

Abstract
Leukotrienes (LTs) are pro-inflammatory lipid mediators derived from arachidonic acid (AA) with roles in inflammatory and allergic diseases. The biosynthesis of LTs is initiated by transfer of AA via the 5-lipoxygenase-activating protein (FLAP) to 5-lipoxygenase (5-LO). FLAP inhibition abolishes LT formation exerting anti-inflammatory effects. The soluble epoxide hydrolase (sEH) converts AA-derived anti-inflammatory epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatetraenoic acids (di-HETEs). Its inhibition consequently also counteracts inflammation. Targeting both LT biosynthesis and the conversion of EETs with a dual inhibitor of FLAP and sEH may represent a novel, powerful anti-inflammatory strategy. We present a pharmacophore-based virtual screening campaign that led to 20 hit compounds of which 4 targeted FLAP and 4 were sEH inhibitors. Among them, the first dual inhibitor for sEH and FLAP was identified, N-[4-(benzothiazol-2-ylmethoxy)-2-methylphenyl]-N’-(3,4-dichlorophenyl)urea with IC50 values of 200 nM in a cell-based FLAP test system and 20 nM for sEH activity in a cell-free assay.
In recent years, the “one-drug-hits-one-target” approach has essentially lost ground. Several successfully marketed drugs were shown to actually affect a multiplicity of targets in retrospective. A prominent example is acetylsalicylic acid, which was initially believed to interact solely with cyclooxygenases (COXs), but actually also interferes, among others, with mitogen-activated protein kinases and nuclear factor κB1. Several natural products with so-called privileged structures often affect a certain disease not only via a single target but rather interfere with pathologies at a variety of points of attack, with particular relevance for inflammation2. Drugs with polypharmacological modes of action were shown to be advantageous over combination therapy as they exert lower incidences of side effects and often lead to more resilient therapies3. Therefore, the rational development of chemical structures that contain fragments to inhibit multiple targets, so-called designed multiple ligands (DML), has emerged as a highly interesting field of research with promise for better pharmacotherapies3.
Computational approaches offer a valuable means for rational, tightly structured analysis of target families4 and can be used for drug design focusing on multiple targets. Pharmacophore modeling allows to condense the functionalities of active compounds towards target-specific interaction patterns5. By combining multiple pharmacophore models for different targets in a virtual screening, it is indeed possible to discover structures that contain fragments to affect two or more targets6.
A key biochemical pathway for targeting multiple inflammatory conditions is the arachidonic acid (AA) cascade. AA is released from membrane phospholipids by cytosolic phospholipase A2 (cPLA2) and further transformed via at least three separate routes: COX, lipoxygenases (LOs), and cytochrome P450 (CYP450) pathways. Prostaglandins (PGs) and thromboxane are formed via the COX pathway, whereas pro-inflammatory leukotrienes (LTs) but also specialized pro-resolving lipid mediators (SPMs, i.e. lipoxins, resolvins, protectins, and maresins) are generated via the 5-LO and related LO cascades. Finally, CYP450 monooxygenases transform AA to anti-inflammatory epoxyeicosatrienoic acids (EETs), which are further converted to dihydroxyeicosatetraenoic acids (di-HETEs) by soluble epoxide hydrolase (sEH)7,8. Due to the multitude of pro-inflammatory and pro-resolving mediators produced from one substrate (i.e. AA) in at least these three branches, blocking a single branch by a selective drug may cause redirection/shunting and amplification of alternative pathways, eventually even connected to increased adverse effects. Therefore, smart polypharmacological approaches promise better effectiveness with even fewer side effects7.
5-LO is a reasonable target to inhibit the biosynthesis of pro-inflammatory LTs. LTB4 and cysteinyl-LTs are derived from LTA4, a mediator that is synthesized from AA by 5-LO. While several 5-LO inhibitors were developed, only zileuton has become a marketed drug so far9. In cellulo, 5-LO requires the 5-LO-activating protein (FLAP) for formation of LTA410. FLAP, a nuclear membrane-anchored protein with apparently no enzymatic activity, is supposed to transfer liberated AA to 5-LO. Pharmacological or genetic inhibition of FLAP abolished 5-LO product formation in vivo11,12.
Only comparatively few chemical scaffolds have been reported as FLAP inhibitors such as MK-886, an indole-class compound13, and a series of quinolone-based inhibitors14, but in both cases research was discontinued. Recently however, FLAP has regained attention as a drug target, most prominently with GSK2190915, a novel promising indole-based derivative that completed phase II trials for the treatment of asthma15. In 2015 research on FLAP inhibitors received another boost with the development of a series of oxadiaozole-containing FLAP inhibitors, shown by Takahashi et al.16 and the discovery of AZD6642, another potent FLAP inhibitor17.
Due to the notion that the arterial wall of hypercholesterolemic patients is in a state of chronic inflammation, LTs have also been implicated in cardiovascular conditions, and the FLAP coding gene ALPOX5AP was revealed as a key gene for coronary heart disease in familial hypercholesterolemia patients18,19.
Upon formation of EETs from AA by CYP ω-oxidases, they are rapidly degraded by sEH to the inactive corresponding di-HETEs20. Therefore, sEH inhibition may lead to elevated EET levels thereby counteracting inflammation. In contrast to FLAP inhibitors, a broad variety of sEH inhibitors is found in the literature. They all display highly specific sEH interaction patterns around an amide or a urea functionality and are therefore ideally suited for pharmacophore modeling. In a recent publication, we presented a series of sEH pharmacophore models with the ability to prospectively identify new sEH inhibitors21.
Targeting both LT synthesis via inhibition of 5-LO and the conversion of EETs by suppressing sEH with a combination of two inhibitors led to an enhanced anti-inflammatory effect compared to single treatment22. Recently, a series of dual sEH/5-LO inhibitors, discovered by a DML approach, were reported with promising results23. FLAP was shown to assist 5-LO at the nuclear membrane also in the formation of anti-inflammatory lipoxin A4 and resolvin D124, while cytosolic 5-LO (distant from FLAP and the nuclear membrane) was suggested to form lipoxin A4 in a FLAP-independent manner25. Based on promising results from pre-clinical and clinical studies with FLAP inhibitors versus 5-LO inhibitors, FLAP might be the superior target to interfere with LT biosynthesis26. But so far, there are no dual sEH/FLAP inhibitors available. In this study, we pursued a pharmacophore model-based virtual screening approach leading to potentially novel, powerful compounds that target sEH and FLAP with anti-inflammatory potential.
Results
We first focused on the development and validation of ligand-based pharmacophore models for FLAP based on published FLAP inhibitors. Our aim was to combine the new FLAP inhibitor models with the previously developed sEH inhibitor models21 to identify potential dual FLAP/sEH inhibitors in a prospective virtual screening.
The FLAP models were generated based on a concise dataset of 11 active compounds from literature. Since FLAP “activity” can only be determined via analysis of cellular 5-LO product formation and is therefore difficult to distinguish from 5-LO activity itself, it was crucial to comprise the dataset only from compounds that were experimentally verified as specific FLAP inhibitors, either by use of crystallization, radio ligand assays, or by unambiguously excluding 5-LO as a target. An overview of the dataset is given in the Supplementary Information Table S1, compounds S1–S11.
Two ligand-based pharmacophore models were generated as follows:
Model FLAP1 (see Fig. 1A) was generated by aligning compounds S10 and S11, two indole-based FLAP inhibitors that are distinguished by a quinoline moiety. The model was refined and finally found 8 out of the 11 FLAP inhibitors (including the molecules it was generated from) as described in the Supplementary Information.
Figure 1.

(A) Pharmacophore model FLAP1: This model was generated by aligning compounds S10 and S11 (Table S1, Supplementary Information). It consists of two aromatic features (blue rings), two hydrophobic features (yellow spheres), a hydrogen-bond acceptor feature (HBA, red sphere), a negative ionizable feature (red star), and a coat of exclusion volumes (X-vols, grey spheres). (B) Model FLAP2. It consists of two hydrophobic features, two aromatic features, two HBA features, and an X-vols coat.
The selectivity of the models was investigated by screening them against a drug-like virtual library (12,775 compounds)27, which yielded 138 hits for this model.
The second model FLAP2 (Fig. 1B) was based on an alignment of supplementary compounds S7–S9, two substituted 2,2-bisaryl-bicycloheptanes (S7, S9) and one 1,1-bisaryl-cyclopentane (S8)28. The model was refined and finally found 9 out of the 11 FLAP inhibitors (including the structures it was generated from) and 3 hits in the virtual library.
Together, the two models found all 11 active compounds within the dataset. For experimental validation, both models were set to screen the commercial SPECS virtual library (www.specs.net). FLAP1 retrieved 204 virtual hits, while FLAP2 found 833. To ensure structural diversity among the hits, they were clustered into ten different structural categories and from each cluster the compound with the highest geometric pharmacophore fit value (assigned by LigandScout) was selected for testing. This lead to a total of 20 compounds selected for experimental testing.
The bioactivity of the selected compounds against FLAP was evaluated using a well-established bioassay, based on intact human neutrophils that were pre-incubated with the test compounds (10 min) and stimulated with Ca2+-ionophore A23187 for another 10 min, followed by RP-HPLC analysis of formed 5-LO products29. To exclude interference of the hits with 5-LO and thus, to discriminate between FLAP and direct 5-LO inhibition, the compounds were tested for suppression of isolated 5-LO in a cell-free assay (in the absence of FLAP). Out of the 10 identified hits by model FLAP1, three compounds (Fig. 2) were active on FLAP: 1, a substituted pyrrole, 2, a dicyclopentanaphthoquinolizine (47.8 (1) and 59.9% (2) remaining 5-LO product formation at 10 μM, respectively), and 3, a substituted benzimidazole (23.5% remaining 5-LO product formation at 10 μM). However, 2 also inhibited 5-LO directly in the cell-free assay and thus may not necessarily act on FLAP. Together, these data reflect a true positive hit rate of 20% for model FLAP1. For model FLAP2, three out of 10 compounds (4, 5) were significantly active against FLAP (i.e., <60% remaining 5-LO product formation at 10 μM, without affecting 5-LO directly) leading to a hit rate of 30%. Strikingly, compound 5 turned out as a highly potent inhibitor of 5-LO product biosynthesis (IC50 = 200 nM, Fig. 3A) in intact cells that is mediated by FLAP. Note that 5 failed to inhibit isolated 5-LO in the cell-free assay (Fig. 3B), supporting 5 as FLAP inhibitor. Compounds 5 and 6 were hardly active against FLAP.
Figure 2. Chemical structures of bioactive compounds.

Figure 3. Concentration-response curves for inhibition of FLAP-dependent 5-LO product formation and sEH activity.

(A) FLAP-dependent 5-LO product formation in intact PMNL (B) inhibition of 5-LO activity, cell-free assays, and (C) inhibition of sEH activity by compound 5. Data, means ± SEM, n = 4.
Seven compounds that were identified by model FLAP2 were predicted as potential sEH inhibitors. A complete list of the selected structures and the respective fitting models can be found in Table S2 in the Supplementary Information. A cell-free assay was applied to determine the inhibitory potency of the compounds against human sEH21. Three of the compounds (7, 8 and 9) did not inhibit sEH (IC50 > 30 μM, Table 1) and 3 compounds (10, 11 and 6) moderately inhibited sEH activity with IC50 of 3 to 12 μM (Table 1). Only 5 potently interfered with sEH (IC50 = 20 nM, Fig. 3C) being even somewhat superior over AUDA (IC50 = 30 nM), a reference sEH inhibitor30. Together, our two ligand-based pharmacophore model virtual screening campaign identified 5 as potential dual FLAP/sEH inhibitor and biological evaluation revealed highly potent, dual inhibition of FLAP and sEH with IC50 values of 200 and 20 nM, respectively.
Table 1. Overview on test substances and biological test results.
| Compound | Model FLAP | Model sEH | 5-LO product formation in intact neutrophils |
sEH activity | ||
|---|---|---|---|---|---|---|
| Remaining activity (% of control ± SEM) at |
||||||
| 1 μM | 10 μM | IC50 [μM] FLAP | IC50 [μM] sEH | |||
| 1 | Flap1 | n.f.a | 104.4 ± 2.8 | 47.8 ± 1.6 | ~10 | n.d.b |
| 2 | Flap1 | n.f. | 102.0 ± 4.6 | 59.9 ± 2.0 | >10 | n.d. |
| 3 | Flap1 | n.f. | 95.1 ± 3.6 | 23.5 ± 1.1* | >1 | n.d. |
| 4 | Flap2 | n.f. | 67.5 ± 3.1 | 6.6 ± 3.0 | >1 | n.d. |
| 5 | Flap2 | 1 and 8 | 1.6 ± 1.6** | 1.7 ± 1.7** | 0.2 ± 0.04 | 0.02 ± 0.007 |
| 6 | Flap2 | 2, 3 and 4 | 83.5 ± 3.8 | 71.3 ± 11.8 | 18 ± 0.5 | 11.4 ± 0.5 |
| 7 | Flap2 | 2 | 102.2 ± 10.1 | 105.3 ± 3.2 | >10 | >100 |
| 8 | Flap2 | 2 | 85.4 ± 4.8 | 56.7 ± 4.2c | >10 | >30 |
| 9 | Flap2 | 4 | 88.5 ± 5.5 | 81.9 ± 8.1 | >10 | >100 |
| 10 | Flap2 | 1 and 4 | 112.6 ± 8.9 | 109.6 ± 7.2 | >10 | 3.0 ± 0.3 |
| 11 | Flap2 | 3 and 4 | 97.3 ± 3.8 | 94.6 ± 2.5 | >10 | 4.7 ± 0.2 |
| 12 | Flap1 | n.f. | 106.0 ± 6.3 | 109.8 ± 5.5 | >10 | n.d. |
| 13 | Flap1 | n.f. | 115.3 ± 7.9 | 86.2 ± 10.2 | >10 | n.d. |
| 14 | Flap1 | n.f. | 107.5 ± 0.3 | 105.7 ± 1.3 | >10 | n.d. |
| 15 | Flap1 | n.f. | 95.8 ± 0.8 | 63.7 ± 6.5 | >10 | n.d. |
| 16 | Flap2 | n.f. | 93.2 ± 4.4 | 90.1 ± 13.0 | >10 | n.d. |
| 17 | Flap1 | n.f. | 107.6 ± 2.7 | 107.4 ± 3.0 | >10 | n.d. |
| 18 | Flap1 | n.f. | 105.4 ± 7.7 | 106.6 ± 4.2 | >10 | n.d. |
| 19 | Flap1 | n.f. | 110.3 ± 4.5 | 108.1 ± 5.6 | >10 | n.d. |
| 20 | Flap2 | n.f. | 84.2 ± 6.3 | 71.9 ± 4.5 | >10 | n.d. |
*p < 0.05, **p < 0.01, one-way ANOVA followed by Bonferroni post hoc test.
an.f.-not found; bn.d.- not determined; cnot tested for direct 5-LO inhibition.
Discussion
Exploiting pharmacophore-based models and virtual screening, we identified the first dual inhibitor for sEH and FLAP with high potency in the nanomolar range. Both model FLAP1 and model FLAP2 were able to identify two active compounds out of ten tested virtual hits that revealed significant bioactivities. A detailed description of the hits and their orientation in the pharmacophore models is given in the Supplementary Information. Of note, the two virtual hit compounds identified by model FLAP2 also inhibited sEH (5 and 6), and out of the 10 test compounds, two more were active on sEH (10 and 11) without activity against FLAP.
The most intriguing structure discovered in this work is the dual FLAP/sEH inhibitor 5 that potently interfered with both sEH (IC50 = 20 nM, Fig. 3) and FLAP (IC50 = 200 nM, Fig. 3). To immediately identify such a potent compound by a virtual screening campaign is remarkable and unusual, highlighting the fortunate success of our efforts. Compound 5 contains an urea moiety that is supposedly responsible for the high activity against sEH (see Fig. 4B) and enables the characteristic HBA and HBD interactions with ASP335, Tyr383, and Tyr46619. Urea containing structures have been shown to be highly effective on sEH21. The requirements for FLAP inhibition are fulfilled by the benzothiazole moiety of the molecule, covering one hydrophobic, one aromatic, and a HBA feature of the pharmacophore. The second HBA feature is mapped on the ether function connected to the phenyl ring, which covers the second hydrophobic and aromatic feature (Fig. 4A). The urea group and the chlorine substituted phenyl group are not essential for binding in the FLAP model, but do not hinder either, thus enabling the molecule to be dually active against FLAP and sEH at such low concentrations.
Figure 4.

Compound 5 mapped on model FLAP2 (A) and within the binding pocket of sEH mapping pharmacophore model 1 based on pdb entry 3ant43 (B), compound 6 mapped on model FLAP2 (C) and with the sEH model 4 based on pdb entry 3i1y44 (D).
Compound 6 was only hardly active against FLAP and sEH even at high concentrations (IC50 = 18 and 11.5 μM, respectively). It is composed of a benzothiazole and a benzimidazole connected by a thioether. Similar to the benzothiazole of 5, the benzimidazole of 6 covers one hydrophobic, one aromatic, and one HBA feature of model FLAP2. The second HBA is mapped on the thioether and the second heterocycle also covers one aromatic and hydrophobic feature (see Fig. 4C). In the binding pattern for sEH, the crucial HBA and hydrogen bond donor (HBD) features are covered by the benzimidazole, while the two hydrophobic features are mapped each on one of the heterocycles (Fig. 4D). Although the activity of 6 is not as remarkable as that of 5, and benzimidazoles are known to inhibit sEH31,32,33, it represents an interesting new scaffold for FLAP, due to its low molecular weight and comparatively simple structure. The ligand efficiency (LE) is defined by Formula (1)34, where MW is the molecular weight in g/mol.
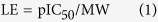 |
The LE for compound 5 is 0.010 for sEH and 0.0079 for FLAP. For 6 the results are 0.0065 (sEH) and 0.0059 (FLAP), indicating that as far as LE is concerned, 6 also constitutes a promising drug lead35.
In conclusion, the combination and application of two independently created pharmacophore model collections for the two pro-inflammatory targets allowed us to identify a completely novel and highly potent dual inhibitor of FLAP/sEH that will be further pharmacologically characterized. The synthesis of derivatives can be employed to experimentally verify the key structural elements for the activity on both enzymes. Moreover, a synthetic route for 5 was developed to provide more material for biological tests. A complete description of the synthesis is given in the Supplementary Information.
Methods
Molecular modeling
In ligand-based pharmacophore modeling, conformations of active molecules are computationally aligned and common pharmacophore features are placed where physicochemical functionalities overlap36. Conformations for this process were calculated with OMEGA37,38, which is implemented in LigandScout 3.12. The pharmacophore models were created using LigandScouts “create shared pharmacophore” function and optimized by adding an X-vol coat. Virtual screening was also performed in LigandScout, generating a 3D conformational library with 25 conformers per entry under omega-fast settings from the SPECS virtual library (202,920 compounds, version 2012). Default settings were used for virtual screening. Clustering the virtual hits for structural diversity was performed via ECFP4 in Accelrys’ Discovery Studio using the “find diverse molecules” protocol, using default settings, and requiring the program to cluster the ligands into ten groups.
Biological testing
Cells
Human neutrophils were freshly isolated from leukocyte concentrates obtained from the Institute of Transfusion Medicine, University Hospital Jena, as described39. Donors (healthy adult volunteers) were informed about the aim of the study and gave written consent. The protocol for experiments was approved by the ethical commission of the University Hospital in Jena. All methods were performed in accordance with the relevant guidelines and regulations. Briefly, neutrophils were isolated by dextran sedimentation, centrifugation on lymphocyte separation medium (LSM 1077, PAA, Coelbe, Germany) and hypotonic lysis of erythrocytes. Neutrophils were resuspended in PBS containing glucose (0.1%) to a final cell density of 5 × 106 cells/ml.
5-LO purification and cell-free 5-LO activity test
E. coli (BL21) was transformed with pT3–5-LO plasmid (kindly provided by Dr. Olof Radmark, Karolinska Institute, Stockholm, Sweden), and recombinant 5-LO protein was expressed at 30 °C as described40. Cells were lysed in 50 mM triethanolamine/HCl pH 8.0, 5 mM EDTA, 1 mM phenylmethanesulphonyl fluoride, soybean trypsin inhibitor (60 μg/ml), and lysozyme (1 mg/ml), homogenized by sonication (3 × 15 s), and centrifuged at 40,000 × g for 20 min at 4 °C. 5-LO was purified from the 40,000 × g supernatant (S40) on an ATP-agarose column. Aliquots of semi-purified 5-LO were diluted with ice-cold PBS containing 1 mM EDTA, pre-incubated with the test compounds or vehicle (0.1% DMSO) on ice for 10 min. 5-LO product formation was initiated by addition of 20 μM AA and the reaction was stopped after 10 min at 37 °C. 5-LO metabolites were analysed by RP-HPLC as described. 5-LO products include the all-trans isomers of LTB4 as well as 5(S)-hydroperoxy-6-trans-8,11,14-cis-eicosatetraenoic acid (5-HPETE) and its corresponding alcohol 5(S)-hydroxy-6-trans-8,11,14-cis-eicosatetraenoic acid (5-HETE)41.
5-LO product formation in intact neutrophils
Freshly isolated neutrophils (5 × 106 cells/ml) were suspended in PGC buffer (PBS pH 7.4, CaCl2 1 mM, glucose 0.1%), pre-incubated with the test compounds or vehicle (0.1% DMSO) for 10 min at 37 °C and stimulated with 2.5 μM Ca2+-ionophore A23187 for additional 10 min at 37 °C. The reaction was stopped by one volume (1 ml) of MetOH and 5-LO products (LTB4 and its trans-isomers as well as 5-HPETE and 5-HETE) were analyzed by HPLC as described above.
sEH purification and activity test
Human recombinant sEH was expressed and purified as described21,42. Briefly, Sf9 insect cells were cultured in suspension at 27 °C and infected with the recombinant baculovirus (kindly provided by Dr. B. Hammock, University of California, Davis, CA). After 72 h, cells were harvested and disrupted in buffer (50 mM NaHPO4, pH 8, 300 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM PMSF, 10 μg/ml leupeptin, and 60 μg/ml soybean trypsin inhibitor) by sonication (3 × 10 sec at 4 °C) and centrifuged for 1 h at 100,000 × g and 4 °C. sEH was purified from the supernatant by affinity chromatography utilizing benzylthio-sepharose and elution by 4-fluorochalcone oxide in PBS containing 1 mM DTT and 1 mM EDTA. The eluted enzyme solution was dialyzed, concentrated using Millipore Amicon-Ultra-15 centrifugal filter units and wash buffer, and the purity was verified by SDS-PAGE.
Enzyme activity of sEH was determined by a fluorescence-based assay using the non-fluorescent compound PHOME (Cayman Chemical, Ann Arbor, MI), which is converted by sEH to the fluorescent 6-methoxy-naphtaldehyde. Test compound or vehicle were pre-incubated with sEH in assay buffer (25 mM Tris HCl, pH 7, 0.1 mg/ml BSA) for 10 min at room temperature. PHOME (50 μM) was added and incubated for 60 min in the dark. The reaction was stopped by ZnSO4 and the fluorescence was monitored (λem 465 nm, λex 330 nm). Potential fluorescence or quenching by the tested compounds was determined by adding the tests compounds to the assay in the absence of sEH enzyme, and any autofluorescence was subtracted from the read out when applicable; fluorescence quenching by the test compounds was not observed.
Statistics
Data are expressed as mean ± S.E.M. IC50 values were calculated by nonlinear regression using GraphPad Prism Version 6 software (San Diego, CA) one site binding competition. Statistical evaluation of the data was performed by one-way ANOVA followed by a Bonferroni post hoc test for multiple comparison. A p value < 0.05 (*) was considered significant.
Additional Information
How to cite this article: Temml, V. et al. Discovery of the first dual inhibitor of the 5-lipoxygenase-activating protein and soluble epoxide hydrolase using pharmacophore-based virtual screening. Sci. Rep. 7, 42751; doi: 10.1038/srep42751 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This project was supported by the Austrian Science Fund, Network project “Drugs from Nature Targeting Inflammation” (S10703 and S10711) and the OeAD (project CZ 14/2013). D.S. holds an Ingeborg Hochmair Professorship at the University of Innsbruck.
Footnotes
The authors declare no competing financial interests.
Author Contributions D.S., H.S., U.G. and O.W. designed and supervised the study. V.T., Z.K. and B.W. performed the molecular modeling and virtual screening part. B.M. developed a synthesis route for 5 and its derivatives and analyzed these compounds. E.R., G.S., J.G. and U.G. tested the compounds in biological assays. All authors interpreted the results and contributed to the writing of the manuscript.
References
- Amann R. & Peskar B. A. Anti-inflammatory effects of aspirin and sodium salicylate. Eur. J. Pharmacol. 447, 1–9 (2002). [DOI] [PubMed] [Google Scholar]
- Koeberle A. & Werz O. Multi-target approach for natural products in inflammation. Drug Discov. Today 19, 1871–1882 (2014). [DOI] [PubMed] [Google Scholar]
- Morphy R. & Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 48, 6523–6543 (2005). [DOI] [PubMed] [Google Scholar]
- Anighoro A., Bajorath J. & Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 57, 7874–7887 (2014). [DOI] [PubMed] [Google Scholar]
- Wolber G., Seidel T., Bendix F. & Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 13, 23–29 (2008). [DOI] [PubMed] [Google Scholar]
- Schuster D. 3D pharmacophores as tools for activity profiling. Drug Discov. Today. Technol. 7, 203–270 (2010). [DOI] [PubMed] [Google Scholar]
- Meirer K., Steinhilber D. & Proschak E. Inhibitors of the arachidonic acid cascade: interfering with multiple pathways. Basic Clin. Pharmacol. Toxicol. 114, 83–91 (2014). [DOI] [PubMed] [Google Scholar]
- Stables M. J. & Gilroy D. W. Old and new generation lipid mediators in acute inflammation and resolution. Prog. Lipid Res. 50, 35–51 (2011). [DOI] [PubMed] [Google Scholar]
- Werz O. & Steinhilber D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol. Therapeut. 112, 701–718 (2006). [DOI] [PubMed] [Google Scholar]
- Abramovitz M. et al. 5-lipoxygenase-activating protein stimulates the utilization of arachidonic acid by 5-lipoxygenase. Eur. J. Biochem. 215, 105–111 (1993). [DOI] [PubMed] [Google Scholar]
- Byrum R. S., Goulet J. L., Griffiths R. J. & Koller B. H. Role of the 5-lipoxygenase–activating protein (FLAP) in murine acute inflammatory responses. J. Exp. Med. 185, 1065–1076 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. F., Ferguson A. D., Mosley R. T. & Hutchinson J. H. What’s all the FLAP about? 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol. Sci. 29, 72–78 (2008). [DOI] [PubMed] [Google Scholar]
- Gillard J. et al. L-663,536 (MK-886) (3-1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl-2,2-dimethylpropanoic acid), a novel orally active leukotriene biosynthesis inhibitor. Can. J. Physiol. Pharm. 67, 456–464 (1989). [DOI] [PubMed] [Google Scholar]
- Müller-Peddinghaus R. et al. BAY X1005, a new inhibitor of leukotriene synthesis: in vivo inflammation pharmacology and pharmacokinetics. J. Pharmacol. Exp. Ther. 267, 51–57 (1993). [PubMed] [Google Scholar]
- Snowise N. G., Clements D., Ho S.-Y. & Follows R. M. A. Addition of a 5-lipoxygenase-activating protein inhibitor to an inhaled corticosteroid (ICS) or an ICS/long-acting beta-2-agonist combination in subjects with asthma. Curr. Med. Res. Opin. 29, 1663–1674 (2013). [DOI] [PubMed] [Google Scholar]
- Takahashi H. et al. Synthesis, SAR, and series evolution of novel oxadiazole-containing 5-lipoxygenase activating protein inhibitors: discovery of 2-[4-(3-{(R)-1-[4-(2-amino-pyrimidin-5-yl)-phenyl]-1-cyclopropyl-ethyl}-[1,2,4]oxadiazol-5-yl)-pyrazol-1-yl]-N,N-dimethyl-acetamide (BI 665915). J.Med. Chem. 58(4), 1669–1690 (2015). [DOI] [PubMed] [Google Scholar]
- Lemurell M. et al. Discovery of AZD6642, an inhibitor of 5-lipoxygenase activating protein (FLAP) for the treatment of inflammatory diseases. J.Med. Chem. 58(2), 897–911 (2015). [DOI] [PubMed] [Google Scholar]
- van der Net J. B. et al. Arachidonate 5-lipoxygenase-activating protein (ALOX5AP) gene and coronary heart disease risk in familial hypercholesterolemia. Atherosclerosis 203, 472–478 (2009). [DOI] [PubMed] [Google Scholar]
- Helgadottir A. et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat. Genet. 36, 233–239 (2004). [DOI] [PubMed] [Google Scholar]
- Schmelzer K. R. et al. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. USA 102, 9772–9777 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltenberger B. et al. Discovery of potent soluble epoxide hydrolase (sEH) inhibitors by pharmacophore-based virtual screening. J. Chem. Inf. Model. 56, 747–762 (2016). [DOI] [PubMed] [Google Scholar]
- Liu J.-Y. et al. Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem. Pharmacol. 79, 880–887 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirer K. et al. Synthesis and structure–activity relationship studies of novel dual inhibitors of soluble epoxide hydrolase and 5-lipoxygenase. J. Med. Chem. 56, 1777–1781 (2013). [DOI] [PubMed] [Google Scholar]
- Lehmann C. et al. Lipoxin and resolvin biosynthesis is dependent on 5-lipoxygenase activating protein. FASEB J. 29(12), 5029–5043 (2015). [DOI] [PubMed] [Google Scholar]
- Fredman G. et al. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl. Acad. Sci. USA 111, 14530–14535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen D., Davidsson Ö. & Whatling C. Recent advances for FLAP inhibitors. Bioorg. Med. Chem. Lett. 25, 2607–2612 (2015). [DOI] [PubMed] [Google Scholar]
- Schuster D. et al. Pharmacophore modeling and in silico screening for new P450 19 (aromatase) inhibitors. J. Chem. Inf. Model 46, 1301–1311 (2006). [DOI] [PubMed] [Google Scholar]
- Macdonald D. et al. Substituted 2,2-bisaryl-bicycloheptanes as novel and potent inhibitors of 5-lipoxygenase activating protein. Bioorg. Med. Chem. Lett. 18, 2023–2027 (2008). [DOI] [PubMed] [Google Scholar]
- Banoglu E. et al. Identification of novel benzimidazole derivatives as inhibitors of leukotriene biosynthesis by virtual screening targeting 5-lipoxygenase-activating protein (FLAP). Bioorg. Med. Chem. 20, 3728–3741 (2012). [DOI] [PubMed] [Google Scholar]
- Jones P. D. et al. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal. Biochem. 343, 66–75 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano Y., Yamaguchi T. & Tanabe E., Structural insights into binding of inhibitors to soluble epoxide hydrolase gained by fragment screening and X-ray crystallography. Bioorg. Med. Chem. 22(8), 2427–2434 (2014). [DOI] [PubMed] [Google Scholar]
- Buscató E. et al. From a multipotent stilbene to soluble epoxide hydrolase inhibitors with antiproliferative properties. ChemMedChem. 8(6), 919–923 (2013). [DOI] [PubMed] [Google Scholar]
- Xue Y. et al. Fragment screening of soluble epoxide hydrolase for lead generation—structure-based hit evaluation and chemistry exploration. ChemMedChem. 11(5), 497–508 (2016). [DOI] [PubMed] [Google Scholar]
- Shultz M. D. Setting expectations in molecular optimizations: strengths and limitations of commonly used composite parameters. Bioorg.Med. Chem. Lett. 23, 5980–5991 (2013). [DOI] [PubMed] [Google Scholar]
- Abad-Zapatero C. Ligand efficiency indices for effective drug discovery. Expert Opin. Drug Discov. 2, 469–488 (2007). [DOI] [PubMed] [Google Scholar]
- Yang S.-Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today 15, 444–450 (2010). [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D., Skillman A. G., Warren G. L., Ellingson B. A. & Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 50, 572–584 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C. D. & Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. (again) :) 52, 2919–2936 (2012). [DOI] [PubMed] [Google Scholar]
- Pergola C. et al. ERK-mediated regulation of leukotriene biosynthesis by androgens: A molecular basis for gender differences in inflammation and asthma. Proc. Natl. Acad. Sci. USA 105, 19881–19886 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L., Szellas D., Rådmark O., Steinhilber D. & Werz O. Phosphorylation- and stimulus-dependent inhibition of cellular 5-lipoxygenase activity by nonredox-type inhibitors. FASEB J 17, 949–951 (2003). [DOI] [PubMed] [Google Scholar]
- Steinhilber D., Herrmann T. & Roth H. J. Separation of lipoxins and leukotrienes from human granulocytes by high-performance liquid chromatography with a Radial-Pak cartridge after extraction with an octadecyl reversed-phase column. J. Chromatogr. B Biomed. Sci. Appl. 493, 361–366 (1989). [DOI] [PubMed] [Google Scholar]
- Wixtrom R. N., Silva M. H. & Hammock B. D. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated Sepharose gels. Anal. Biochem. 169, 71–80 (1988). [DOI] [PubMed] [Google Scholar]
- Tanaka D. et al. A practical use of ligand efficiency indices out of the fragment-based approach: ligand efficiency-guided lead identification of soluble epoxide hydrolase inhibitors. J. Med. Chem. 54, 851–857 (2011). [DOI] [PubMed] [Google Scholar]
- Eldrup A. B. et al. Structure-based optimization of arylamides as inhibitors of soluble epoxide hydrolase. J. Med. Chem. 52, 5880–5895 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.