaffold N50 length of 703 Kb (Table 1).
Table 1.
Summary of the narrow‐leafed lupin cv. Tanjil genome assembly. The assembly comprises scaffolds, the majority of which have been placed into pseudochromosomes based on genetic map and synteny data
| Assembly statistics | Total length (bp) | Average length (bp) | Maximum length (bp) | Minimum length (bp) | N50 | N50 length | Total # sequences | Total unknown N bases |
|---|---|---|---|---|---|---|---|---|
| Contigs | 810 353 784 | 758 | 922 429 | 100 | 4 246 | 45 646 | 1 068 669 | 0 |
| Scaffolds | 609 123 749 | 42 362 | 4 089 732 | 200 | 232 | 703 185 | 14 379 | 4 078 848 |
| Pseudochromosomes | 470 424 067 | 23 521 203 | 36 457 581 | 16 251 777 | 8 | 24 697 652 | 20 | 3 351 285 |
| Unplaced scaffolds | 138 780 182 | 10 239 | 1 472 692 | 200 | 610 | 45 366 | 13 554 | 808 063 |
The NLL genome is highly repetitive (57% of the genome) (Table S2), with over half its repeats (32% of the genome) matching known transposable elements (TEs) (Table 2). Typical of most eukaryotes, TEs were most commonly long terminal repeats (LTRs) retrotransposons (28%), with DNA LTRs, long interspersed nuclear elements (LINE) and short interspersed nuclear elements (SINE) TEs comprising a relatively small proportion (4.8%, 2.7% and 0.1%, respectively). Noncoding RNA was estimated to comprise 0.1% of the genome (Table S3), the majority being ribosomal RNA (0.035%) and transfer RNA (0.012%), with predicted snRNA and miRNA representing 0.009% and 0.006%. Analysis of divergence between known TEs (Jurka et al., 2005) indicated a peak at ~30%, however the same analysis applied to de novo repeats produced a bimodal distribution with an additional less divergent peak at ~10% (Figure S2).
Table 2.
Summary of transposon content in the narrow‐leafed lupin cv. Tanjil genome assembly
| Repbase TEs | TE Proteins | De novo | Combined TEs | % in Genome | Length (bp) | % in Genome | ||
|---|---|---|---|---|---|---|---|---|
| Length (bp) | % in Genome | Length (bp) | % in Genome | Length (bp) | ||||
| DNA | 8 983 926 | 1.47 | 7 351 979 | 1.20 | 23 429 353 | 3.83 | 29 084 889 | 4.76 |
| LINE | 8 299 104 | 1.35 | 10 841 081 | 1.77 | 13 051 653 | 2.13 | 16 438 300 | 2.69 |
| LTR | 79 075 250 | 12.95 | 90 533 453 | 14.83 | 154 738 027 | 25.35 | 172 348 763 | 28.23 |
| SINE | 66 384 | 0.01 | 0 | 0 | 483 328 | 0.08 | 544 025 | 0.09 |
| Other | 3917 | 0.000642 | 0 | 0 | 0 | 0 | 3917 | 0.000642 |
| Unknown | 0 | 0 | 2988 | 0.00049 | 134 545 040 | 22.04 | 134 548 028 | 22.04 |
| Total | 95 943 148 | 15.71 | 108 715 411 | 17.81 | 319 388 057 | 52.32 | 331 905 409 | 54.37 |
A total of 33 076 protein‐coding genes were annotated (Figure S3) after combining evidence from transcriptome alignments derived from five different tissue types (leaf, stem, root, flower and seed), protein homology, and in silico gene prediction (Table 3). Additionally, peptide data from proteomics analysis of leaf, seed, stem and root samples were mapped to both the translated gene annotations and the 6‐frame translation of the whole‐genome assembly (Bringans et al., 2009) (Table S4). Proteogenomic comparison of peptide‐mapping versus gene annotation supported between 94 and 1134 annotations per tissue type (Table S4), and provided valuable information on tissue localization for the products of these genes. InterPro terms were the most informative functional annotation assigned to NLL proteins with 26 580 (80.4%) proteins annotated (Table S5). Comparing gene counts for Interpro terms in NLL to other plant species (source: PLAZA 3.0 (Van Bel et al., 2011)) via Fisher's exact test, numerous Interpro terms were over‐represented in NLL and were often significantly higher than most species, excepting G. max (Data S1). However, in a few cases, NLL InterPro terms were more abundant versus all species including G. max. These included tyrosine protein kinases, photosystem II cytochrome b559, porins and microtubule‐associated proteins. The NLL assembly was also depleted in genes with InterPro terms corresponding to NBS–LRR proteins, DNA helicases, peptidase C48, hAT transposases and certain transcription factors.
Table 3.
Summary of predicted protein‐coding gene annotations of narrow‐leafed lupin and their supporting evidence types
| Gene set | Number | Average transcript length (bp) | Average CDS length (bp) | Average exon per gene | Average exon length (bp) | Average intron length (bp) | |
|---|---|---|---|---|---|---|---|
| De novo | AUGUSTUS | 34 525 | 2 983.98 | 1 252.69 | 5.49 | 228.06 | 385.36 |
| GENSCAN | 29 436 | 10 570.55 | 1 367.30 | 6.22 | 219.52 | 1 760.19 | |
| A. thaliana | 48 717 | 2 815.40 | 968.47 | 3.72 | 260.12 | 678.23 | |
| Homolog | C. cajan | 46 735 | 2 422.52 | 929.51 | 3.90 | 238.24 | 514.57 |
| C. arietinum | 42 856 | 4 349.29 | 1 125.34 | 4.04 | 278.37 | 1 059.62 | |
| G. max | 39 433 | 3 648.01 | 1 245.43 | 4.55 | 273.19 | 675.09 | |
| M. truncatula | 61 321 | 2 454.66 | 843.16 | 3.10 | 271.33 | 764.67 | |
| P. vulgaris | 68 168 | 1 936.06 | 786.66 | 3.23 | 242.93 | 513.54 | |
| EST | 1 795 | 2 134.32 | 606.22 | 3.16 | 191.27 | 704.40 | |
| GLEAN | 32 413 | 3 568.05 | 1 305.97 | 5.58 | 233.78 | 493.22 | |
| RNA‐seq | 49 946 | 2 309.00 | 803.54 | 4.01 | 199.94 | 498.02 | |
| Final set | 33 076 | 3 673.44 | 1 289.14 | 5.52 | 233.52 | 488.41 |
Chromosome‐level analysis of the NLL genome assembly
Enhanced genetic map data were used to place NLL scaffolds in a chromosomal context. Fluidigm assays yielded 469 transcriptome‐derived SNPs that were polymorphic in the RIL population (n = 153) derived from a domestic (83A:476) by wild (P27255) cross. An additional 8668 DArTSeq molecular markers, including 4767 presence/absence variants (DArT_PAV markers) and 3901 SNPs (DArT_SNP markers) were also applied. When combined with 830 previously reported sequence‐associated marker loci and seven trait loci, a total of 9972 loci (Data S2) were used to generate the improved map (Data S3), which comprised 20 linkage groups that correspond to the haploid chromosome complement of NLL (Lesniewska et al., 2011). The genetic map covers 2500.8 cM, with an average interval size of 0.85 cM between 2959 nonredundant framework loci (Table S6). This map incorporated for the first time a small orphan cluster of markers into linkage group 20 (Kamphuis et al., 2015) and has evenly distributed linkage group lengths (cM) (Figure S4A) and average interval sizes (cM) (Figure S4B).
A combination of 7707 markers physically mapped unambiguously to the scaffold assembly, including the following new markers: 3492 DArTSeq markers with presence–absence polymorphism, 2975 DArTSeq markers with SNP polymorphism, 555 Fluidigm markers with SNP polymorphism and 685 other previously reported PCR‐based markers (Gao et al., 2011; Kamphuis et al., 2015; Kroc et al., 2014) (Data S2). Twenty pseudomolecule sequences, ranging from 16.2 to 36.5 Mb, were built from 825 scaffolds. The pseudomolecule assemblies total 470 424 067 bp (77.2% of the full assembly length) (Table 1). Of these scaffolds, 820 were anchored to linkage groups and provisionally ordered and oriented using the high‐density marker resource, and five were added on the basis of synteny comparisons, using all‐by‐all dot plot comparisons between the NLL pseudomolecules and remaining unplaced scaffolds, and five other legume genomes (Glycine max, Lotus japonicas, Medicago truncatula and Phaseolus vulgaris; Data S6). The five added scaffolds comprised 2 004 769 bp or 0.4% of the pseudomolecule length. Additionally, when marker resolution was insufficient to confidently order and orient scaffolds (primarily in pericentromeric regions, where recombination rates are very low), synteny with the species above was considered in the scaffold order and orientation, under the assumption that discontinuities in genomic synteny that occur precisely at NLL scaffold boundaries are likely due to misorientation or local misplacement.
Genome assembly validation and comparison to previous draft assembly
To validate the quality of the genome assembly a CEGMA analysis (Parra et al., 2007) was conducted to identify whether the majority of core eukaryotic genes are present in the assembly. This showed 235 complete and eight partial core eukaryotic genes were present in the assembly which equates to 98.0% or 243 genes of the gene set of 248 genes (for details and the missing protein KOG id's see Data S4). The transcriptome data for five different tissue types was aligned to the NLL assembly and for four of the five datasets 98.5%–99.0% of reads mapped back to the assembly (Table S7), suggesting the majority of the gene‐rich space of the NLL genome is captured in the assembly. For the root transcriptome 89.1% aligned back to the assembly, which could be due to contamination from the soil or soil microbes. Furthermore, of the 33 076 genes in the predicted gene set of the current assembly, 1.8% (596 genes) are absent in the previous draft assembly from 2013% and 47.5% (15 703 genes) had partial hits, whereas 50.7% (16 777 genes) had 100% complete alignment in the previous assembly. In conclusion, over 98% of the gene‐rich space is captured in our assembly and it is a significant improvement of the fragmented draft assembly from (Yang et al., 2013b) which had a scaffold N50 of 7319 scaffolds compared to 232 scaffolds for this assembly (Table 1).
CoReFinder (Collapse/Repeat Finder) was applied to 20 pseudochromosomes and unplaced scaffolds greater than 10 kb, and a total of 14 923 collapsed regions of 3 462 044 bp (0.58% of the genome) were identified (Data S5). In addition, a total of 66 301 repeated regions of 23 699 757 bp (3.89% of the genome) were identified. A copy number estimate of the repeated regions was also performed and ranged from 1.58 (pseudochromosome NLL‐01) to 171.60 (Scaffold_486) (Data S5). In conclusion, the assembly captures the majority of the gene space (~98%) and shows a low level of collapsed genes.
Comparative genomics across legume species
Resequencing of additional NLL lines at 51.5–59.2× coverage (Table S8) allowed comparisons of sequence variation across the NLL lines Unicrop (early domesticated cultivar), 83A:476 and P27255 (wild accession), relative to the pseudochromosomes of the reference cv. Tanjil (Figure 1). This indicated that the wild P27255 was significantly divergent across all regions of the genome with 216 167 indels and 3 053 917 SNPs (Table S8). In contrast, domesticated lines exhibited lower levels of diversity overall with 47 113 indels and 606 035 SNPs for line 83A:476 and 81 375 indels and 1 099 966 SNPs for cultivar Unicrop. Several trait‐associated markers (anthracnose and phomopsis resistance, flowering time, bitterness, pod shattering) could be mapped onto pseudochromosomes, facilitating ‘reverse‐genetic’ nomination of candidate genes for disease resistance and domestication traits (Table S9).
Figure 1.
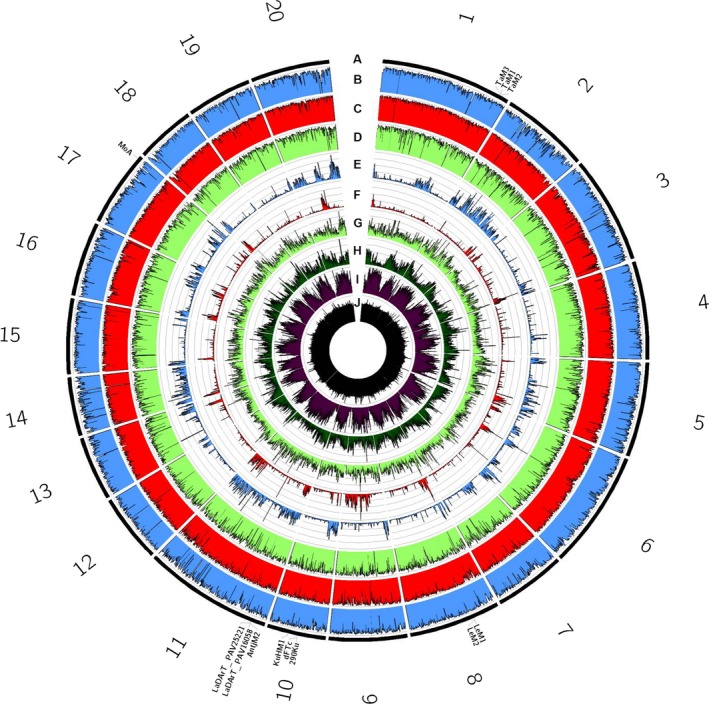
Summary of sequence variability in narrow‐leafed lupin lines Unicrop, 83A:476 and P27255, relative to pseudochromosomes (corresponding to linkage groups) of the reference genome of cv. Tanjil. (A) Pseudochromosomes (black), with sequence‐based genetic markers relevant to this study highlighted. (B–D) Per cent of 100‐Kb windows covered by ≥5× read depth for resequencing data from lines Unicrop (b, blue), 83A:476 (C, red) and P27255 (D, light green). (E–G) Density of polymorphic sequence sites ranging from 0 to 20 000 variants/Mb calculated within 100‐Kb windows, for lines Unicrop (E, blue), 83A:476 (F, red) and P27255 (G, light green). (H) Per cent of 100‐kb windows representing annotated genes in cv. Tanjil (dark green). (i) Per cent of 100‐kb windows represented annotated repetitive DNA in cv. Tanjil (purple). (j) Per cent G:C content ranging from 0% to 50%, calculated in 100‐Kb windows, in cv. Tanjil (black).
Comparison of orthologous gene content across multiple plant species highlighted a significant proportion of proteins that are conserved between NLL and four other legume species (Figure S5, Table S10). Among these species, lupin possesses a relatively high number of expanded paralogous genes (Figure S6), second only to Glycine max—likely due in both cases to independent whole‐genome duplication (WGD) and whole‐genome triplication in the Glycine and Lupinus lineages, respectively.
We find clear evidence of a whole‐genome triplication (WGT) in the genistoid lineage. This is inferred on the basis of synteny comparisons between NLL and itself and between NLL and other sequenced legume genomes (Data S6). Dot plots between NLL and another legume genome frequently show three strong, overlapping synteny blocks when these are viewed with respect to the other legume genome, or two blocks in the NLL self‐comparison (with the third copy visible as the NLL self‐match on the main diagonal). For the genomes Lotus japonicus, Medicago truncatula and Phaseolus vulgaris, the proportions of the NLL genome with a ‘synteny coverage depth’ of three with respect to the other genome are 21.4%, 21.0% and 13.2%, respectively (Table S11), while in comparisons going the ‘other way’ (with respect to NLL), the proportion of the genome with synteny coverage depth of three is negligible: 0.62%, 1.08%, 1.73%. In contrast, the proportion of those genomes with coverage depth at two (with respect to NLL) is high (14.2%, 27.5% and 28.6%), as expected, due to the papilionoid WGD (Table S11). In comparisons with Glycine max, the proportion of the genome with a ‘synteny coverage depth’ of three is 12.7% with respect to Glycine, while going the other way (with respect to NLL), the fourfold synteny coverage depth is greater than the threefold coverage depth (22.9% vs. 12.6%), as expected due to the additional WGD in the Glycine lineage.
Divergence times between Lupinus and other papilionoid legumes were calculated based on accumulation of synonymous changes between orthologous gene pairs between species (Figure S7), using a known species phylogeny and rooting the tree at the papilionoid WGD. The galegoid clade, containing Lupinus, is known to have originated near the base of the papilionoid subfamily (Lavin et al., 2005). If the papilionoid WGD immediately preceded the papilionoid radiation (Cannon et al., 2015), at ~58 Mya (Lavin et al., 2005), then we estimate the genistoid lineage separated from the other papilionoid legumes at ~54.6 Mya, and the whole‐genome triplication to have occurred in the genistoid lineage at ~24.6 Mya (Figure S7; Data S7).
These time estimates assume constant rates of synonymous nucleotide changes before and after the WGD. Additional taxon sampling in the Genisteae would be needed to refine the WGT timing; however, it is clear that the genistoid WGT is considerably older than the Glycine WGD, as Ks values for the WGT and WGD peaks are more than twofold greater in Lupinus than in Glycine (0.3 vs. 0.12).
From Ks analyses, we infer that the Lupinus lineage has accumulated point mutations at a rate similar to Lotus and Glycine, but more slowly than for Phaseolus or Medicago. This is apparent in papilionoid WGD peaks present at ~0.7 to 1.0, in self‐comparisons between paralogs (Figure 2). Furthermore, a WGT is evident in the genistoid lineage at around Ks ~0.3. This compares with the Glycine WGD peak at 0.12 and the papilionoid WGD at ~0.74 in Lupinus or ~0.68 in Glycine. If the papilionoid WGD occurred at ~58 Mya (Cannon et al., 2015; Lavin et al., 2005), then, assuming constant rates in this lineage, the genistoid WGT would have occurred at around 24.6 Mya.
Figure 2.
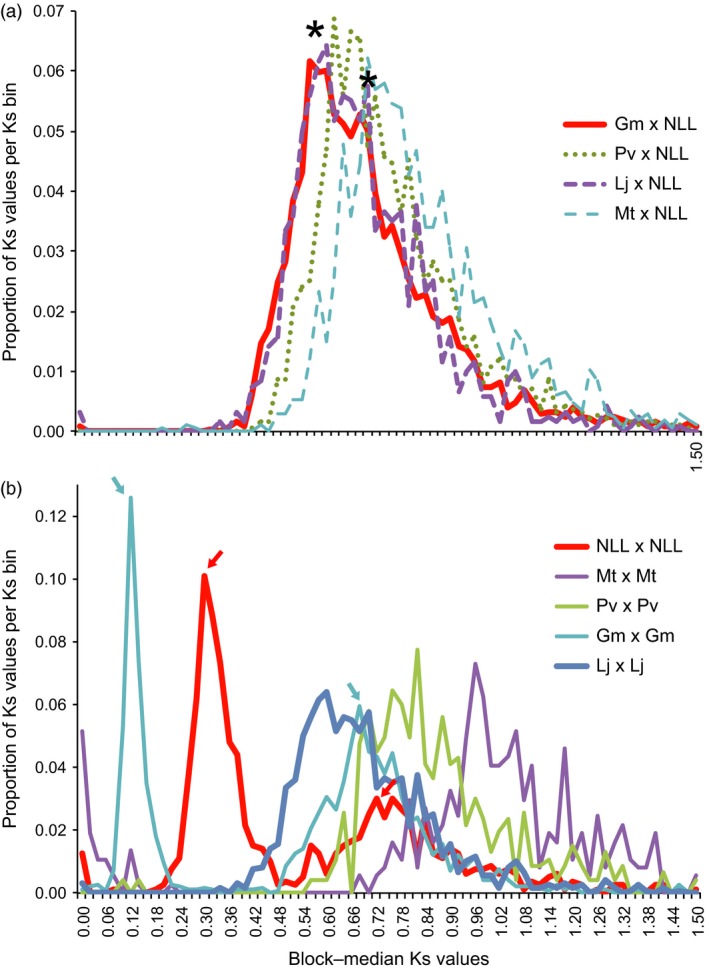
Synonymous substitution (Ks) analysis, showing proportion of values per Ks bin. Ks values are medians from synteny blocks for the indicated comparisons, and values in these plots are scaled to the total number of Ks counts for each comparison. (a) Orthologous comparisons between narrow‐leafed lupin (NLL) and Glycine max (Gm, red line), Phaseolus vulgaris (Pv, green dots), Lotus japonicus (Lj, purple dashed line) and Medicago truncatula (Mt, blue dashed lines). Asterisks show a primary peak for the speciation‐derived orthologs, and a probable smaller secondary peak for the papilionoid whole‐genome duplication‐derived ‘old orthologs’. (b) Paralogous genome self‐comparisons for narrow‐leafed lupin, Medicago truncatula, Phaseolus vulgaris, Glycine max and Lotus japonicus. Coloured arrows show two peaks in both Glycine max and narrow‐leafed lupin: the first peak in each case represents independent whole‐genome duplications in these lineages (Glycine at ~11 Mya and Lupinus at ~24 Mya), and the second peaks correspond to the shared papilionoid whole‐genome duplication. See Figure S7 and Data S7 for additional rate and date estimations.
Synteny comparisons with other sequenced legume genomes show extended regions of homology on all chromosomes, retained since divergence from the common ancestor of Lupinus and the other papilionoid species, which occurred ~55 Mya. For example, blocks spanning more than 6.4 million bases remain between soya bean and NLL (Table S12; Data S6, Data S7). Comparisons between NLL and soya bean generally show at least threefold synteny for NLL synteny viewed on soya bean as the reference, and at least fourfold synteny for soya bean synteny viewed on NLL as the reference, as both soya bean and NLL experienced the papilionoid WGD at ~58 Mya, and independent WGD at ~11 and WGT 24.6 Mya, respectively (see all‐by‐all chromosome dot plots for the NLL chromosomes compared to other NLL chromosomes and soya bean chromosomes in Data S6, and synteny depth coverage in Table S11). However, blocks are more degraded in NLL than soya bean. From the soya bean self‐comparison, in the recent and papilionoid WGDs, the longest remaining blocks are 12.8 million and 3.46 million bases, respectively, while from the NLL self‐comparison, in the recent and papilionoid WGDs, the blocks are 5.6 and 1.4 million bases, respectively. Average block lengths follow similar patterns, with the average ‘old’ (papilionoid) blocks from soya bean being 1.47 times longer than in NLL. The somewhat greater degradation in gene order in NLL is consistent with greater loss of paralogous genes (and decreased total gene count) in NLL than in soya bean.
Relating NLL gene content to industry‐relevant phenotypes
Analysis of the annotated gene set using InterPro and Go‐terms (Data S8) coupled with the dense reference genetic map (Data S3) allowed the nomination of candidate genes for phenotypes segregating in the recombinant inbred line (RIL) population. A major disease pressure on lupins, including NLL, is anthracnose (caused by Colletotrichum lupini). The cultivar Tanjil is resistant to anthracnose, and a single dominant resistance gene (Lanr1) maps to linkage group 11 (Kamphuis et al., 2015; Yang et al., 2013b). Using our new genetic map (Data S3), we refined the location of Lanr1 to a single scaffold (Scaffold_133), between flanking markers LaDArT_PAV20595 and LaDArT_PAV25221 (Table S9). This region spans 388 kb, harbours 5 cosegregating markers and contains 41 predicted genes (Lup005013.1‐Lup005054.1) including an NLR resistance gene (Lup005042.1). Alignment of Lup005042.1 sequence from the four parents of the two RIL populations used to fine‐map the location of Lanr1 showed complete conservation for resistant lines Tanjil and 83A:476, but considerable divergence to susceptible lines Unicrop and P27255 (Figure S8), thus making Lup005042.1 a good candidate for Lanr1.
Legumes typically undergo important symbiotic relationships with other organisms. This includes associations with beneficial bacteria to form rhizobium–legume symbiosis (RLS) and with beneficial fungi to form arbuscular mycorrhizal symbiosis (AMS). Some of the genes required for a successful association are shared by both types of symbioses, and it is believed that the evolutionary younger RLS recruited part of the genetic programme of the more ancient AMS (Parniske, 2008). Around 80% of land plants can form AMS, but some lineages have lost this ability along with some of the genes required to establish this relationship (Delaux et al., 2014). Among legumes, lupins are unique, because they can form RLS but are unable to form AMS. This has been used to identify genes that are shared between both symbiotic associations (Bravo et al., 2016; Delaux et al., 2014). The NLL genome was screened for the presence of AMS genes and was found to include 20 of 38 characterized mycorrhizal association genes. These included genes involved in rhizobium–legume symbiosis, or the biosynthesis, regulation or transport of plant hormones. However, NLL lacked key genes required specifically for AM symbiosis (in italics in Table 4) but not nodulation, including SbtM1, SbtM3, HA1, EXO70I, RAM2, PT4, STR1, STR2, RAM1, ERF1, RAD1, DIP1, FatM, KIN2, KIN3, KIN5, RFCb and CYT733A1 (Table 4; Data S9). The only exception was PP2AB'1, which so far is known only to be required for AMS (Charpentier et al., 2014), but may play other, yet to be discovered roles in lupin biology. During nodulation, lupins become infected by rhizobia via intercellular penetration rather than through intracellular infection threads, as do most other legumes (González‐Sama et al., 2004). Short‐infection‐thread‐like structures have been observed in cortical cells, but their importance is not clear (González‐Sama et al., 2004; James et al., 1997; Tang et al., 1992). Despite this, all genes known to be required for rhizobial infection were present in NLL (Table 4), suggesting fundamentally conserved mechanisms underlying different infection modes.
Table 4.
Overview of genes associated with arbuscular mycorrhizal and rhizobial associations in the genomes of Medicago truncatula and narrow‐leafed lupin
| Symbiotic component | Gene product | Medicago | Lupin | Referencea |
|---|---|---|---|---|
| NUP85 | Nucleoporin | Medtr1g006690 | Lup020970.1 | 1 |
| NUP133 | Nucleoporin | Medtr5g097260 | Lup029707.1 | 2 |
| NENA | Nucleoporin | Medtr6g072020 | Lup022917.1 | 3 |
| MCA8 | Calcium pump | Medtr7g100110 | Lup028615.1 Lup006231.1 Lup028310.1 Lup018698.1 | 4 |
| DELLA1 | Transcriptional regulators | Medtr3g065980 | Lup023873.1 Lup029445.1 | 5 |
| DELLA2 | Transcriptional regulators | TC182493 | Lup007545.1 Lup009138.1 | 5 |
| CCD7 | Carotenoid cleavage dioxygenase | Medtr7g045370 | Lup003751.1 | 6 |
| CCD8 | Carotenoid cleavage dioxygenase | Medtr3g109610 Medtr7g063800 | Lup028507.1 | 6 |
| PDR1 | ABC transporter | Medtr3g107870 Medtr1g011640 Medtr1g011650 | Lup013990.1 Lup001244.1 | 7 |
| D27 | Carotenoid isomerase | Medtr1g471050 Medtr7g095920 | Lup011456.1 Lup018644.1 | 8 |
| SUT2 | Sucrose transporter | Medtr8g468330 | Lup016593.1 | 9 |
| DMI1 (Pollux) | Cation channel | Medtr2g005870 | Lup014919.1 | 10 |
| NSP1 | GRAS transcription factor | Medtr8g020840 | Lup007304.1 | 11 |
| NSP2 | GRAS transcription factor | Medtr3g072710 | Lup012083.1 | 12 |
| DMI3 (CCaMK) | Calcium/calmodulin‐dependent protein kinase | Medtr8g043970 | Lup001774.1 | 13 |
| DMI2 (SYMRK/NORK) | Receptor‐like kinase | Medtr5g030920 | Lup025527.1 | 14 |
| Castor | Cation channel | Medtr7g117580 | Lup029273.1 | 15 |
| VAPYRIN | MSP and ANK repeat‐containing protein | Medtr6g027840 | Lup000011.1 Lup001531.1 | 16 |
| IPD3 (Cyclops) | Coiled‐coil domain containing protein | Medtr5g026850 | Lup027672.1 | 17 |
| NFP | LysM receptor‐like kinase | Medtr5g019040 | Lup012981.1 | 18 |
| PP2AB'1 | Protein phosphatase 2A | Medtr1g112940 | Lup024672.1 | 19 |
| LYK3 | LysM receptor kinase | Medtr5g086130 | Lup018960.1 | 20 |
| ERN1 | Transcription factor | Medtr7g085810 | Lup000007.1 | 21 |
| ERN2 | Transcription factor | Medtr6g029180 | Lup009942.1 | 22 |
| NIN | Transcription factor | Medtr5g099060 | Lup029716.1 | 23 |
| NF‐YA1 | Transcription factor | Medtr1g056530 | Lup000323.1 | 24 |
| NF‐YA2 | Transcription factor | Medtr7g106450 | Lup019646.1 | 25 |
| RPG | Coiled‐coil protein | Medtr1g090807 | Lup001677.1 | 26 |
| LIN | E3 ubiquitin ligase | Medtr1g090320 | Lup001700.1 | 27 |
| PUB1 | E3 ubiquitin ligase | Medtr5g083030 | Lup029507.1 | 28 |
| SUNN | LRR receptor kinase | Medtr4g070970 | Lup003404.1 | 29 |
| NPL | Pectate lyase | Medtr3g086320 | Lup011017.2 | 30 |
| CRE1 | Cytokinin receptor | Medtr8g106150 | Lup008799.1 | 31 |
| FLOT4 | Flotillin | Medtr3g106430 | Lup030707.1 | 32 |
| SYP132A | Syntaxin | Medtr2g088700 | Lup029417.1 Lup030298.1 | 33 |
| SbtM1 | Subtilisin‐like protease | Medtr5g011320 | 34 | |
| SbtM3 | Subtilisin‐like protease | Medtr5g011340 | 34 | |
| HA1 | ATPase | Medtr8g006790 | 35,36 | |
| Exo70 | Exocyst complex protein | Medtr1g017910 | 37 | |
| RAM2 | GPAT | Medtr1g040500 | 38 | |
| PT4 | Phosphate transporter | Medtr1g028600 | 39 | |
| STR1 | Half‐ABC transporter | Medtr8g107450 | 40 | |
| STR2 | Half‐ABC transporter | Medtr5g030910 | 40 | |
| RAM1 | GRAS transcription factor | Medtr7g027190 | 41 | |
| ERF1 | Transcription factor | Medtr7g009410 | 42 | |
| RAD1 | GRAS transcription factor | Medtr4g104020 | 43 | |
| DIP1 | GRAS transcription factor | Medtr8g093070 | 44 | |
| FatM | Acyl‐(ACP) thioesterase | Medtr1g109110 | 45 | |
| KIN2 | Protein kinase | Medtr4g129010 | 45 | |
| KIN3 | Protein kinase | Medtr7g116650 | 45 | |
| KIN5 | Serine–threonine protein kinase | Medtr3g104900 | 45 | |
| RFCb | Replication factor C | Medtr3g118160 | 45 | |
| CYT733A1 | P450 enzyme | Medtr6g034940 | 45 |
References for each of the functionally characterized genes in relation to either AM symbiosis or Rhizobia symbiosis can be found in Data S10.
We also examined genes involved in flowering time as early flowering is an important trait in NLL (Berger et al., 2013). Most genes and gene families prominent in flowering time control and light signalling in other dicot species were represented in NLL (Table S13), with notable exceptions. These included the FLC clade of vernalization‐responsive MADS‐domain proteins, which appears to be broadly absent from legume genomes including NLL (Hecht et al., 2005). Other genes appeared absent from NLL despite their presence in other papilionoid legumes (Table S13). These included the red light photoreceptor gene PHYE, which is present in Medicago and soya bean but absent in pea (Hecht et al., 2005; Platten et al., 2005; Schmid et al., 2003; Yant et al., 2010), suggesting it may have been lost more than once during legume evolution. A more striking case is the FT family which appears to consist of three relatively ancient clades in papilionoid legumes, FTa, FTb and FTc, with the FTa clade further divided into FTa1 and FTa3 subclades (Hecht et al., 2011). Genes in the FTa1 and FTb clades have significant roles in flowering time control in other legumes (Hecht et al., 2011; Kong et al., 2010; Laurie et al., 2011; Zhai et al., 2014), but both groups of genes are absent from the NLL draft assembly, which has only duplicated copies of FTa3 and FTc genes. Furthermore, microsynteny analysis between chickpea, common bean, medicago, soya bean and NLL showed conservation of the genes flanking the Ft genes in legumes with the FTa1, FTa2 and FTb absent in the NLL genome assembly, whereas FTc1 and FTa3 are present in the Tanjil assembly (Figure S9). The FTa1 and FTb clade genes are also not found in any of the comprehensive NLL transcriptome datasets (Kamphuis et al., 2015). This implies that the strong vernalization response of NLL (Berger et al., 2012) involves a mechanism distinct from that in Medicago truncatula where FTa1 is the major target (Laurie et al., 2011).
Conclusion
The comprehensive draft assembly of NLL (cultivar Tanjil) is the first representative of the genistoid clade of Papilionoideae legumes and will support further whole‐genome analysis of other species in this important clade. Resequencing of additional lupin lines and in‐depth transcriptome sequencing revealed widespread polymorphisms that were used to generate a dense reference genetic map. These resources are accessible through the Lupin Genome Portal (http://www.lupinexpress.org) which includes interactive BLAST, GBrowse and CMap interfaces (Donlin, 2009; Priyam et al., 2015; Youens‐Clark et al., 2009) and provides a platform for genome‐wide association studies and genomics‐based breeding approaches. The knowledge of germplasm diversity, and capacity for reverse genetics facilitated by the dense genetic map and pseudomolecule assembly can accelerate future breeding of elite cultivars. This will fortify efforts to improve lupins as human health food crops and increase yield stability and productivity of lupins for farmers worldwide.
Experimental procedures
Library preparation and sequencing
Paired‐end Illumina gDNA libraries of 100 bp length and 170, 500 and 800 bp insert sizes were generated (82.2× coverage). This was complemented by mate‐paired libraries of 50 bp read length and 2, 4, 10, 20 and 40 Kb insert sizes generating a total of 150.41 Gb or 162.8× coverage. Illumina sequence reads were trimmed for adapter and low‐quality sequences via CutAdapt v1.1 (min length 25 bp, rounds 3, match length 5 bp) (Martin, 2011). Mate‐paired libraries were filtered for contaminating paired‐end reads by merging pairs of reads with overlapping 3′ sequences via FLASH v1.2.2 (Magoč and Salzberg, 2011). Additional RNA‐seq Illumina data used in this project to complement genome sequence data were described in a previous study (Kamphuis et al., 2015). The total genome size for narrow‐leafed lupin was estimated by performing a 17‐mer frequency analysis of genomic paired‐end libraries via Kmerfreq (Liu et al., 2013), using the following equation: total genome size = (K‐mer frequency/primary peak depth).
Genetic mapping
To assign scaffolds to linkage groups, we developed additional transcriptome‐derived SNP markers compatible with the Fluidigm microfluidic array platform as previously described (Kamphuis et al., 2015) to add to the 1475 loci of the previous reference genetic map. These new Fluidigm SNP assays (469) were used to genotype the same 153 recombinant inbred lines (RILs) developed from a cross between 83A:476 (an Australian breeding line) and P27255 (a wild accession from Morocco) used previously to generate a genetic map (Nelson et al., 2006). Additionally, DArTSeq analysis (Diversity Arrays Technology, Canberra) was performed, resulting in a further 3901 SNP‐polymorphic markers and 4765 markers polymorphic by allele presence/absence. These new markers, together with the 830 previously reported STS markers, giving a total of 9972 markers and seven phenotypic trait loci (Kamphuis et al., 2015), generated an improved genetic map prepared with the aid of MultiPoint 3.1 (MultiQTL Ltd., Haifa, Israel) using the approach detailed in our previous study (Kamphuis et al., 2015).
De novo genome assembly and validation
Paired‐end Illumina data were assembled via SOAPdenovo2 (Luo et al., 2012) producing an initial assembly that was further scaffolded by SSPACE2 v2.0 (Boetzer et al., 2011), progressing iteratively through paired‐end (170, 500 and 800 bp) and mate‐paired (2, 4, 10, 20 and 40 Kb) sequence libraries in order of increasing insert size. Five rounds of scaffolding were performed for each insert library, followed by five rounds of gap‐closing via BGI GapCloser (Luo et al., 2012) using paired‐end sequences only. Further scaffolding was performed using BAC‐end sequence data (insert size ~100 Kb) (Gao et al., 2011), via Bambus (range 50–400 000 insert) (Pop et al., 2004). The length of assembly ‘gaps’ (i.e. unknown stretches represented by runs of >10 ‘N’ bases) was corrected to a uniform 100 bp. Scaffolds were screened for simple repeats via RepeatMasker (‐no_is ‐norna ‐noint) (Smit et al., 1996–2010) and tandem repeats finder (2 7 7 80 10 50 50 ‐f ‐d ‐m ‐h) (Benson, 1999). Sequences <200 bp length or with >=50% repetitive, simple repeat or unknown N bases were removed from the assembly as per GenBank requirements. The assembly was then validated versus the new genetic map generated in this study. Sequence‐based genetic markers were mapped to scaffolds via ePCR (Schuler, 1997) and BLASTN ((Altschul et al., 1990); e ≤ 1‐e05). Marker location on scaffolds was determined preferentially by ePCR (max. 2 gaps, 2 mismatches, amplicon range 10–1000 bp for markers designed for the Fluidigm platform, 10–5000 bp for other markers), where in silico PCR produced a single amplicon for the match with the minimum possible hamming distance (mismatches+gaps). Where in silico PCR could not determine an unambiguous marker location, the locations of unambiguous top BLASTN hits for known marker amplicon sequences to the scaffolds were used instead. Scaffold joins were compared to marker order on the genetic map, and where a conflict was found, preliminary scaffolds were split on all ‘gaps’ located between conflicted markers. Whole‐genome alignments to the Glycine max genome assembly using promer and mummerplot (Kurtz et al., 2004), generally filtering at 90% identity and requiring maximum unique matches (–mum), were also used to manually split scaffolds where ‘macrosynteny’ was observed and this did not conflict with genetic map data. This final filtered and validated set of scaffolds was then assembled into pseudochromosomes based on the 20 linkage groups of the genetic map. Where possible, scaffolds were assigned to linkage groups in the order of their constituent markers on the map, reverse complemented if indicated by two or more markers. Synteny versus G. max was also used to manually place scaffolds on the map, particularly where abrupt disruption of synteny corresponded to neighbouring scaffold termini. Scaffolds placed on linkage groups were subsequently joined by uniform unknown gap lengths of 100 bp to form pseudochromosomes.
Annotation of genes and other genome features
Transcriptome sequences for cv Tanjil were previously assembled by Kamphuis et al. (2015), and in this study additional transcriptomes for cv Unicrop and P27255 were generated by the same method. Annotation of gene structure in the cv Tanjil reference genome was predicted de novo using AUGUSTUS (Stanke et al., 2006) and GENSCAN (Burge and Karlin, 1997). Further support for gene annotations was provided through alignment to the genome assembly of EST sequences derived from GenBank EST records listed under the taxon ‘Fabaceae’ (Benson et al., 2013), and homology to proteins of Arabidopsis thaliana (Initiative, 2000), Cajanus cajan (Varshney et al., 2012), Cicer arietinum (Varshney et al., 2013), Glycine max (Schmutz et al., 2010), Medicago truncatula (Young et al., 2011) and Phaseolus vulgaris (Schmutz et al., 2014). De novo predictions were combined and curated with supporting homology and EST evidence via GLEAN (Elsik et al., 2007). RNA‐seq data were aligned to the genome via TopHat (Trapnell et al., 2009), assembled transcripts via Cufflinks (Trapnell et al., 2010) and predicted open reading frames according to transcript alignments. GLEAN results were aggregated with RNA‐seq‐supported gene models to produce the final gene set. Functional annotations were assigned to genes based on searches against Interpro (Quevillon et al., 2005), KEGG (Kanehisa and Goto, 2000), GO (Ashburner et al., 2000) and UniProt (The UniProt Consortium, 2013).
Repetitive DNA regions were predicted in the genome for both transposable elements (TEs) and tandem repeats. Annotation of TEs was based on homology and de novo methods. The homology approach used RepeatMasker v3.30 (Smit et al., 1996–2010) (with RepeatProteinMasker) to identify repeats matching known repeat sequences in Repbase v16.10 (Jurka et al., 2005). The de novo method predicted repetitive DNA via Repeatmodeler v1.0.5 (Smit and Hubley, 2010). Tandem repeats were predicted using Tandem Repeats Finder v4.04 (Benson, 1999).
Proteogenomics
Samples of NLL were obtained from leaf, seed, stem and root tissues and protein extracted and subjected to iTRAQ by Proteomics International using to the iTRAQ protocol (Sciex, USA).
Spectral data were analysed using ProteinPilot™ 4.0 Software (Sciex) against query and decoy databases generated from both translated gene annotation and six‐frame‐translated open reading frames. The database of potential open reading frames was generated by obtaining the six‐frame translation of scaffolds via EMBOSS getorf (between stop codons, ≥10 aa in length). The spectral data were exported as XML files with proteogenomic mapping of peptides to scaffold and pseudochromosome sequences performed with CDSmapper (http://sourceforge.net/projects/cdsmapper/).
Comparative genomics
Analysis of variation across the cv Tanjil genome
SNP and indel sequence variation was assessed across a panel of cultivars relative to the cv. Tanjil reference genome. NGS reads were aligned to the cv Tanjil reference genome via bowtie v 2.0.5 (–very‐sensitive) (Langmead and Salzberg, 2012), and variants were called via the Genome Analysis Tookit 3.4‐46 (McKenna et al., 2010). GATK was used to perform read deduplication via Markduplicates, then variant calling with HaplotypeCaller (–stand_call_conf 20 –stand_emit_conf 20 –min_pruning 5), producing variant data in VCF format (Danecek et al., 2011). Genome comparisons were visualized using Circos v0.67‐1 (Krzywinski et al., 2009).
Orthologous gene clusters were predicted via OrthoMCL (Li et al., 2003) comparing translated annotations of NLL to protein datasets from C. cajan (Varshney et al., 2012), C. arietinum (Varshney et al., 2013), G. max (Schmutz et al., 2010), M. truncatula (Young et al., 2011), P. vulgaris (Schmutz et al., 2014) and A. thaliana (Initiative, 2000).
Analysis of rates of silent‐site substitutions was carried out by searching all peptides against all others for the species Lupinus angustifolius, Glycine max (v 2.0), Lotus japonicus (v 3.0), Medicago truncatula (v 4.0) and Phaseolus vulgaris (v 1.0). Top respective matches were retained between each species per chromosome pairing (allowing for multiple total hits between two species for a given query gene), and within each species (for analysis of whole‐genome duplications). Then in‐frame alignments of coding sequences were made for each retained peptide alignment. From alignments of coding sequences, values for Ks, Ka and Ka/Ks were calculated using the ‘codeml’ method from the PAML package (Yang, 2007). Also from protein alignments, synteny blocks were inferred using DAGchainer (Haas et al., 2004). From the per‐gene‐pair alignments and the synteny blocks, median Ks values for blocks were calculated and used for Ks histogram peaks (Figure 2).
Ages of species divergences and whole‐genome duplications (Figure S7) were calculated from modal Ks peaks (Data S7), by treating initially unknown branch lengths in the known species/duplication tree as variables in a set of equations. The species/duplication tree was rooted at the papilionoid whole‐genome duplication, which predated the main papilionoid radiation (Cannon et al., 2015). A time of 58 Mya for the initial papilionoid radiation was assumed (Lavin et al., 2005). There were 11 unknown branch lengths in the tree in Figure S7, and sufficient data from the modal distances between and within species comparisons to solve for these unknowns algebraically.
To evaluate evidence for a whole‐genome triplication (WGT), synteny blocks were identified using DAGchainer, and synteny coverage depth was calculated using the BEDTools v2.25.0 (Quinlan and Hall, 2010) ‘coverage’ function to make comparisons between other genomes and NLL as the reference, or between NLL and each other genome as the reference. Coverage of synteny blocks was calculated at each nucleotide position using the ‐d option and summarized per coverage depth level.
For visual dot plot assessments of NLL compared with itself and with other legume genomes, we used promer and mummerplot from the MUMmer package (Kurtz et al., 2004), (v3.23) to make comparisons of translated nucleotide sequence, on genomic sequence that was masked for all except exonic sequence. The promer results were filtered to require at least 80% identity.
Genome assembly validation and comparison to previous draft assembly
The quality of the Tanjil draft assembly was evaluated using the default parameters of CEGMA (Core Eukaryotic Genes Mapping Approach) v 2.5 (Parra et al., 2007).
CoReFinder (Collapse/Repeat Finder) is a differential comparative read mapping pipeline, which identifies and discriminates between collapsed and repeated regions in genome assemblies. Paired‐end reads with insert sizes of 170 bp, 500 bp and 800 bp totalling a coverage of 138.63× were aligned to the assembly using SOAPaligner v2.21 (Li et al., 2009b), with mapped reads reported in three ways via the ‘−r’ parameter: −r0 (reads that map uniquely), −r1 (reads that map to more than one location, but only one random hit is reported) and −r2 (report all hits) and converted to sorted .bam files using SAMtools v1.2 (Li et al., 2009a). The .bam files were then split into pseudochromosomes/scaffolds using BamTools v2.4.0 (Barnett et al., 2011), such that for each pseudochromosome/scaffold there were three .bam files corresponding to each mapping. The per‐base coverage was calculated for each .bam file using BEDTools v2.25.0 (Quinlan and Hall, 2010). The BEDTools output was merged such that each pseudochromosome/scaffold had a single tab‐delimited output file consisting of the name of the pseudochromosome/scaffold, the position, the per‐base coverage for −r0, the per‐base coverage for −r1 and the per‐base coverage for −r2.
For each pseudochromosome/scaffold, a custom R script was used on the tab‐delimited file to mine for collapsed and repeated regions by iterating through each position in the file. Any region where the median per‐base coverage of −r0, −r1 and −r2 was greater than twice the overall median coverage was flagged as ‘coll’ (collapsed). Any region where the coverage for −r0 was between 0 and 2, the coverage for −r1 was greater or equal to 2, and the coverage for −r2 was 0.5 times the overall median was marked as ‘rnc’ (repeated, non‐collapsed). Regions that were marked as ‘coll’ or ‘rnc’ that were within 100 bp of each other were merged using BEDTools.
NGS reads were aligned to the previous draft assembly and the current Tanjil assembly via bowtie v 2.0.5 (–very‐sensitive) (Langmead and Salzberg, 2012), and RNASeq datasets for the five tissue types were aligned to the assembly using TopHat v 2.0.9 (–b2‐very‐fast −r 50 –mate‐std‐dev 200 ‐i 20 ‐I 4000‐g 20 ‐report‐secondary‐alignments ‐m 0–min‐coverage‐intron 20 –coverage‐search –microexon‐search) to determine and compare the coverage of the various paired‐end, mate‐pair and RNASeq datasets in the two Tanjil draft assemblies.
Comparison of gene function across Lupinus and other plant taxa
To observe general variation in gene function, functional annotations were assigned to the proteins of NLL cv Tanjil via Interproscan (Quevillon et al., 2005) and compared to those assigned in other plant species available from the PLAZA Dicots v3.0 and Monocots v3.0 databases (Van Bel et al., 2011). Fisher's exact test was applied to the number of genes assigned an Interpro term in NLL versus Glycine max, or the average of various groups of species: legumes, dicots, monocots and all available Viridiplantae (Data S1). In the Supplementary Data File provided, further filtering has been applied requiring an expansion in NLL (gene count fold change > 1) and a P‐value of ≤ 0.05.
To focus on variation in gene content relevant to arbuscular mycorrhiza and rhizobia association, a protein database was constructed which included the predicted proteins of NLL cv. Tanjil from this study, and 50 other land plant species (Bravo et al., 2016). This database was queried with proteins known to be involved in arbuscular mycorrhizal symbiosis in Medicago truncatula via BLASTP (Altschul et al., 1990), and the top 200 matches were used to create phylogenies. The protein models were aligned using MAFFT v7.205 (Katoh et al., 2002) with default values, and columns of the alignment that contained more than 50% gaps were eliminated. A phylogenetic tree was generated with FastTree v2.1.5 (Price et al., 2010) using the wag model of amino acid evolution. The presence or absence of NLL true orthologs was assessed through visual analysis of the topology of the phylogenies generated.
Accession code
Genome sequence assembly and annotation data can be found in GenBank under BioProject ID: PRJNA299755 and is also available for download and interrogation via BLAST and GBrowse from the Lupin Genome Portal (http://www.lupinexpress.org).
Author Contributions
JKH, MY, LGK, GG, MNN, SB, RF, L‐LG, SL, AM, JJ and KBS contributed to generation of genome sequence, transcriptome sequences, BAC‐end sequences, genetic mapping and physical mapping data and development of the genome browser; JKH, MY, LGK, GG, SC and JJ worked on genome assembly; JKH, MY and JJ contributed to the genome annotation; JKH, MY, LGK, MNN, AB, SB, RF, L‐LG, MJH, SL, C‐WL, AM, JM, JW and JJ contributed to the gene function analysis; JKH, MY, LGK, MNN, SC, GG, WH, PEB, BH, DE and JJ worked on genome analysis and comparative genomics; JKH, LGK and KBS wrote the manuscript with input from MY, MNN, CAA, SB, SC, MJH, AM, GM, JM, JM, DE and JJ; KBS; CAA, GM and KBS conceived and directed the project.
Supporting information
Figure S1 K‐mer‐based estimation of genome size based on frequency analysis of 17‐mers in paired‐end libraries. A primary coverage peak in 17‐mer frequency was observed at 39× coverage, which corresponded to a frequency of 37 098 706 666 and 44 885 658 200 bases. Using the equation (K‐mer frequency/Peak depth) the K‐mer frequency analysis estimated a total genome size of 951 248 889 bp or 951.2 Mb. This estimate is consistent with C‐value based estimates of 924 Mb.
Figure S2 Summary of sequence divergence (100% – sequence identity) across DNA transposon and retrotransposon families (LTR, LINE and SINE) predicted in the narrow‐leafed lupin cv. Tanjil genome assembly relative to: (a) a representative repeat sequence in Repbase; or (b) de novo repeat family consensus sequence.
Figure S3 Length distributions for gene annotation sub‐feature including mRNA (a), coding sequence (CDS) (b), exons (c) and introns (d).
Figure S4 (a) Length (cM) of the 20 linkage groups of the 83A:476 × P27255 RIL genetic map using 2959 non‐redundant molecular markers*. (b) Average interval size (cM) of the 83A:476 × P27255 RIL genetic map using 2959 non‐redundant molecular markers*. *Out of the 9972 markers genotyped in the RIL population (n = 153) 2959 had unique (non‐redundant) centiMorgan positions in the genetic map. Of the 9972 markers 7707 physically mapped unambiguously to unique locations in the pseudomolecule assembly and were used to orientate and assign scaffolds to pseudomolecules.
Figure S5 Venn diagram showing shared orthologous groups in narrow‐leafed lupin and other sequenced plant genomes.
Figure S6 Protein orthology comparison of narrow‐leafed lupin (L. angustifolius) and other sequenced plant species.
Figure S7 Estimation of divergence time for narrow‐leafed lupin (L. angustifolius) and selected other sequenced legume species. Red numbers are estimated divergence times from the present day in millions of years (Mya), derived relative to an assumed time of 58 Mya for the origin of the papilionoid clade and papilionoid whole‐genome duplication at effectively the same time. Black numbers on the branches are rates of synonymous‐site changes (Ks), calculated from modal Ks values between all paralogous genes between the species included above (Data S6). Asterisks mark whole‐genome duplications/triplication.
Figure S8 Alignment of translated amino‐acid sequences corresponding to narrow‐leafed lupin cv. Tanjil locus Lup005042.1 for anthracnose‐resistant (Colletotrichum lupini) NLL lines 83A:476 and Tanjil and susceptible NLL lines Unicrop and P27255. The translated amino‐acid sequences for 83A:476, P27255 and Unicrop were derived from the re‐sequencing data for these accessions/cultivars.
Figure S9 Schematic representation of microsynteny analysis of the FTa, FTb, and FTc gene clusters in barrel medic (M. truncatula), chickpea (C. arietinum), common bean (Phaseolus vulgaris), soybean (G. max) and narrow‐leafed lupin (L. angustifolius) showing that FTa1, FTa2 and FTb genes are absent in these microsyntenic regions of NLL. (a) FTb region microsynteny (b) FTa1/2 and FTc1/2 region microsynteny; (c) FTa3/4 region microsynteny.
Table S1 Summary of the total amount of sequence data generated for the L, angustifolius cv. Tanjil genome assembly and the average coverage per paired‐end and mate‐pair library, assuming an estimated genome size of 924 Mb based on C value prediction.
Table S2 Summary of repetitive DNA regions predicted within the narrow‐leafed lupin genome.
Table S3 Summary of non‐coding RNA genes predicted within the narrow‐leafed lupin cv. Tanjil genome assembly.
Table S4 Summary of proteomics analyses applied to four tissues of narrow‐leafed lupin and the number of peptides and proteins identified at a 99% confidence interval.
Table S5 Summary of functional annotations assigned to gene annotations in narrow‐leafed lupin cv. Tanjil.
Table S6 Summary statistics for 20 narrow‐leafed lupin linkage groups NLL‐01 to NLL‐20 comprising 9965 molecular markers and 7 trait loci.
Table S7 Read alignment of narrow‐leafed lupin cultivar Tanjil RNASeq data for five different tissue types, paired‐end and mate‐pair data to the genome.
Table S8 Overview of the coverage and variants (insertions/deletions and SNPs) identified for the three re‐sequenced narrow‐leafed lupin lines.
Table S9 Location of domestication traits and disease resistance genes in the genome assembly of narrow‐leafed lupin cv. Tanjil.
Table S10 Summary of orthologous gene families in narrow‐leafed lupin and other sequenced plant species.
Table S11 Synteny coverage depth for NLL vs. other genomes and for other genomes vs. NLL. Each row begins with a coverage depth (0, 1, 2, etc.). Percentages for a given coverage depth and a species indicate the proportion of the genome with the indicated synteny coverage depth, with respect to the indicated reference genome. For example, in table 11A, 21.4% of the Lotus genome is covered by three synteny features with NLL.
Table S12 Values under “WGD Ks peaks” are the Ks bin values for the mode in Ks plots from the indicated species pairs, corresponding to the papilionoid whole‐genome duplication (WGD) in that Ks plot. Plots can be seen in Figure 2 and in Supplemental Data File 5. For example, for Glycine‐Glycine, this would be the second modal peak in the orthologous plot, Figure 2B (the most recent peak being the one from the WGD within the Glycine lineage). For Glycine‐Lupinus, the WGD peak is older than the speciation peak. This can be seen in Figure 2A, second asterisk. Values under “Speciation Ks peaks” are also inferred from Figure 2 and in Supplemental Data File 6. For example, for Glycine‐Lupinus, the speciation peak can be seen in Figure 2A, first asterisk. Values under “Recent independent WGD peaks” are seen in Figure 2B for Glycine‐Glycine and Lupinus‐Lupinus.
Table S13 Summary of genes and gene families prominent in flowering time control and light signalling in other dicot species present or absent in the narrow‐leafed lupin genome.
Data S1 Predicted gene family expansions and contractions.
Data S2 Summary of sequence‐based genetic markers and their mapping to assembled scaffold sequences of narrow‐leafed lupin cv. Tanjil.
Data S3 An updated genetic map for narrow‐leafed lupin cv. Tanjil.
Data S4 CEGMA analysis.
Data S5 CoReFinder analysis.
Data S6 Dot plot comparisons between narrow‐leafed lupin and other legumes.
Data S7 Histograms of synonymous‐site changes between paralogous genes between pairs of sequenced legume genomes.
Data S8 Functional annotations assigned to gene annotations of narrow‐leafed lupin.
Data S9 References for Table 4.
Acknowledgements
We thank Elaine Smith, Natalie Fletcher and Hayley Casarotto for technical support; James Miller and Paul Lacaze from Millennium Science for support with the development of the Fluidigm SNP markers; and Joel Geoghegan (Centre for Cancer Biology, Adelaide) for performing the Fluidigm genotyping service. We also thank Bevan Buirchell and the Department of Agriculture and Food, Western Australia (DAFWA), for kindly providing the NLL seeds. This research was undertaken with the assistance of resources provided at the Pawsey Supercomputing Centre, and the NCI Specialised Facility in Bioinformatics through the National Computational Merit Allocation Scheme supported by the Australian Government. The mass spectrometry analyses were performed in facilities provided by the Lotterywest State Biomedical Facility‐Proteomics node, Harry Perkins Institute for Medical Research. This project was funded by the Grains Research and Development Corporation (GRDC), the Commonwealth Scientific and Industrial Research Organisation (CSIRO) and the University of Western Australia (UWA). The authors declare no conflict of interests.
References
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Ashburner, M. , Ball, C.A. , Blake, J.A. , Botstein, D. , Butler, H. , Cherry, J.M. , Davis, A.P. et al (2000) Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett, D.W. , Garrison, E.K. , Quinlan, A.R. , Stromberg, M.P. and Marth, G.T. (2011) BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27, 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, D.A. , Cavanaugh, M. , Clark, K. , Karsch‐Mizrachi, I. , Lipman, D.J. , Ostell, J. and Sayers, E.W. (2013) GenBank. Nucleic Acids Res. 41, D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, J.D. , Buirchell, B. , Luckett, D.J. and Nelson, M.N. (2012) Domestication bottlenecks limit genetic diversity and constrain adaptation in narrowleafed lupin (Lupinus angustifolius L.). Theoret. Appl. Genet. 124, 637–652. [DOI] [PubMed] [Google Scholar]
- Berger, J.D. , Clements, J.C. , Nelson, M.N. , Kamphuis, L.G. , Singh, K.B. and Buirchell, B. (2013) The essential role of genetic resources in narrow‐leafed lupin improvement. Crop Pasture Sci. 64, 361–373. [Google Scholar]
- Boetzer, M. , Henkel, C.V. , Jansen, H.J. , Butler, D. and Pirovano, W. (2011) Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Bravo, A. , York, T. , Pumplin, N. , Mueller, L.A. and Harrison, M.J. (2016) Genes conserved for arbuscular mycorrhizal symbiosis identified through phylogenomics. Nat. Plants, 2, 15208. [DOI] [PubMed] [Google Scholar]
- Bringans, S. , Hane, J. , Casey, T. , Tan, K.‐C. , Lipscombe, R. , Solomon, P. and Oliver, R. (2009) Deep proteogenomics; high throughput gene validation by multidimensional liquid chromatography and mass spectrometry of proteins from the fungal wheat pathogen Stagonospora nodorum . BMC Bioinformat. 10, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge, C. and Karlin, S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Cannon, S.B. , McKain, M.R. , Harkess, A. , Nelson, M.N. , Dash, S. , Deyholos, M.K. , Peng, Y. et al (2015) Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol. Biol. Evol. 32, 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier, M. , Sun, J. , Wen, J. , Mysore, K.S. and Oldroyd, G.E. (2014) Abscisic acid promotion of arbuscular mycorrhizal colonization requires a component of the PROTEIN PHOSPHATASE 2A complex. Plant Physiol. 166, 2077–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxford, A.E. , Rogers, T. , Caligari, P.D. and Wilkinson, M.J. (2008) High‐resolution melt analysis to identify and map sequence‐tagged site anchor points onto linkage maps: a white lupin (Lupinus albus) map as an exemplar. New Phytol. 180, 594–607. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaux, P.‐M. , Varala, K. , Edger, P.P. , Coruzzi, G.M. , Pires, J.C. and Ané, J.‐M. (2014) Comparative phylogenomics uncovers the impact of symbiotic associations on host genome evolution. PLoS Genet. 10, e1004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin, M.J. (2009) Using the generic genome browser (GBrowse). Curr. Protocols Bioinformat. Chapter 9 Unit 9.9 doi:10.1002/0471250953.bi0909s28. [DOI] [PubMed] [Google Scholar]
- Drummond, C.S. , Eastwood, R.J. , Miotto, S.T. and Hughes, C.E. (2012) Multiple continental radiations and correlates of diversification in Lupinus (Leguminosae): testing for key innovation with incomplete taxon sampling. Syst. Biol. 61, 443–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsik, C.G. , Mackey, A.J. , Reese, J.T. , Milshina, N.V. , Roos, D.S. and Weinstock, G.M. (2007) Creating a honey bee consensus gene set. Genome Biol. 8, R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO . (2013) Statistical Yearbook 2013: World Food and Agriculture. Rome: Food and Agriculture Organization of the United Nations; 289. [Google Scholar]
- Foley, R.C. , Gao, L.‐L. , Spriggs, A. , Soo, L.Y. , Goggin, D.E. , Smith, P.M. , Atkins, C.A. et al (2011) Identification and characterisation of seed storage protein transcripts from Lupinus angustifolius . BMC Plant Biol. 11, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley, R.C. , Jimenez‐Lopez, J.C. , Kamphuis, L.G. , Hane, J.K. , Melser, S. and Singh, K.B. (2015) Analysis of conglutin seed storage proteins across lupin species using transcriptomic, protein and comparative genomic approaches. BMC Plant Biol. 15, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L.‐L. , Hane, J. , Kamphuis, L. , Foley, R. , Shi, B.‐J. , Atkins, C. and Singh, K. (2011) Development of genomic resources for the narrow‐leafed lupin (Lupinus angustifolius): construction of a Bacterial Artificial Chromosome (BAC) library and BAC‐end sequencing. BMC Genom. 12, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladstones, J . (1970) Lupins as crop plants. Field Crop Abstracts, 23, 123–147. [Google Scholar]
- González‐Sama, A. , Lucas, M.M. , De Felipe, M.R. and Pueyo, J.J. (2004) An unusual infection mechanism and nodule morphogenesis in white lupin (Lupinus albus). New Phytol. 163, 371–380. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Wortman, J.R. and Salzberg, S.L. (2004) DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics, 20, 3643–3646. [DOI] [PubMed] [Google Scholar]
- Hecht, V. , Foucher, F. , Ferrándiz, C. , Macknight, R. , Navarro, C. , Morin, J. , Vardy, M.E. et al (2005) Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol. 137, 1420–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht, V. , Laurie, R.E. , Vander Schoor, J.K. , Ridge, S. , Knowles, C.L. , Liew, L.C. , Sussmilch, F.C. et al (2011) The pea GIGAS gene is a FLOWERING LOCUS T homolog necessary for graft‐transmissible specification of flowering but not for responsiveness to photoperiod. Plant Cell, 23, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, C. and Eastwood, R. (2006) Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proc. Natl Acad. Sci. 103, 10334–10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Initiative, A.G. (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796. [DOI] [PubMed] [Google Scholar]
- James, E. , Minchin, F. , Iannetta, P. and Sprent, J. (1997) Temporal relationships between nitrogenase and intercellular glycoprotein in developing white lupin nodules. Ann. Bot. 79, 493–503. [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kamphuis, L.G. , Hane, J.K. , Nelson, M.N. , Gao, L. , Atkins, C.A. and Singh, K.B. (2015) Transcriptome sequencing of different narrow‐leafed lupin tissue types provides a comprehensive uni‐gene assembly and extensive gene‐based molecular markers. Plant Biotechnol. J. 13, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. and Goto, S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzak, A. , Šafář, J. , Janda, J. , Doležel, J. , Wolko, B. and Naganowska, B. (2006) The bacterial artificial chromosome (BAC) library of the narrow‐leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 11, 396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K.I. and Miyata, T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res., 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, F. , Liu, B. , Xia, Z. , Sato, S. , Kim, B.M. , Watanabe, S. , Yamada, T. et al (2010) Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiol. 154, 1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroc, M. , Koczyk, G. , Święcicki, W. , Kilian, A. and Nelson, M.N. (2014) New evidence of ancestral polyploidy in the Genistoid legume Lupinus angustifolius L. (narrow‐leafed lupin). Theoret. Appl. Genet. 127, 1237–1249. [DOI] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , Jones, S.J. et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, S. , Phillippy, A. , Delcher, A.L. , Smoot, M. , Shumway, M. , Antonescu, C. and Salzberg, S.L. (2004) Versatile and open software for comparing large genomes. Genome Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambers, H. , Clements, J.C. and Nelson, M.N. (2013) How a phosphorus‐acquisition strategy based on carboxylate exudation powers the success and agronomic potential of lupines (Lupinus, Fabaceae). Am. J. Bot. 100, 263–288. [DOI] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie, R.E. , Diwadkar, P. , Jaudal, M. , Zhang, L. , Hecht, V. , Wen, J. , Tadege, M. et al (2011) The Medicago FLOWERING LOCUS T homolog, MtFTa1, is a key regulator of flowering time. Plant Physiol. 156, 2207–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin, M. , Herendeen, P.S. and Wojciechowski, M.F. (2005) Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst. Biol. 54, 575–594. [DOI] [PubMed] [Google Scholar]
- Lee, Y.P. , Mori, T.A. , Sipsas, S. , Barden, A. , Puddey, I.B. , Burke, V. , Hall, R.S. et al (2006) Lupin‐enriched bread increases satiety and reduces energy intake acutely. Am. J. Clin. Nutrit. 84, 975–980. [DOI] [PubMed] [Google Scholar]
- Lesniewska, K. , Książkiewicz, M. , Nelson, M.N. , Mahé, F. , Aïnouche, A. , Wolko, B. and Naganowska, B. (2011) Assignment of 3 genetic linkage groups to 3 chromosomes of narrow‐leafed lupin. J. Hered. 102, 228–236. [DOI] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al (2009a) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. , Yu, C. , Li, Y. , Lam, T.W. , Yiu, S.M. , Kristiansen, K. and Wang, J. (2009b) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics, 25, 1966–1967. [DOI] [PubMed] [Google Scholar]
- Liu, B. , Shi, Y. , Yuan, J. , Hu, X. , Zhang, H. , Li, N. , Li, Z. et al (2013) Estimation of genomic characteristics by analyzing k‐mer frequency in de novo genome projects. arXiv:1308.2012.
- Luo, R. , Liu, B. , Xie, Y. , Li, Z. , Huang, W. , Yuan, J. , He, G. et al (2012) SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. GigaScience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni, C. , Sessa, F. , Accardo, E. , Vanoni, M. , Morazzoni, P. , Scarafoni, A. and Duranti, M. (2004) Conglutin γ, a lupin seed protein, binds insulin in vitro and reduces plasma glucose levels of hyperglycemic rats. J. Nutrit. Biochem. 15, 646–650. [DOI] [PubMed] [Google Scholar]
- Magoč, T. and Salzberg, S.L. (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27, 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet. J. 17, 10–12. [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganowska, B. , Wolko, B. , Sliwinska, E. and Kaczmarek, Z. (2003) Nuclear DNA content variation and species relationships in the genus Lupinus (Fabaceae). Ann. Bot. 92, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M.N. , Phan, H.T. , Ellwood, S.R. , Moolhuijzen, P.M. , Hane, J. , Williams, A. , Clare, E. et al (2006) The first gene‐based map of Lupinus angustifolius L.‐location of domestication genes and conserved synteny with Medicago truncatula . Theoret. Appl. Genet. 113, 225–238. [DOI] [PubMed] [Google Scholar]
- Nelson, M.N. , Moolhuijzen, P.M. , Boersma, J.G. , Chudy, M. , Lesniewska, K. , Bellgard, M. , Oliver, R.P. et al (2010) Aligning a new reference genetic map of Lupinus angustifolius with the genome sequence of the model legume, Lotus japonicus . DNA Res. 17, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke, J.A. , Yang, S.S. , Miller, S.S. , Bucciarelli, B. , Liu, J. , Rydeen, A. , Bozsoki, Z. et al (2013) An RNA‐seq transcriptome analysis of orthophosphate‐deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol. 161, 705–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parniske, M. (2008) Arbuscular mycorrhiza: the mother of plant root endosymbioses. Nat. Rev. Microbiol. 6, 763–775. [DOI] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Parra‐González, L. , Aravena‐Abarzua, G. , Navarro‐Navarro, C. , Udall, J. , Maughan, J. , Peterson, L. , Salvo‐Garrido, H. et al (2012) Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies. BMC Genom. 13, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten, J.D. , Foo, E. , Elliott, R.C. , Hecht, V. , Reid, J.B. and Weller, J.L. (2005) Cryptochrome 1 contributes to blue‐light sensing in pea. Plant Physiol. 139, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop, M. , Kosack, D.S. and Salzberg, S.L. (2004) Hierarchical scaffolding with Bambus. Genome Res. 14, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. and Arkin, A.P. (2010) FastTree 2–approximately maximum‐likelihood trees for large alignments. PLoS ONE, 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyam, A. , Woodcroft, B.J. , Rai, V. , Munagala, A. , Moghul, I. , Ter, F. , Gibbins, M.A. , Moon, H. , Leonard, G. , Rumpf, W. and Wurm, Y . (2015) Sequenceserver: a modern graphical user interface for custom BLAST databases. bioRXiv, doi:10.1101/033142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevillon, E. , Silventoinen, V. , Pillai, S. , Harte, N. , Mulder, N. , Apweiler, R. and Lopez, R. (2005) InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, M. , Uhlenhaut, N.H. , Godard, F. , Demar, M. , Bressan, R. , Weigel, D. and Lohmann, J.U. (2003) Dissection of floral induction pathways using global expression analysis. Development, 130, 6001–6012. [DOI] [PubMed] [Google Scholar]
- Schmutz, J. , Cannon, S.B. , Schlueter, J. , Ma, J. , Mitros, T. , Nelson, W. , Hyten, D.L. et al (2010) Genome sequence of the palaeopolyploid soybean. Nature, 463, 178–183. [DOI] [PubMed] [Google Scholar]
- Schmutz, J. , McClean, P.E. , Mamidi, S. , Wu, G.A. , Cannon, S.B. , Grimwood, J. , Jenkins, J. et al (2014) A reference genome for common bean and genome‐wide analysis of dual domestications. Nat. Genet. 46, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler, G.D. (1997) Sequence mapping by electronic PCR. Genome Res. 7, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secco, D. , Shou, H. , Whelan, J. and Berkowitz, O. (2014) RNA‐seq analysis identifies an intricate regulatory network controlling cluster root development in white lupin. BMC Genom. 15, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, A. and Hubley, R . (2010) RepeatModeler Open‐1.0. http://www.repeatmasker.org.
- Smit, A. , Hubley, R. and Green, P . 1996. –2010. RepeatMasker Open‐3.0. http://www.repeatmasker.org.
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. and Morgenstern, B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, C. , Robson, A. , Dilworth, M. and Kuo, J. (1992) Microscopic evidence on how iron deficiency limits nodule initiation in Lupinus angustifolius L. New Phytol. 121, 457–467. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium . (2013) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucl. Acids Res. 41, D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Baren, M.J. , Salzberg, S.L. et al (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bel, M. , Proost, S. , Wischnitzki, E. , Movahedi, S. , Scheerlinck, C. , Van de Peer, Y. and Vandepoele, K. (2011) Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 158, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney, R.K. , Chen, W. , Li, Y. , Bharti, A.K. , Saxena, R.K. , Schlueter, J.A. , Donoghue, M.T. et al (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource‐poor farmers. Nat. Biotechnol. 30, 83–89. [DOI] [PubMed] [Google Scholar]
- Varshney, R.K. , Song, C. , Saxena, R.K. , Azam, S. , Yu, S. , Sharpe, A.G. , Cannon, S. et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Straub, D. , Yang, H. , Kania, A. , Shen, J. , Ludewig, U. and Neumann, G. (2014) The regulatory network of cluster‐root function and development in phosphate‐deficient white lupin (Lupinus albus) identified by transcriptome sequencing. Physiol. Plant. 151, 323–338. [DOI] [PubMed] [Google Scholar]
- Williams, W. (1979) Studies on the development of lupins for oil and protein. Euphytica, 28, 481–488. [Google Scholar]
- Yang, Z. (2007) PAML4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol., 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, H. , Tao, Y. , Zheng, Z. , Shao, D. , Lo, Z. , Sweetingham, M.W. , Buirchell, B.J. et al (2013a) Rapid development of molecular markers by next‐generation sequencing linked to a gene conferring phomopsis stem blight disease resistance for marker‐assisted selection in lupin (Lupinus angustifolius L.) breeding. Theoret. Appl. Genet. 126, 511–522. [DOI] [PubMed] [Google Scholar]
- Yang, H. , Tao, Y. , Zheng, Z. , Zhang, Q. , Zhou, G. , Sweetingham, M.W. , Howieson, J.G. et al (2013b) Draft genome sequence, and a sequence‐defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE, 8, e64799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant, L. , Mathieu, J. , Dinh, T.T. , Ott, F. , Lanz, C. , Wollmann, H. , Chen, X. et al (2010) Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2 . Plant Cell, 22, 2156–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youens‐Clark, K. , Faga, B. , Yap, I.V. , Stein, L. and Ware, D. (2009) CMap 1.01: a comparative mapping application for the Internet. Bioinformatics, 25, 3040–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, N.D. , Debellé, F. , Oldroyd, G.E. , Geurts, R. , Cannon, S.B. , Udvardi, M.K. , Benedito, V.A. et al (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature, 480, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, H. , Lu, S. , Liang, S. , Wu, H. , Zhang, X. , Liu, B. , Kong, F. et al (2014) GmFT4, a homolog of FLOWERING LOCUS T, is positively regulated by E1 and functions as a flowering repressor in soybean. PLoS ONE, 9, e89030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 K‐mer‐based estimation of genome size based on frequency analysis of 17‐mers in paired‐end libraries. A primary coverage peak in 17‐mer frequency was observed at 39× coverage, which corresponded to a frequency of 37 098 706 666 and 44 885 658 200 bases. Using the equation (K‐mer frequency/Peak depth) the K‐mer frequency analysis estimated a total genome size of 951 248 889 bp or 951.2 Mb. This estimate is consistent with C‐value based estimates of 924 Mb.
Figure S2 Summary of sequence divergence (100% – sequence identity) across DNA transposon and retrotransposon families (LTR, LINE and SINE) predicted in the narrow‐leafed lupin cv. Tanjil genome assembly relative to: (a) a representative repeat sequence in Repbase; or (b) de novo repeat family consensus sequence.
Figure S3 Length distributions for gene annotation sub‐feature including mRNA (a), coding sequence (CDS) (b), exons (c) and introns (d).
Figure S4 (a) Length (cM) of the 20 linkage groups of the 83A:476 × P27255 RIL genetic map using 2959 non‐redundant molecular markers*. (b) Average interval size (cM) of the 83A:476 × P27255 RIL genetic map using 2959 non‐redundant molecular markers*. *Out of the 9972 markers genotyped in the RIL population (n = 153) 2959 had unique (non‐redundant) centiMorgan positions in the genetic map. Of the 9972 markers 7707 physically mapped unambiguously to unique locations in the pseudomolecule assembly and were used to orientate and assign scaffolds to pseudomolecules.
Figure S5 Venn diagram showing shared orthologous groups in narrow‐leafed lupin and other sequenced plant genomes.
Figure S6 Protein orthology comparison of narrow‐leafed lupin (L. angustifolius) and other sequenced plant species.
Figure S7 Estimation of divergence time for narrow‐leafed lupin (L. angustifolius) and selected other sequenced legume species. Red numbers are estimated divergence times from the present day in millions of years (Mya), derived relative to an assumed time of 58 Mya for the origin of the papilionoid clade and papilionoid whole‐genome duplication at effectively the same time. Black numbers on the branches are rates of synonymous‐site changes (Ks), calculated from modal Ks values between all paralogous genes between the species included above (Data S6). Asterisks mark whole‐genome duplications/triplication.
Figure S8 Alignment of translated amino‐acid sequences corresponding to narrow‐leafed lupin cv. Tanjil locus Lup005042.1 for anthracnose‐resistant (Colletotrichum lupini) NLL lines 83A:476 and Tanjil and susceptible NLL lines Unicrop and P27255. The translated amino‐acid sequences for 83A:476, P27255 and Unicrop were derived from the re‐sequencing data for these accessions/cultivars.
Figure S9 Schematic representation of microsynteny analysis of the FTa, FTb, and FTc gene clusters in barrel medic (M. truncatula), chickpea (C. arietinum), common bean (Phaseolus vulgaris), soybean (G. max) and narrow‐leafed lupin (L. angustifolius) showing that FTa1, FTa2 and FTb genes are absent in these microsyntenic regions of NLL. (a) FTb region microsynteny (b) FTa1/2 and FTc1/2 region microsynteny; (c) FTa3/4 region microsynteny.
Table S1 Summary of the total amount of sequence data generated for the L, angustifolius cv. Tanjil genome assembly and the average coverage per paired‐end and mate‐pair library, assuming an estimated genome size of 924 Mb based on C value prediction.
Table S2 Summary of repetitive DNA regions predicted within the narrow‐leafed lupin genome.
Table S3 Summary of non‐coding RNA genes predicted within the narrow‐leafed lupin cv. Tanjil genome assembly.
Table S4 Summary of proteomics analyses applied to four tissues of narrow‐leafed lupin and the number of peptides and proteins identified at a 99% confidence interval.
Table S5 Summary of functional annotations assigned to gene annotations in narrow‐leafed lupin cv. Tanjil.
Table S6 Summary statistics for 20 narrow‐leafed lupin linkage groups NLL‐01 to NLL‐20 comprising 9965 molecular markers and 7 trait loci.
Table S7 Read alignment of narrow‐leafed lupin cultivar Tanjil RNASeq data for five different tissue types, paired‐end and mate‐pair data to the genome.
Table S8 Overview of the coverage and variants (insertions/deletions and SNPs) identified for the three re‐sequenced narrow‐leafed lupin lines.
Table S9 Location of domestication traits and disease resistance genes in the genome assembly of narrow‐leafed lupin cv. Tanjil.
Table S10 Summary of orthologous gene families in narrow‐leafed lupin and other sequenced plant species.
Table S11 Synteny coverage depth for NLL vs. other genomes and for other genomes vs. NLL. Each row begins with a coverage depth (0, 1, 2, etc.). Percentages for a given coverage depth and a species indicate the proportion of the genome with the indicated synteny coverage depth, with respect to the indicated reference genome. For example, in table 11A, 21.4% of the Lotus genome is covered by three synteny features with NLL.
Table S12 Values under “WGD Ks peaks” are the Ks bin values for the mode in Ks plots from the indicated species pairs, corresponding to the papilionoid whole‐genome duplication (WGD) in that Ks plot. Plots can be seen in Figure 2 and in Supplemental Data File 5. For example, for Glycine‐Glycine, this would be the second modal peak in the orthologous plot, Figure 2B (the most recent peak being the one from the WGD within the Glycine lineage). For Glycine‐Lupinus, the WGD peak is older than the speciation peak. This can be seen in Figure 2A, second asterisk. Values under “Speciation Ks peaks” are also inferred from Figure 2 and in Supplemental Data File 6. For example, for Glycine‐Lupinus, the speciation peak can be seen in Figure 2A, first asterisk. Values under “Recent independent WGD peaks” are seen in Figure 2B for Glycine‐Glycine and Lupinus‐Lupinus.
Table S13 Summary of genes and gene families prominent in flowering time control and light signalling in other dicot species present or absent in the narrow‐leafed lupin genome.
Data S1 Predicted gene family expansions and contractions.
Data S2 Summary of sequence‐based genetic markers and their mapping to assembled scaffold sequences of narrow‐leafed lupin cv. Tanjil.
Data S3 An updated genetic map for narrow‐leafed lupin cv. Tanjil.
Data S4 CEGMA analysis.
Data S5 CoReFinder analysis.
Data S6 Dot plot comparisons between narrow‐leafed lupin and other legumes.
Data S7 Histograms of synonymous‐site changes between paralogous genes between pairs of sequenced legume genomes.
Data S8 Functional annotations assigned to gene annotations of narrow‐leafed lupin.
Data S9 References for Table 4.