

Summary
NanoString nCounter is a recently developed platform that can make direct multiplexed measurement of gene expression using color-coded probe pairs (1, 2). We have found that this platform is uniquely suitable for quantification of pathogen gene expression during infection, where pathogen RNA comprises a tiny portion of total RNA isolated from the infected tissue. Here, we describe a protocol that we have successfully applied to a number of pathogens across multiple infection models, including both invasive and mucosal infection by Candida albicans, and lung infection by Aspergillus fumigatus and Cryptococcus neoformans.
Keywords: Pathogen, infection, RNA isolation, gene expression, in vivo profiling, nanoString
1. Introduction
What genes does a pathogen express during infection to cope with the host environment? Which regulatory pathways are responsible for the onset and retreat of such responses? These are among the most important questions in microbial pathogenesis. However, for most mammalian pathogens, gene expression profiling studies have been limited by the technical difficulty to accurately quantify pathogen gene transcripts from infected tissues, even in light of new genome-wide technologies (3). Host RNA constitutes an overwhelming portion (usually > 99%) of the total RNA isolated from infected tissue samples. This poses a challenge for most expression profiling technologies: it contributes to high background on microarrays, and it dominates sequence reads from RNA-Seq. Pathogen cell isolation from infected tissue, in theory, can help to enrich for pathogen RNA, but it poses additional problems: it requires large quantities of infected tissue; the procedure can be tedious; and most importantly, it is difficult to conserve the native state or integrity of RNA during the lengthy process. Here we describe an in vivo gene expression profiling protocol that is fast, extremely sensitive and highly reproducible (Note 1). We developed this protocol during our investigation of the fungal pathogen Candida albicans in a murine model of hematogenously disseminated candidiasis. Using this protocol, we have documented time courses of dynamically regulated C. albicans gene expression during kidney infection, and discovered unexpected features of the gene expression response to antifungal drug treatment in vivo (4). We have successfully applied this protocol to a number of other tissue types, pathogens and infection models (5–7).
2. Materials
2.1 RNA isolation from infected tissue
GentelMACS dissociator and M-type homogenization tube (Miltenyi Biotec)
Minibeadbeater
Tabletop centrifuge
Zirconia beads
Phenol:Chloroform:Isoamyl Alcohol 25:24:1
RNeasy kit, including buffers RLT, RW, and RPE, and RNeasy spin columns
BioPhotometer
2.2 NanoString profiling
NanoString nCounter system (nanoString)
NanoString nCounter master kit (nanoString)
NanoString custom built codeset: The investigator selects the high priority genes and nanoString will design the probes and synthesize the codeset reaction mix. For in vivo profiling studies, it is important to remind the nanoString codeset design team to avoid probe sequences that could cross hybridize to host genes.
Thermal cycler
MultiExperiment Viewer 4 software (8)
3. Methods
3.1 RNA isolation from infected tissue (using mouse kidney as an example)
Male Balb/c mice weighting 20–22g were used for all studies. Three mice per experimental group were inoculated intravenously with 1 × 106 yeast-phase C albicans cells. The animals were sacrificed and kidneys were harvested at specific time points post-infection. The right kidney was snap frozen in liquid nitrogen and stored at −80°C in a screw cap tube for later RNA extraction (Note 2).
Prepare the following reagents before removing kidneys from the −80°C freezer: add 2-mercaptoethanol (1% V/V) to buffer RLT; label M-tubes and chill on ice; label 2 ml screw cap tubes, and add approximately 300 ul Zirconia beads.
Remove kidneys from −80°C freezer and put on ice. Add 1.2 ml of buffer RLT with 2-mercaptoethanol to each kidney (Note 3). Decant kidney with buffer into an M-tube.
Homogenize the kidney in M-tube using gentelMACS dissociator on pre-loaded setting RNA_02.01.
Centrifuge the M-tube at 1000g for 1 minute in a tabletop centrifuge at room temp.
Transfer 600 ul of homogenate from M-tube to the screw cap tube containing Zirconia beads. Save the remaining homogenate on ice.
Add 600 ul Phenol:Chloroform:Isoamyl Alcohol 25:24:1 to the tubes from the previous step.
Close the lids tightly and vortex on mini-beadbeater for 3 minutes in a 4°C cold room.
Centrifuge the tubes at 15000g for 5 minutes in a 4°C cold room.
Carefully transfer the aqueous phase to a new 1.5 ml microfuge tube, mix well with equal volume of 70% ethanol, then load onto the RNeasy spin column.
Wash the spin column once with 700 ul buffer RW, followed by twice with 500 ul buffer RPE. Centrifuge one extra minute in a dry collection tube to remove remaining liquid in the spin column.
Elute RNA with 50 ul of H2O, and measure the RNA concentration using a BioPhotometer (Note 4).
3.2 NanoString profiling (12 reactions)
Thaw the nanoString reporter codeset (green cap tube) and capture codeset (gray cap tube) on ice.
Add 130 ul hybridization buffer to the reporter codeset (green), invert to mix and spin down.
Add 20 ul of the mix to each of the 12 reaction tubes.
Add 10 ug of total tissue RNA (in a volume of 5ul) to each tube (Note 5). Mix by pipetting.
Add 5 ul capture codeset to each tube. Mix by pipetting.
Incubate the reaction at 65°C in a thermal cycler overnight (12–18 hours).
Take out one sealed sample cartridge (−20°C) and two prep plates (4°C). Let them warm to room temperature.
Centrifuge the prep plates at 670g for 2 minutes in a tabletop centrifuge.
Set up the nanoString prep station following on-screen instructions.
Remove the reactions from the thermal cycler and immediately load on the prep station. Select to run the high sensitivity program (3 hours).
When the prep station program is complete, remove the cartridge, seal the lanes with a clear tape (provided by nanoString), apply mineral oil to the bottom of the cartridge (for generation one nCounter only, later generations do not require this step), then load the cartridge onto the nanoString digital analyzer.
Set up the nanoString digital analyzer following on-screen instructions.
Select the high resolution (600 fields) option, run the scanning program (~4.5 hours).
Upon receiving the results (by email), import raw data into nSolver software (provided by nanoString). The software will automatically check data quality and raise flags if the quality of the data falls out of the normal range. Perform technical adjustment using the built-in function (optional, follow instructions in the software), then export the data as an excel file (Note 6).
Normalize the data using one of the following methods (Note 7): total counts from all genes in the codeset; one or a few internal control genes; geometric mean of highly expressed genes.
Calculate the mean expression values for each gene (if the experiment was done in replicates or triplicates), then calculate the ratio of expression levels among different experimental groups (Note 8).
Visualize the datasets in heap maps by a clustering program such as MultiExperiment Viewer (8).
Figure 1.
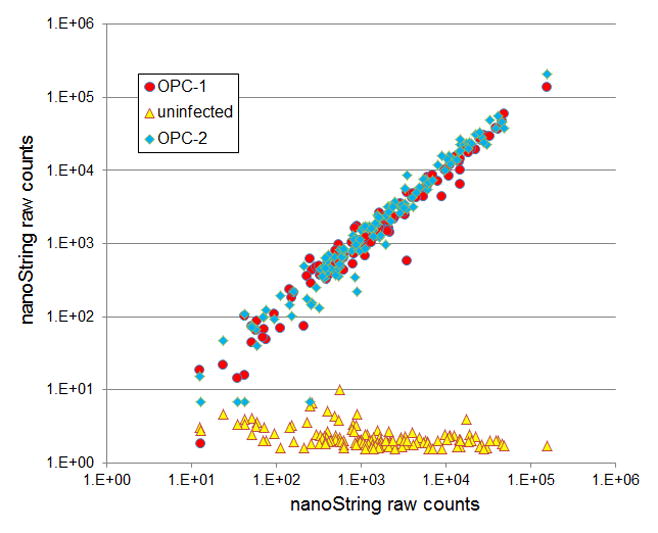
Raw nanoString counts for 135 C. albicans environmental response genes. NanoString probe counts for infected (red and blue data points) and uninfected (yellow) mouse tongue samples are presented as scatter plots for the 2 days postinfection time point. Adapted from (7).
Figure 2.
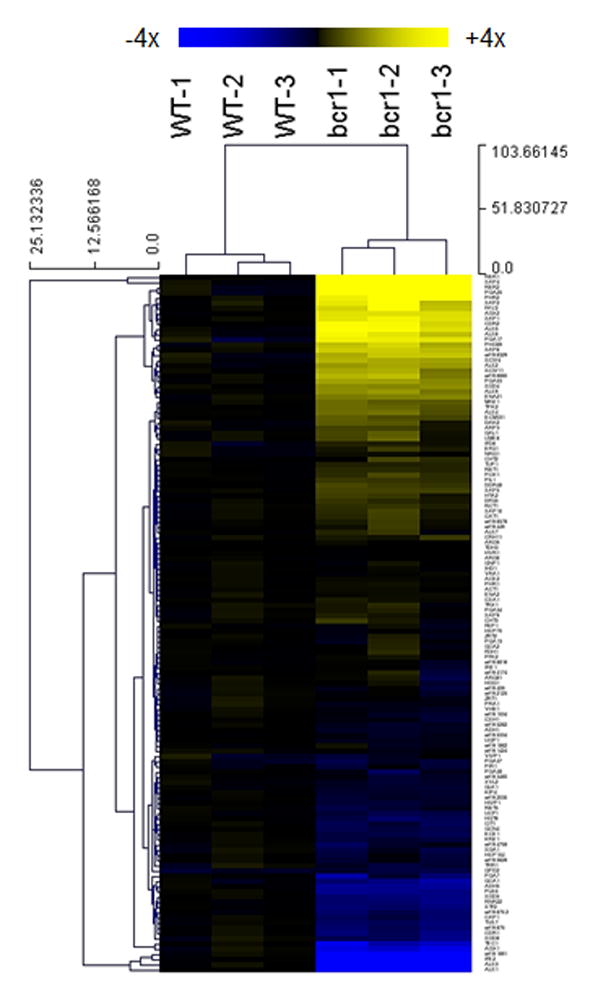
Expression of C. albicans environmentally responsive genes during OPC. Expression levels for 135 C. albicans genes at 2 days postinfection in the mouse OPC model are presented in a heat map format. Expression levels were normalized to the mean value of the wildtype. Color saturation represents the extent of the expression change, with full saturation at 4 fold up- or down-regulation. Adapted from (7).
Table 1.
Three methods to calculate normalization factors.
| Methods | OPC sample 1 | OPC sample 2 | OPC sample 3 | Sample 1–3 average |
|---|---|---|---|---|
| Total counts for all genes | 196429 | 469794 | 152306 | 272843 |
| Normalization factor based on total counts (method 1) | 1.39 | 0.58 | 1.79 | |
| Counts for TDH3 | 10813 | 21362 | 6677 | 12951 |
| Normalization factor based on TDH3 (method 2) | 1.20 | 0.61 | 1.94 | |
| Geometric mean for 20 most highly expressed genes | 5602 | 14292 | 4172 | 8022 |
| Normalization factor based on geometric mean (method 3) | 1.43 | 0.56 | 1.92 |
Acknowledgments
This work was supported in part by NIH grants R21 DE023311 (APM), R56 AI111836 (APM & SGF), R01 AI054928 (SGF), and R01 DE017088 (SGF).
Footnotes
. This protocol requires total processing time of less than 48 hours, from tissue to expression profiling data. The hands-on time is around 4 hours for 12 samples. While it is feasible to perform RNA extraction for 12 samples at the same time, we recommend doing 6 samples at a time to ensure quality. On the other side, we recommend to run a full nanoString cartridge (12 samples) at a time when possible, both to save material costs and to improve consistency of data.
. We first tried using RNAlater (Life Technologies) to preserve tissue samples before RNA isolation. We found RNAlater solution adds viscosity to tissue homogenate and adversely affects quality and quantity of RNA recovery. We then tried snap-freezing tissues in liquid nitrogen followed by storage at −80°C before RNA isolation. We were able to consistently recover RNA with high quality and high yield from snap-frozen tissues.
. One key variable in this protocol is the amount of buffer RLT added to the tissue at the very first step. Too much buffer will dilute the homogenate and lead to lower RNA concentration, while too little buffer may lead to formation of viscous gels after the phenol chloroform extraction step and leave no aqueous phase for RNA recovery. We have empirically determined the optimal volume of buffer to use for the following tissues (assuming typical sizes): one mouse kidney (1.2 ml), one mouse tongue (1.0 ml), one mouse lung (2.0 ml).
. To improve the recovery of RNA, add the first round eluate back to the column, and elute again. We can routinely recover approximately 100 ug of total tissue RNA from one RNeasy spin column (~2 ug/ul × 50 ul). If larger amount of RNA is needed, a second prep can be made from the remaining tissue homogenate. Do not dispose of the remaining homogenate until RNA concentration has been measured, just in case.
. Depending on the infection model, the inoculum size and the pathogen strain, the percentage of pathogen RNA in total tissue RNA varies from 0–2%, and typically falls within the 0.05–0.5% range. For example, 10 ug of total tissue RNA from C. albicans infected kidney could generate nanoString raw counts equal to that of 10 ng of pure C. albicans RNA from an in vitro culture (10 ng/ 10 ug = 0.1%).
. One main challenge for pathogen gene expression profiling in vivo is to get enough reads from pathogen RNA. Given the low percentage of pathogen transcripts in total RNA, we have to use large amount of total RNA. The nanoString platform has a unique advantage in this perspective: it is so specific in recognizing the target RNA that the overwhelming amount of host RNA does not cause a significant level of noise. As shown in Figure 1 (adapted from (7)), raw counts from uninfected tissue sample were all below 10, while raw counts from infected tissue samples ranged between <10 to >105. Only 4 out of 135 genes fell below the noise levels.
. Because the percentage of pathogen RNA in total tissue RNA varies in a wide range, we do not really know the quantity of pathogen RNA in a given amount of total RNA that we use for hybridization with nanoString probes. Therefore, normalization using pathogen genes is a critical step before the expression profiles can be compared among different samples. There are three commonly used methods for normalization. 1. Use total counts from all genes in the codeset. 2. Use one or a few “housekeeping” genes. 3. Use the geometric mean (Nth root of the product of N numbers) of highly expressed genes. Each method has its pros and cons. For a large codeset (>100 genes) containing “randomly” selected probes (such as all genes in the genome that encode a protein with a DNA-binding domain), using total counts for normalization can be a good choice, because the total counts of a large number of unrelated genes may faithfully reflect the amount of RNA input. For a small codeset (<100 genes) containing probes focused on a specific process (such as hyphal growth, given that hyphal growth genes tend to be co-regulated), choosing one or a few housekeeping genes as control for normalization is essential. TDH3, a robustly expressed metabolic gene, has served well as control for many of our experiments. The third method is a hybrid of method 1 and 2, with an emphasis for equal contribution from highly expressed genes. In table 1, we use oropharyngeal candidiasis (OPC) experiment data from Fanning et al. (7) to demonstrate how the normalization factors are calculated and to what extent these different normalization methods could affect the outcome of expression profiling. In table 1, row 2, total counts for all genes for sample 1–3 are 196429, 469794 and 152306, and the average for the 3 samples is 272843. Divide the average total counts by sample 1 total counts, we get the normalization factor for sample 1 as 273943/196429 = 1.39. Following the same calculations we can generate normalization factors for each sample by all three methods. In the case shown in Table 1, normalization factors based on the three methods are within 15% difference from each other, hence unlikely to have a significant influence on the interpretation of the profiling data.
. Expression data generated using this protocol are highly reproducible (Figure 1, comparing red and blue dots; and Figure 2, comparing among biological triplicates in a heat map). The nanoString platform is extremely sensitive and has a dynamic range encompassing the biological expression levels. We were able to quantify transcripts level for >95% of genes in our codeset in the OPC model (Figure 1 and Figure 2), and were able to discern dynamic gene expression changes in a time course study of kidney infection (4).
References
- 1.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26(3):317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 2.Malkov VA, Serikawa KA, Balantac N, Watters J, Geiss G, Mashadi-Hossein A, Fare T. Multiplexed measurements of gene signatures in different analytes using the Nanostring nCounter Assay System. BMC Res Notes. 2009;2:80. doi: 10.1186/1756-0500-2-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol. 2012;10(9):618–30. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- 4.Xu W, Solis NV, Ehrlich RL, Woolford CA, Filler SG, Mitchell AP. Activation and Alliance of Regulatory Pathways in C. albicans during Mammalian Infection. PLoS Biol. 2015;13(2) doi: 10.1371/journal.pbio.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Meara TR, Xu W, Selvig KM, O’Meara MJ, Mitchell AP, Alspaugh JA. The Cryptococcus neoformans Rim101 transcription factor directly regulates genes required for adaptation to the host. Mol Cell Biol. 2013;34(4):673–84. doi: 10.1128/MCB.01359-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng S, Clancy CJ, Xu W, Schneider F, Hao B, Mitchell AP, Nguyen MH. Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J Infect Dis. 2013;208(9):1529–37. doi: 10.1093/infdis/jit335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fanning S, Xu W, Solis N, Woolford CA, Filler SG, Mitchell AP. Divergent targets of Candida albicans biofilm regulator Bcr1 in vitro and in vivo. Eukaryot Cell. 2012;11(7):896–904. doi: 10.1128/EC.00103-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeed AI1, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–8. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]