

Abstract
Huntington’s disease (HD) is an incurable neurodegenerative disorder that is characterized by motor dysfunction, cognitive impairment, and behavioral abnormalities. It is an autosomal dominant disorder caused by a CAG repeat expansion in the huntingtin gene, resulting in progressive neuronal loss predominately in the striatum and cortex. Despite the discovery of the causative gene in 1993, the exact mechanisms underlying HD pathogenesis have yet to be elucidated. Treatments that slow or halt the disease process are currently unavailable. Recent advances in induced pluripotent stem cell (iPSC) technologies have transformed our ability to study disease in human neural cells. Here, we firstly review the progress made to model HD in vitro using patient-derived iPSCs, which reveal unique insights into illuminating molecular mechanisms and provide a novel human cell-based platform for drug discovery. We then highlight the promises and challenges for pluripotent stem cells that might be used as a therapeutic source for cell replacement therapy of the lost neurons in HD brains.
Keywords: Huntington’s disease, Induced pluripotent stem cells, Stem cell models, Drug discovery, Cell replacement therapy
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by abnormal expansion of triplet CAG repeats in the huntingtin gene (HTT), which encodes an expanded polyglutamine (polyQ) stretch in the huntingtin protein (Htt) [1]. This toxic mutant protein (mHtt) leads to progressive and prominent degeneration of the GABAergic projection neurons in the striatum and ultimately more widespread loss of other brain regions [2]. Typically, HD manifests at a mean age of 40 years, with death occurring 15–20 years from the onset [3]. It is characterized by distinct clinical features, including chorea, dystonia and parkinsonism, cognitive decline, and behavioral abnormalities [3]. HD shows a stable prevalence of approximately 5–7 diseased individuals per 100,000 among most Caucasian populations, with many more people at risk of the disease [1, 4]. HD is fully penetrant when individuals carry 40 or more CAG repeats, while incomplete penetrance happens with 36–39 repeats [5]. Mutations between 28 and 35 CAG repeats show instability during paternal inheritance [1, 5]. Although the causative gene and mutation was identified more than 20 years ago, the precise pathophysiological mechanisms by which this mutation leads to degeneration of the target neurons and disease onset remain elusive. Presently, there is no effective disease-modifying treatment to delay or halt the fatal progression of disease, and current management is aimed primarily at the alleviation of symptoms [6].
Recently, a revolutionary discovery by Yamanaka et al. has demonstrated that mouse and human fibroblasts could be reprogrammed into self-renewing pluripotent cells displaying many properties of embryonic stem cells (ESCs) by retrovirally introducing four specific transcription factors (Oct3/4, Sox2, Klf4, and c-Myc) [7, 8]. Oct3/4 proves to be the most important factor, the expression of which is highly specific for pluripotent stem cells and cannot be replaced by other Oct family members to generate induced pluripotent stem cells (iPSCs) [9]. In contrast, the other three factors are expressed in other cells and can be replaced by other family members [10]. Sox2 is expressed in neural stem/progenitor cells and plays a vital role in repressing neuronal differentiation, but the forced expression of Oct3/4 rescues the pluripotency of Sox2-null ESCs [11, 12]. Klf4 is expressed in skin, stomach, intestine, and skeletal muscle, while c-Myc is ubiquitously expressed. Either c-Myc- or Klf4-deficient mice survive until birth, indicating that other factors compensate to maintain pluripotency [10]. In recent years, the breakthrough of iPSCs has promoted a tremendous increase in interest regarding the application of iPSC technology to regenerative medicine and human disease modeling [13]. In particular, iPSCs have shown great potential as a therapeutic strategy for incurable diseases of the central nervous system, ranging from brain cancers to neurodegenerative disorders, such as HD, Parkinson’s disease (PD), Alzheimer’s disease, and amyotrophic lateral sclerosis [14–17]. Here, we review the application prospects of iPSC technology in HD, focusing particularly on the use of iPSCs in disease modeling, drug discovery, and cell replacement therapy.
Neuropathology and Molecular Pathogenesis of Huntington’s Disease
The normal function of Htt remains poorly understood at present [18]. As a ubiquitously expressed and conserved protein across species, Htt is fundamental for post-implantation development and may play a significant role in normal functioning of the basal ganglia [19]. Wild-type Htt upregulates the transcriptional level of brain-derived neurotrophic factor (BDNF), which is particularly important for the survival and maturation of striatal neurons predominantly affected in HD [18, 20]. And the levels of BDNF protein and BDNF mRNA were found consistently reduced in HD striatum [20]. Although mHtt is expressed throughout the body, neuropathological alterations in HD are markedly selective, with prominent cell loss and atrophy in the caudate and putamen [21]. GABAergic medium-sized spiny neurons (MSNs) in the striatum are the most vulnerable [22]. Other brain areas significantly affected in HD patients include the cerebral cortex, hippocampus, substantia nigra, thalamus, hypothalamus, and cerebellum [21]. Another hallmark pathological feature of HD is the appearance of inclusion bodies containing mHtt+ aggregates [23]. Initial studies of HD brains reported inclusions mainly in the nucleus, while subsequent works also identified mHtt+ aggregates in the cytoplasm and neuropil, even in extracellular matrix and blood vessels [24, 25].
The production of toxic mHtt protein has several pathophysiological consequences for the affected neurons in HD [3, 21]. By interacting with mitochondria, mHtt might impair oxidative metabolism and mitochondrial calcium handling, leading to reduced ATP levels, defective Ca2+ uptake, and enhanced oxidative stress [26]. Striatal MSNs are selectively vulnerable to the toxicity of excitatory amino acids, in particular glutamate and its analogues [27]. Importantly, mHtt expression alters excitatory synaptic activity in the striatum by reducing glutamate uptake and enhancing signaling at N-methyl-D-aspartate receptors [28]. The gene-expression profiles between postmortem human HD brains and animal models are remarkably comparable, supporting the idea that altered transcription is a crucial mechanism in HD pathogenesis [29, 30]. Expanded polyQ repeats enter the nucleus and mediate transcriptional dysregulation through their interaction with cellular transcription factors, which may ultimately induce neuronal dysfunction and cell death in HD [31–33]. Moreover, mHtt aggregates are produced continuously beyond the ability of cells to degrade these proteins by proteasome and autophagy pathways, resulting in increasing aggregate accumulation [34–38]. All these pathogenic mechanisms eventually lead to apoptotic or necrotic cell death, responsible for abnormal neuropathological features of HD [39].
Current Therapeutics for Huntington’s Disease
Despite the fact that the HD gene was identified over 20 years ago, no curable therapy for the disease is currently available. The majority of current pharmacological therapeutics are solely symptomatic treatment of the dominant motor, psychiatric, and cognitive aspects of HD [40]. Tetrabenazine remains the only drug approved by the US Food and Drug Administration for the treatment of chorea associated with HD and has shown clear efficacy for reducing chorea, though it has a risk of potentially deleterious effects [41]. Antipsychotic agents, including olanzapine, haloperidol, risperidone, and clozapine, are used to treat patients with psychiatric/behavioral disturbances. Donepezil and rivastigmine are traditional therapeutics for enhancing cognitive function. In the past few years, there are a number of clinical trials designed to evaluate potential therapeutic agents, most of which target those aforementioned pathophysiological consequences that occur downstream of protein translation [42]. Several agents are in ongoing phase II/III trials such as cysteamine, resveratrol, pridopidine, laquinimod, and epigallocatechin gallate [6]. However, the majority of clinical studies to date have not demonstrated efficacy, although these agents have yielded promising results in preclinical models [6]. Given that HD is a monogenic disorder, novel approaches aimed at silencing or repairing the mHTT gene, such as antisense oligonucleotides (ASOs), RNA interference (RNAi), ribozymes, DNA enzymes, and genome-editing approaches, are becoming attractive therapeutic options [43]. These strategies directly interfere with the cause of the disease by targeting mHtt at the genomic or post-transcriptional level [44]. Several oligonucleotide-based approaches have been reported as potential allele-selective HD therapeutics [45, 46]. A phase 1/2a clinical study by Isis Pharmaceuticals using ASOs in patients with early-stage HD has now initiated (NCT02519036). Several hurdles, such as off-target effects and lack of effective and nontoxic delivery systems, need to be overcome before these approaches can enter the clinic [43].
Modeling Huntington’s Disease In Vitro with Patient-Specific iPSCs
Illuminating the molecular basis of HD and, ultimately, expediting the discovery of disease-modifying therapies that delay the onset and slow the progression of HD depend on reliable disease models [47]. Both chemically induced and transgenic animal models that recapitulate features of HD have enabled plenty of powerful avenues for research [47, 48]. However, traditional animal models may not be able to precisely mimic the disease process in human cells due to differences between species [49]. The majority of therapies that are effective in animals have failed in human clinical trials [50]. Furthermore, generating and breeding transgenic animals are costly and slow. Thus, a rapider and more human-relevant model system is required to advance research into the HD pathogenesis and drug discovery. The rapid development of iPSC technology opens exciting new opportunities to model HD in vitro with the ability to differentiate patient-derived pluripotent stem cells into susceptible neuronal subtypes.
iPSCs have prominent advantages over ESCs in some respects. First, iPSCs can be derived directly from skin cells of HD patients (Fig. 1), avoiding ethical concerns resulting from blastocyst destruction in the process of obtaining human ESCs [51]. Besides, iPSCs generated from individual patients would be a better source for cell replacement therapy than ESCs that would inevitably lead to immune rejection issues [52]. Furthermore, patient-derived iPSCs harbor all disease-associated genetic components that render GABAergic neurons susceptible to the disease. Therefore, they represent the most genetically precise model and might be helpful to further investigate genetic factors related to HD pathogenesis [53, 54]. Moreover, gene-silencing technologies based on patient-specific iPSCs may offer a way to correct this monogenic disorder, paving the road for personalized medicine [44, 55, 56].
Fig. 1.
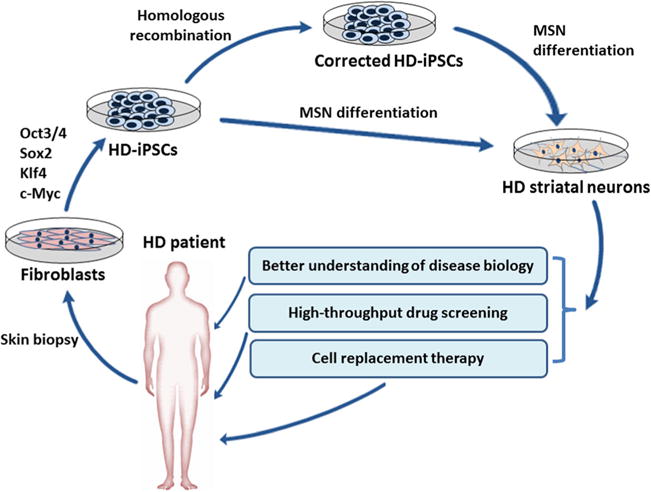
The generation and application of iPSCs in HD research. HD patient-specific iPSCs can be obtained by reprogramming of skin fibroblasts. Established iPSCs can be used as a tool for better understanding the molecular basis of HD. iPSC technology can also be coupled with high-throughput screening that provides a more efficacious platform to assess novel drug candidates aimed at stopping or slowing disease process. Moreover, HD-iPSCs can be differentiated into specific cell types predominantly affected in the disease (striatal MSNs). Emerging gene therapies make the genetic correction of HD-iPSCs become feasible, paving the way for autologous transplantation strategies of healthy iPSCs or iPSC-derived neural cells
In principle, in vitro disease modeling comprises differentiating control and disease-specific iPSCs/ESCs into GABAergic neurons and comparing these target cells for disease-relevant phenotypes [53]. To date, iPSC lines that have been generated from HD patients indeed exhibited robust pathologic phenotypes that replicate many features of this disorder (Table 1). The first study on the feasibility of reprogramming patient fibroblasts into iPSCs was reported by Park et al. [57]. However, initial phenotypic differences between HD-iPSC lines carrying 72 CAG repeats and controls were not significant. Afterwards, Zhang et al. reported an increase in caspase-3/7 activity in neural stem cells (NSCs) generated from HD-iPSCs upon growth factor withdrawal when subjecting iPSCs to a neuronal differentiation paradigm [58]. This group also observed mitochondrial dysfunction and decreased BDNF transcription upon growth factor deprivation in HD-iPSC-derived NSCs [55]. Another study revealed differentially expressed protein patterns in comparative proteomic analysis [59]. Specifically, a total of 26 upregulated oxidative stress-associated proteins and downregulated cytoskeleton-associated proteins were identified in HD-iPSCs at undifferentiated stages. Furthermore, HD-iPSC lines exhibited more DNA damage-mediated apoptosis and reduced neuronal differentiation efficiency and neurite length [59]. Moreover, multiple molecular pathways that are characteristically dysregulated in HD were also present in undifferentiated pluripotent HD-iPSCs, including dysregulation of the MAPK and Wnt signaling pathways and altered expression of p53 [60]. Apart from iPSCs generated from aforementioned heterozygous HTT mutations, Camnasio et al. for the first time generated iPSCs from two homozygous HD individuals who carried HTT expansions between 39 and 44 CAGs, and observed enhanced lysosomal activity in HD-iPSCs and their derivatives. However, no distinguishable differences in reprogramming, growth rate, caspase activation, or neuronal differentiation were observed between normal and mutant genotypes [61].
Table 1.
Human iPSC-based models of Huntington’s disease
| Donor cell | HTT mutation | Phenotype in iPSCs and/or generated neural cells | Reference |
|---|---|---|---|
| HD patient fibroblast | 72 CAG repeats | No apparent difference from control iPSCs | [57] |
| HD patient fibroblast | 72 CAG repeats | Enhanced caspase-3/7 activity upon growth factor deprivation in HD-iPSC-derived NSCs | [58] |
| HD patient fibroblast | 72 CAG repeats | Increased caspase-3/7 activity, mitochondrial dysfunction, and decreased BDNF transcription upon growth factor removal in HD-iPSC-derived NSCs | [55] |
| HD patient fibroblast | 72 CAG repeats | Differentially expressed protein patterns in proteomic analysis, upregulated oxidative stress-related proteins and downregulated cytoskeleton-associated proteins, more DNA damage-mediated apoptosis, reduced neuronal differentiation efficiency and neurite length | [59] |
| HD patient fibroblast | 71 and 109 CAG repeats | Dysregulation of the MAPK-ERK signaling pathway, altered expression of oxidative stress-related proteins and p53 in undifferentiated pluripotent HD-iPSCs | [60] |
| HD patient fibroblast | 2 homozygous genotypes (between 39 and 44 CAG repeats) and 1 heterozygous genotype (45 CAG repeats) | No differences in reprogramming, growth rate, caspase activation, or neuronal differentiation; increased lysosomal activity in HD-iPSCs and derived neurons | [61] |
| HD patient fibroblast | 180, 109, and 60 CAG repeats | Transcriptional changes involved in signaling, cell cycle, axonal guidance, and neuronal development; changes in the actin cytoskeleton; decreased cell-cell adhesion; impaired energy metabolism; altered electrophysiological properties; increased susceptibility to cell stressors such as BDNF withdrawal and glutamate | [62] |
| HD patient fibroblast | 109 and 50 CAG repeats | Elevated cytoplasmic vacuolation which increased over time in HD-iPSCs derived astrocytes | [64] |
| HD patient fibroblast | 72 CAG repeats | mHtt aggregate formation upon proteasome inhibition or at later stages of transplantation into rat brains | [66] |
| HD patient fibroblast | 180, 109, and 60 CAG repeats | Contained more neural progenitor cells after differentiation, enhanced vulnerability to BDNF withdrawal in the juvenile-onset HD lines | [63] |
In 2012, the HD Consortium reported a total of 14 iPSC lines derived from HD patients and controls, and uncovered a series of CAG-repeat-expansion-associated phenotypes [62]. Whole-transcript expression profiling revealed many transcriptional changes involved in signaling, cell cycle, axonal guidance, and neuronal development, which were consistent with previously described disturbances in HD pathogenesis. Furthermore, CAG-repeat-associated alterations in actin cytoskeleton, cell-cell adhesion, ATP/ADP levels, and electrophysiological properties have been observed in derived neural cells. Moreover, HD-derived lines exhibited increased susceptibility to cell stressors such as BDNF withdrawal and glutamate, which was verified by a more recent study in the juvenile-onset HD lines [62, 63].
Not only has the neuronal lineage derived from HD-iPSCs been intensively studied, an independent group has also differentiated iPSCs into an astrocytic lineage. Surprisingly, the diseased astrocytes displayed obvious cytoplasmic vacuolation that increased over time under fundamental conditions without additional stressors [64]. This finding was consistent with those seen in peripheral blood lymphocytes of HD patients [64, 65].
Although iPSC models have reproduced many features of HD, the spontaneous formation of mHtt+ aggregates has not been observed in human iPSCs. A recent study indicated that treatment of in vitro culture with a proteasome inhibitor MG132 could induce mHtt aggregation in HD-iPSCs [66]. Interestingly, this group also observed mHtt+ aggregates at 33 weeks after transplantation of HD neural progenitor cells (NPCs) into rat brains, suggesting that additional stressors and cellular age may be key factors in developing protein aggregates in human HD-iPSCs.
The endogenous HD mutations persist in all cell types. Almost all neural cells derived from HDi PSCs contain the same CAG expansion as the parental fibroblasts, and the size of CAG repeats did not augment during the course of reprogramming, long-term growth in vitro, and neuronal differentiation [58, 61]. The HD Consortium indicated that the normal and expanded CAG repeat alleles exhibited only slight instability upon differentiation for most of the HD-derived lines [62]. One line displayed complete stability of the short allele; however, the long CAG repeat increased to 118 from 110 after 26 passages of NSCs [62].
Creation of iPSC lines from patients with this monogenic disorder not only allows experiments on disease phenotypes in vitro but also opens an opportunity to repair gene defects ex vivo. An et al. reported that HD-iPSCs could be corrected by replacement of the expanded CAG repeat with a normal repeat using homologous recombination, and the correction persisted in differentiated MSNs in vitro and in vivo [55]. In order to improve the efficiency of recombination, they extended this work by adopting a CRISPR based genome-editing approach [67].
Using Human iPSC Lines for Drug Discovery
Despite the fact that the causative gene for HD has been identified for more than 20 years, current therapeutics available to HD patients are mainly palliatives and disease-modifying therapies have not yet been established [41]. Development of new drugs is an expensive and time-consuming process, while the majority of drug candidates that are efficacious in animals have failed in clinical trials due to safety and efficacy issues [68]. The final goal of in vitro disease modeling with human pluripotent stem cells is to find better therapeutic targets and more effective drugs that would benefit a large number of patients. A recent study used human ESC/iPSC-derived dopaminergic neurons as a platform to screen a group of compounds as potential neuroprotective agents in PD [69]. Of the 44 compounds known to work in rodent systems, only 16 showed neuroprotection in neurotoxin-induced dopaminergic cell models, highlighting the importance of using disease-relevant human neurons for such assays [69]. iPSCs offer numerous advantages over the traditional methods in drug discovery. This human model may eventually reveal how mHtt triggers molecular events that ultimately result in motor, cognitive, and behavioral disturbances of HD patients. Recently, a particular interest has been focused on the discovery of small molecules that are able to inhibit the toxic effects of mHtt and attenuate neurodegeneration [70]. Human iPSC-based models have the potential as power tools for high-throughput drug screening, bioinformatics, and global gene-expression analyses, accelerating the pace of drug discovery and reducing drug attrition rate [49, 71].
Furthermore, patient- and disease-specific iPSCs have the potential for indicating mutation-triggered molecular events and compensatory processes, thus providing valuable data on the efficacy and safety of the tested drugs [71, 72]. One of the future directions for personalized medicine is that a potential therapy might be tested first in iPSCs from a HD patient to determine its efficacy and safety. If the therapy becomes approved, the same approach could be adopted to determine the diseased individuals suitable for this therapy, thereby averting potential adverse effects in patients who do not benefit from it [53, 73].
Pluripotent Stem Cells for Cell Replacement Therapy
As HD advances, a late-stage intervention might be replenishment of lost neurons by cell replacement therapy, thus reversing disease phenotypes and slowing the progression of neurodegeneration [74, 75]. Early pioneer clinical studies transplanted fetal striatal tissues into the striatum of HD patients, providing evidence that the allografts led to short-term motor and cognitive improvements [76–78]. Intractable issues associated with fetal grafts include their limited source and prolonged immunosuppression, along with the controversial ethical concerns [79]. Recently, an increasing interest is focused on alternative approaches of using pluripotent stem cells as a more favorable source to replace dying or damaged neurons in neurodegenerative disorders [14]. ESCs or iPSCs are able to differentiate into the target cell types affected in disease and provide a readily obtainable source of graft material. Furthermore, iPSCs have the ability to generate patient-specific neural precursors, thus eliminating possible problems of immunological rejection. However, the HD-iPSC-derived cells still carry the causative mutation which would produce toxic mutant proteins and lead to ultimate cell death. Of note, emerging gene therapies such as RNAi, ASOs, and genome-editing approaches are capable of silencing or repairing the mHTT gene [44]. Therefore, transplantation of the corrected neural cells back into the patient brain would then abate immune rejection, replenish lost cells, and rescue functional deficiencies [56, 75].
Nowadays, pluripotent stem cell transplantation in the context of HD is largely carried out in preclinical animal models of HD (Table 2). A few human ESC/iPSC transplantations in rodent HD models have shown success in substituting for damaged neural cells [80–82]. Aubry et al. first directed human ESCs into neural, neuronal, and striatal differentiation in vitro before transplantation, and then observed that the grafted striatal progenitors successfully differentiated into mature GABAergic neurons in vivo [80]. Another study by Ma et al. also demonstrated the ability of human ESCs to differentiate into DARPP-32+ GABAergic cells [81]. Furthermore, transplantation of these GABAergic neurons and their progenitors into the striatum of chemical-lesioned mice led to the generation of large populations of mature GABAergic neurons [81]. These human GABA neurons were found to integrate with host neurons and correct locomotion deficits of HD mice, further substantiating the therapeutic potential of human ESC-derived cells [81]. Delli Carri et al. employed a new differentiation protocol which simulated the normal neurodevelopment of the ventral telencephalon to induce both human ESCs and iPSCs to give rise to NPCs [82]. In addition, they differentiated NPCs into GABAergic neurons which not just expressed typical MSN neuronal markers but also carried dopamine and adenosine receptors [82]. When grafted into the striatum of chemical-lesioned rats, human pluripotent stem cell-derived NPCs successfully survived and differentiated toward a MSN fate, leading to a restoration of apomorphine-induced rotation behavior [82]. Recently, the efficacy and safety of rodent iPSC grafts have also been evaluated in HD animals. These grafted rodent stem cells were able to differentiate into DARPP-32+ neurons in the lesioned striatum and ameliorate the corresponding striatal atrophy [83, 84]. Transplantation of mouse iPSCs also improved recovery of learning and memory deficits induced by quinolinic acid [84]. Moreover, glucose metabolism of the injured striatum by microPET/CT scanning was enhanced at 4–6 weeks post-transplantation [84]. It is noteworthy that these positive benefits are mostly present in lesioned rodent models of HD, which are far from accurately modeling the disease’s main pathological features. Actually, in the past few years, we have seen many failures in clinical trials on potential therapies that showed efficacy in animal studies of HD [50]. In view of the limitations of the animal models used, preclinical data need to be rigorously and objectively assessed before translation into clinically relevant therapies.
Table 2.
Pluripotent stem cell transplantation in preclinical animal models of Huntington’s disease
| Species of grafted material | Stem cell class | Concentration of grafted cells | Animal model | Outcome | How long post-transplantation outcomes were observed | Reference |
|---|---|---|---|---|---|---|
| Human | ESC | 25,000–100,000 cells/μl | Quinolinic acid-lesioned adult OFA and nude rats, weight 220–260 g | Differentiated into mature GABAergic MSNs in vivo | 13–21 weeks | [80] |
| Human | ESC | 50,000 cells/μl | Quinolinic acid-lesioned adult male SCID mice, 10 weeks of age | Generated large populations of DARPP-32+ GABA neurons which made connections with host neurons, corrected the motor deficits | 4, 8, 12, 16 weeks | [81] |
| Human | ESC/iPSC | 250,000 cells/μl | Quinolinic acid-lesioned female Lister hooded rats | Differentiated into DARPP-32+ neurons, restored apomorphine-induced rotation behavior | 3, 6, and 9 weeks | [82] |
| Sprague Dawley rat | iPSC | 200,000 cells/μl | 3-Nitropropionic acid-lesioned male and female Sprague Dawley rats, 7.5–8.5 weeks of age | Differentiated into DARPP-32+ neurons; preserved motor function; reversed histological changes including striatum atrophy, lateral ventricle enlargement, and an increase in striosome size | 4–10 weeks | [83] |
| C57BL/6 mouse | iPSC | 50,000 cells/μl | Quinolinic acid-lesioned adult male Sprague Dawley rats, weight 250–280 g | Survived and migrated into the lesioned striatum, differentiated into neurons and astrocytes, enhanced glucose metabolism, improved learning and memory function as well as striatal atrophy | 1, 2, 4, and 6 weeks | [84] |
Although iPSCs have the ability to differentiate into the desired neuron types, recent evidence suggests that the functional benefits of stem cell therapies may be mediated by secretory molecules in addition to cell replenishment. Many types of stem cells produce a variety of growth factors, cytokines, and chemokines in an autocrine/paracrine manner, playing important roles in neuronal survival, neurogenesis, and mitochondrial activation [85]. A number of investigators have further examined the potential benefits of genetically engineered stem cells that could overexpress trophic factors like BDNF and glial-derived neurotrophic factor [86]. Recently, a group transformed iPSCs into NPCs engineered to overexpress BDNF. Importantly, intracerebroventricular transplantation of these neural cells reversed the immune impact caused by lipopolysaccharide and blunted the stressor-induced corticosterone response [87]. This combination of iPSCs and trophic factors overexpression could potentially stimulate neurogenesis and repair, and contribute to neuroprotection, thus offering great potential in disease-modifying treatment of HD.
However, the therapeutic promise of cell replacement therapy in HD is debatable. During the long-term follow-up of HD patients treated with fetal striatal grafts, functional benefits in transplanted patients have not been robust or sustainable, and some cases even showed progressive deterioration over time [88, 89]. Cicchetti et al. reported that neural transplants in HD patients underwent disease-like neuronal degeneration with a preferential loss of striatal projection neurons [90]. Furthermore, the group described the presence of mHtt+ aggregates in striatal fetal allografts in HD patients following transplantation [25]. Jeon et al. studied the in vivo effects of HD patient-derived iPSCs following transplantation in either chemical-lesioned rats or transgenic HD mice [66, 91]. Interestingly, the grafted HD-iPSC-derived neural precursors generated GABA neurons efficiently, and no mHtt+ aggregates were detected at 12 weeks post-transplantation [66, 91]. However, when the grafted cells were analyzed at 33 weeks, there were clear signs of HD pathology [66]. In recent years, there have been accumulating in vitro evidences for cell-to-cell transfer of mHtt oligomers/aggregates [92–95]. Most recently, Pecho-Vrieseling et al. reported transneuronal propagation of mHtt protein pathology in the corticostriatal pathway, which is early and severely affected in the HD brain [96]. Surprisingly, another study observed that neuronal Htt aggregates were able to access and initiate a prion-like conversion of normally soluble, cytoplasmic Htt in the glia of the Drosophila brain [97]. These findings raise uncertainty about cell replacement therapy for the treatment of HD. There remains much debate as to the exploration of cell replacement therapy as a therapeutic strategy for HD.
Conclusion and Future Perspective
Although still in its infancy, the tremendous potential of iPSC technology opens up exciting new opportunities for investigation of neurodegenerative disorders. As iPSCs are generated directly from affected patients, they are likely to represent the most genetically and molecularly accurate model of the disease. Therefore, using iPSC lines for disease modeling may bridge the gaps between animal models and human neural cells, helping elucidate the molecular basis of HD. Furthermore, iPSC technology could be coupled with high-throughput screening that provides a faster and more efficacious platform to assess a number of former and novel drug candidates aimed at stopping or slowing disease process. However, many tasks remain to be fulfilled to enable iPSC technologies to accurately model HD and to develop new therapeutics. It is unclear what the readout can be for the screening in the case of HD because the spontaneous formation of mHtt+ aggregates has not been detected in iPSC-derived cells from HD patients. Thus, the next step will necessarily involve developing an accurate assay system for disease-associated phenotypes. For future research involving iPSC-based systems to model disease, it may be beneficial to focus on molecular changes in HD-iPSCs that occur in patients prior to cell death or the onset of symptoms. Unraveling such reversible phenotypes in iPSCs that appear early in the course of HD, for example, alterations in gene transcription, and adapting these cells for drug development assays may be key to finding pharmacological interventions that can prevent neurodegeneration long before the devastating late-stage consequences occur.
iPSCs not only are invaluable tools for disease modeling and drug discovery in HD but also have emerged with great potential in areas of cell replacement therapy. Further gene-silencing technologies based on patient-specific iPSCs may offer an opportunity to correct this monogenic disorder, paving the way for personalized medicine. Although promising, successful implementation of iPSC-based therapy is far from becoming a reality. Nowadays, pluripotent stem cell transplantation in the context of HD is largely carried out in rodent models, and there are still enormous hurdles to be overcome as the field moves forward. Replacing complete neural circuits in the adult brain is clearly challenging. Although several studies in rodents indicate the differentiation of ESC/iPSCs into neurons with MSN properties, detailed mechanisms by which the transplanted cells differentiate into the correct cell type and integrate with host cells remain to be clarified. Most notably, mHtt protein is expressed in all cells of the brain and indeed of the body of patients. Even though striatal transplants were initially effective at early stages, this efficacy would not be sustained once mHtt induced neurodegeneration of the cerebral cortex and other brain regions. Recently, accumulating evidence for cell-to-cell transfer of mHtt raises uncertainty about cell transplantation for the treatment of HD. Therefore, further thoughtful and rigorous attempts are needed before translation of preclinical transplant results into clinic.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China to T.W. (31171211 and 81471305), N.X. (81200983), and J.S.H. (81301082); a grant from China Medical Foundation to N.X. (2012B09); and a grant from Hubei Molecular Imaging Key Laboratory to N.X. (0203201343).
Footnotes
Conflict of Interest The authors declare that they have no competing interests.
References
- 1.Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218–228. doi: 10.1016/s0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 2.Rikani AA, Choudhry Z, Choudhry AM, Rizvi N, Ikram H, Mobassarah NJ, Tulli S. The mechanism of degeneration of striatal neuronal subtypes in Huntington disease. Ann Neurosci. 2014;21(3):112–114. doi: 10.5214/ans.0972.7531.210308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 4.Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord. 2012;27(9):1083–1091. doi: 10.1002/mds.25075. [DOI] [PubMed] [Google Scholar]
- 5.Myers RH. Huntington’s disease genetics. NeuroRx. 2004;1(2):255–262. doi: 10.1602/neurorx.1.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shannon KM, Fraint A. Therapeutic advances in Huntington’s disease. Mov Disord. 2015 doi: 10.1002/mds.26331. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, et al. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26(1):101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 10.Yamanaka S. A fresh look at iPS cells. Cell. 2009;137(1):13–17. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 11.Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. 2007;9(6):625–635. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- 12.Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39(5):749–765. doi: 10.1016/S0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 13.Dametti S, Faravelli I, Ruggieri M, Ramirez A, Nizzardo M, Corti S. Experimental advances towards neural regeneration from induced stem cells to direct in vivo reprogramming. Mol Neurobiol. 2015:1–8. doi: 10.1007/s12035-015-9181-7. [DOI] [PubMed] [Google Scholar]
- 14.Ross CA, Akimov SS. Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum Mol Genet. 2014;23(R1):R17–R26. doi: 10.1093/hmg/ddu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi SS, Lee SR, Kim SU, Lee HJ. Alzheimer’s disease and stem cell therapy. Exp Neurobiol. 2014;23(1):45–52. doi: 10.5607/en.2014.23.1.45. doi:10.5607/en. 2014.23.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao L, Tan L, Jiang T, Zhu XC, Yu JT. Induced pluripotent stem cells for disease modeling and drug discovery in neurodegenerative diseases. Mol Neurobiol. 2015;52(1):244–255. doi: 10.1007/s12035-014-8867-6. [DOI] [PubMed] [Google Scholar]
- 17.Upadhyay G, Shankar S, Srivastava RK. Stem cells in neurological disorders: emerging therapy with stunning hopes. Mol Neurobiol. 2015;52(1):610–625. doi: 10.1007/s12035-014-8883-6. [DOI] [PubMed] [Google Scholar]
- 18.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6(12):919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 19.Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 20.Zuccato C, Cattaneo E. Huntington’s disease. Handb Exp Pharmacol. 2014;220:357–409. doi: 10.1007/978-3-642-45106-5_14. [DOI] [PubMed] [Google Scholar]
- 21.Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90(3):905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- 22.Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):55–69. doi: 10.1007/s00401-007-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp Neurol. 2012;238(1):1–11. doi: 10.1016/j.expneurol.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19(7):2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cicchetti F, Lacroix S, Cisbani G, Vallieres N, Saint-Pierre M, St-Amour I, Tolouei R, Skepper JN, et al. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann Neurol. 2014;76(1):31–42. doi: 10.1002/ana.24174. [DOI] [PubMed] [Google Scholar]
- 26.Brustovetsky N. Mutant huntingtin and elusive defects in oxidative metabolism and mitochondrial calcium handling. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estrada Sanchez AM, Mejia-Toiber J, Massieu L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch Med Res. 2008;39(3):265–276. doi: 10.1016/j.arcmed.2007.11.011. doi:10.1016/j.arcmed. 2007.11.011. [DOI] [PubMed] [Google Scholar]
- 28.Sepers MD, Raymond LA. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov Today. 2014;19(7):990–996. doi: 10.1016/j.drudis.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Cha JH. Transcriptional signatures in Huntington’s disease. Prog Neurobiol. 2007;83(4):228–248. doi: 10.1016/j.pneurobio.2007.03.004. doi:10.1016/j.pneurobio.2007.03. 004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valor L. Transcription, epigenetics and ameliorative strategies in Huntington’s disease: a genome-wide perspective. Mol Neurobiol. 2015;51(1):406–423. doi: 10.1007/s12035-014-8715-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22(5):1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97(12):6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291(5512):2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 34.Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, et al. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448(7154):704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 35.Ortega Z, Lucas JJ. Ubiquitin-proteasome system involvement in Huntington’s disease. Front Mol Neurosci. 2014;7:77. doi: 10.3389/fnmol.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarkar S. Chemical screening platforms for autophagy drug discovery to identify therapeutic candidates for Huntington’s disease and other neurodegenerative disorders. Drug Discov Today Technol. 2013;10(1):e137–e144. doi: 10.1016/j.ddtec.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Margulis J, Finkbeiner S. Proteostasis in striatal cells and selective neurodegeneration in Huntington’s disease. Front Cell Neurosci. 2014;8:218. doi: 10.3389/fncel.2014.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today. 2014;19(7):963–971. doi: 10.1016/j.drudis.2014.02.014. doi:10.1016/j.drudis.2014.02. 014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet. 2014;384(9942):545–555. doi: 10.1016/s0140-6736(14)61010-2. [DOI] [PubMed] [Google Scholar]
- 40.Killoran A, Biglan KM. Current therapeutic options for Huntington’s disease: good clinical practice versus evidence-based approaches? Mov Disord. 2014;29(11):1404–1413. doi: 10.1002/mds.26014. doi:10.1002/mds. 26014. [DOI] [PubMed] [Google Scholar]
- 41.Frank S. Treatment of Huntington’s disease. Neurotherapeutics. 2014;11(1):153–160. doi: 10.1007/s13311-013-0244-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sampaio C, Borowsky B, Reilmann R. Clinical trials in Huntington’s disease: interventions in early clinical development and newer methodological approaches. Mov Disord. 2014;29(11):1419–1428. doi: 10.1002/mds.26021. [DOI] [PubMed] [Google Scholar]
- 43.Aronin N, DiFiglia M. Huntingtin-lowering strategies in Huntington’s disease: antisense oligonucleotides, small RNAs, and gene editing. Mov Disord. 2014;29(11):1455–1461. doi: 10.1002/mds.26020. [DOI] [PubMed] [Google Scholar]
- 44.Godinho BM, Malhotra M, O’;Driscoll CM, Cryan JF. Delivering a disease-modifying treatment for Huntington’s disease. Drug Discov Today. 2015;20(1):50–64. doi: 10.1016/j.drudis.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 45.Ostergaard ME, Kumar P, Nichols J, Watt A, Sharma PK, Nielsen P, Seth PP. Allele-selective inhibition of mutant huntingtin with 2-Thio- and C5- triazolylphenyl-deoxythymidine-modified anti-sense oligonucleotides. Nucleic Acid Ther. 2015 doi: 10.1089/nat.2015.0547. [DOI] [PubMed] [Google Scholar]
- 46.Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–2185. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington’s disease. Nat Rev Neurosci. 2013;14(10):708–721. doi: 10.1038/nrn3570. [DOI] [PubMed] [Google Scholar]
- 48.Figiel M, Szlachcic W, Switonski P, Gabka A, Krzyzosiak W. Mouse models of polyglutamine diseases: review and data table. Part I. Mol Neurobiol. 2012;46(2):393–429. doi: 10.1007/s12035-012-8315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merkle Florian T, Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell. 2013;12(6):656–668. doi: 10.1016/j.stem.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 50.Menalled L, Brunner D. Animal models of Huntington’s disease for translation to the clinic: best practices. Mov Disord. 2014;29(11):1375–1390. doi: 10.1002/mds.26006. [DOI] [PubMed] [Google Scholar]
- 51.Zeng X, Hunsberger JG, Simeonov A, Malik N, Pei Y, Rao M. Concise review: modeling central nervous system diseases using induced pluripotent stem cells. Stem Cells Transl Med. 2014;3(12):1418–1428. doi: 10.5966/sctm.2014-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicoleau C, Viegas P, Peschanski M, Perrier AL. Human pluripotent stem cell therapy for Huntington’s disease: technical, immunological, and safety challenges human pluripotent stem cell therapy for Huntington’s disease: technical, immunological, and safety challenges. Neurotherapeutics. 2011;8(4):562–576. doi: 10.1007/s13311-011-0079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaye JA, Finkbeiner S. Modeling Huntington’s disease with induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:50–64. doi: 10.1016/j.mcn.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becanovic K, Norremolle A, Neal SJ, Kay C, Collins JA, Arenillas D, Lilja T, Gaudenzi G, et al. A SNP in the HTT promoter alters NF-kappaB binding and is a bidirectional genetic modifier of Huntington disease. Nat Nuerosci. 2015;18(6):807–816. doi: 10.1038/nn.4014. [DOI] [PubMed] [Google Scholar]
- 55.An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11(2):253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crane AT, Rossignol J, Dunbar GL. Use of genetically altered stem cells for the treatment of Huntington’s disease. Brain Sci. 2014;4(1):202–219. doi: 10.3390/brainsci4010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134(5):877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang N, An MC, Montoro D, Ellerby LM. Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Currents. 2010;2:Rrn1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chae JI, Kim DW, Lee N, Jeon YJ, Jeon I, Kwon J, Kim J, Soh Y, et al. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem J. 2012;446(3):359–371. doi: 10.1042/bj20111495. [DOI] [PubMed] [Google Scholar]
- 60.Szlachcic WJ, Switonski PM, Krzyzosiak WJ, Figlerowicz M, Figiel M. Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis Model Mech. 2015 doi: 10.1242/dmm.019406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Camnasio S, Delli Carri A, Lombardo A, Grad I, Mariotti C, Castucci A, Rozell B, Lo Riso P, et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis. 2012;46(1):41–51. doi: 10.1016/j.nbd.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 62.Consortium THi. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11(2):264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mattis VB, Tom C, Akimov S, Saeedian J, Ostergaard ME, Southwell AL, Doty CN, Ornelas L, et al. HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Juopperi TA, Kim WR, Chiang CH, Yu H, Margolis RL, Ross CA, Ming GL, Song H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol Brain. 2012;5:17. doi: 10.1186/1756-6606-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagata E, Sawa A, Ross CA, Snyder SH. Autophagosome-like vacuole formation in Huntington’s disease lymphoblasts. Neuroreport. 2004;15(8):1325–1328. doi: 10.1097/01.wnr.0000127073.66692.8f. [DOI] [PubMed] [Google Scholar]
- 66.Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, et al. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30(9):2054–2062. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- 67.An MC, O’Brien RN, Zhang N, Patra BN, De La Cruz M, Ray A, Ellerby LM. Polyglutamine disease modeling: epitope based screen for homologous recombination using CRISPR/Cas9 System. PLoS Curr. 2014;6 doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu DX, Marchetto MC, Gage FH. Therapeutic translation of iPSCs for treating neurological disease. Cell Stem Cell. 2013;12(6):678–688. doi: 10.1016/j.stem.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 69.Peng J, Liu Q, Rao MS, Zeng X. Using human pluripotent stem cell-derived dopaminergic neurons to evaluate candidate Parkinson’s disease therapeutic agents in MPP+ and rotenone models. J Biomol Screen. 2013;18(5):522–533. doi: 10.1177/1087057112474468. [DOI] [PubMed] [Google Scholar]
- 70.Fecke W, Gianfriddo M, Gaviraghi G, Terstappen GC, Heitz F. Small molecule drug discovery for Huntington’s disease. Drug Discov Today. 2009;14(9–10):453–464. doi: 10.1016/j.drudis.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 71.Perrier A, Peschanski M. How can human pluripotent stem cells help decipher and cure Huntington’s disease? Cell Stem Cell. 2012;11(2):153–161. doi: 10.1016/j.stem.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 72.Gunaseeli I, Doss MX, Antzelevitch C, Hescheler J, Sachinidis A. Induced pluripotent stem cells as a model for accelerated patient- and disease-specific drug discovery. Curr Med Chem. 2010;17(8):759–766. doi: 10.2174/092986710790514480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Engle SJ, Puppala D. Integrating human pluripotent stem cells into drug development. Cell Stem Cell. 2013;12(6):669–677. doi: 10.1016/j.stem.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 74.Rosser A, Svendsen CN. Stem cells for cell replacement therapy: a therapeutic strategy for HD? Mov Disord. 2014;29(11):1446–1454. doi: 10.1002/mds.26026. [DOI] [PubMed] [Google Scholar]
- 75.Chen Y, Carter RL, Cho IK, Chan AW. Cell-based therapies for Huntington’s disease. Drug Discov Today. 2014;19(7):980–984. doi: 10.1016/j.drudis.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bachoud-Levi A, Bourdet C, Brugieres P, Nguyen JP, Grandmougin T, Haddad B, Jeny R, Bartolomeo P, et al. Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington’s disease. Exp Neurol. 2000;161(1):194–202. doi: 10.1006/exnr.1999.7239. [DOI] [PubMed] [Google Scholar]
- 77.Bachoud-Levi AC, Remy P, Nguyen JP, Brugieres P, Lefaucheur JP, Bourdet C, Baudic S, Gaura V, et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet. 2000;356(9246):1975–1979. doi: 10.1016/s0140-6736(00)03310-9. [DOI] [PubMed] [Google Scholar]
- 78.Philpott LM, Kopyov OV, Lee AJ, Jacques S, Duma CM, Caine S, Yang M, Eagle KS. Neuropsychological functioning following fetal striatal transplantation in Huntington’s chorea: three case presentations. Cell Transplant. 1997;6(3):203–212. doi: 10.1177/096368979700600303. [DOI] [PubMed] [Google Scholar]
- 79.Antoniades CA, Watts C. Huntington’s disease and cell therapies: past, present, and future. Methods Mol Biol. 2013;1010:19–32. doi: 10.1007/978-1-62703-411-1_2. [DOI] [PubMed] [Google Scholar]
- 80.Aubry L, Bugi A, Lefort N, Rousseau F, Peschanski M, Perrier AL. Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci U S A. 2008;105(43):16707–16712. doi: 10.1073/pnas.0808488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ma L, Hu B, Liu Y, Vermilyea SC, Liu H, Gao L, Sun Y, Zhang X, et al. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10(4):455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Delli Carri A, Onorati M, Lelos MJ, Castiglioni V, Faedo A, Menon R, Camnasio S, Vuono R, et al. Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32+ medium-sized spiny neurons. Development. 2013;140(2):301–312. doi: 10.1242/dev.084608. [DOI] [PubMed] [Google Scholar]
- 83.Fink KD, Crane AT, Leveque X, Dues DJ, Huffman LD, Moore AC, Story DT, Dejonge RE, et al. Intrastriatal transplantation of adenovirus-generated induced pluripotent stem cells for treating neuropathological and functional deficits in a rodent model of Huntington’s disease. Stem Cells Transl Med. 2014;3(5):620–631. doi: 10.5966/sctm.2013-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mu S, Wang J, Zhou G, Peng W, He Z, Zhao Z, Mo C, Qu J, et al. Transplantation of induced pluripotent stem cells improves functional recovery in Huntington’s disease rat model. PLoS One. 2014;9(7):e101185. doi: 10.1371/journal.pone.0101185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Im W, Kim M. Cell therapy strategies vs. paracrine effect in Huntington’s disease. J Mov Disord. 2014;7(1):1–6. doi: 10.14802/jmd.14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Olson SD, Pollock K, Kambal A, Cary W, Mitchell GM, Tempkin J, Stewart H, McGee J, et al. Genetically engineered mesen-chymal stem cells as a proposed therapeutic for Huntington’s disease. Mol Neurobiol. 2012;45(1):87–98. doi: 10.1007/s12035-011-8219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu G, Rustom N, Litteljohn D, Bobyn J, Rudyk C, Anisman H, Hayley S. Use of induced pluripotent stem cell derived neurons engineered to express BDNF for modulation of stressor related pathology. Front Cell Neurosci. 2014;8:316. doi: 10.3389/fncel.2014.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bachoud-Levi AC, Gaura V, Brugieres P, Lefaucheur JP, Boisse MF, Maison P, Baudic S, Ribeiro MJ, et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: a long-term follow-up study. Lancet Neurol. 2006;5(4):303–309. doi: 10.1016/s1474-4422(06)70381-7. [DOI] [PubMed] [Google Scholar]
- 89.Barker RA, Mason SL, Harrower TP, Swain RA, Ho AK, Sahakian BJ, Mathur R, Elneil S, et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J Neurol Neurosurg Psychiatry. 2013;84(6):657–665. doi: 10.1136/jnnp-2012-302441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cicchetti F, Saporta S, Hauser RA, Parent M, Saint-Pierre M, Sanberg PR, Li XJ, Parker JR, et al. Neural transplants in patients with Huntington’s disease undergo disease-like neuronal degeneration. Proc Natl Acad Sci U S A. 2009;106(30):12483–12488. doi: 10.1073/pnas.0904239106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeon I, Choi C, Lee N, Im W, Kim M, Oh SH, Park IH, Kim HS, et al. In vivo roles of a patient-derived induced pluripotent stem cell Line (HD72-iPSC) in the YAC128 model of Huntington’s disease. Int J Stem Cells. 2014;7(1):43–47. doi: 10.15283/ijsc.2014.7.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11(23):2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 93.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11(2):219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Herrera F, Tenreiro S, Miller-Fleming L, Outeiro TF. Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr. 2011;3:Rrn1210. doi: 10.1371/currents.RRN1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P, Zurzolo C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci. 2013;126(Pt 16):3678–3685. doi: 10.1242/jcs.126086. [DOI] [PubMed] [Google Scholar]
- 96.Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, Esposito MS, Botta P, Goldstein C, Bernhard M, et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci. 2014;17(8):1064–1072. doi: 10.1038/nn.3761. [DOI] [PubMed] [Google Scholar]
- 97.Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the drosophila brain. Nat Commun. 2015;6:6768. doi: 10.1038/ncomms7768. [DOI] [PMC free article] [PubMed] [Google Scholar]