

Abstract
Objective:
To interrogate a poly-T variant (rs10524523, ′523) in TOMM40, a gene adjacent to the APOE gene on chromosome 19, in older persons with APOE ε3/3 homozygosity for association with cognitive decline, the clinical hallmark of Alzheimer disease (AD).
Methods:
Data came from participants in 2 cohort studies of aging and dementia who underwent annual clinical evaluations for up to 21 years. APOE and TOMM40′523 genotypes were determined from DNA from blood or brain samples. Linear mixed models compared the rates of decline in cognition among APOE ε3/3 carriers with different ′523 genotypes.
Results:
The 1,170 APOE ε3/3 homozygotes were of European ancestry, were free of dementia at baseline, and had an average age of 78.5 years at baseline. Three major genotypes at the ′523 variant were linked to APOE ε3/3; 26.5% had 2 short poly-Ts (S/S), 48.5% had 1 short and 1 very long poly-T (S/VL), and 24.0% had 2 very long poly-Ts (VL/VL). Participants with '523-S/S had faster decline in global cognition than participants with '523-S/VL or VL/VL (p = 0.002). The same association was observed for episodic memory (p < 0.001) and semantic memory (p = 0.003) but not for working memory, perceptual speed, or visuospatial ability.
Conclusions:
Our data reveal an association of APOE ε3/3-TOMM40′523 haplotypes with cognitive decline in community-based older persons such that the S/S poly-T genotype is related to faster cognitive decline, primarily in the domains of episodic and semantic memory.
APOE is a well-validated susceptibility gene for sporadic late-onset Alzheimer disease (AD).1,2 Compared with the more frequent APOE ε3 allele, ε4 is associated with a higher risk of AD, earlier disease onset, and faster cognitive decline, whereas ε2 is associated with lower risk, later onset, and slower decline.1–5 Recent evidence suggests that a poly-T variant in the adjacent gene of translocase of outer mitochondrial membrane 40 homolog (TOMM40′523) is also associated with age at AD onset.6–8 The variant is in linkage disequilibrium with APOE such that in whites the ε4 allele is almost exclusively linked to a long poly-T repeat. In contrast, the ε3 allele can be linked to either a short or a very long poly-T repeat, raising the possibility that different TOMM40′523 genotypes may differentiate AD susceptibility among ε3/3 carriers, which constitute ≈60% of the older white population.
Reports on the effect of the TOMM40′523 variant in ε3/3 carriers on AD risk and age at disease onset are emerging but have been inconsistent.9–12 Notably, studies that rely on clinical diagnosis of AD dementia may be contaminated by preclinical and subclinical cases in the reference group. The longitudinal measure of cognitive decline, on the other hand, better reflects AD as a continuous process and thus has been increasingly used as a complementary AD endophenotype.13 Knowledge of the relationship between the ′523 variant and cognitive decline is limited. The only 2 studies of which we are aware had relatively few numbers of longitudinal assessments; one had biannual assessments with a mean of ≈6 years,14 and the other had up to 4 assessments in 5-year intervals.15 Here, we investigate the effect of the TOMM40′523 variant on cognitive decline by leveraging genetic and longitudinal cognitive data from >1,000 community-based older persons who were APOE ε3/3 homozygous and were followed annually for up to 21 years. We summarize the distribution of ′523 length variation and examine its association with decline in global cognition and 5 relatively dissociable cognitive domains.
METHODS
Study participants.
Data came from 2 ongoing cohort studies of aging and dementia, the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP).16,17 The 2 studies are conducted by the same team of investigators with similar data collection procedures and share a common core of testing batteries. This design allows combined analyses. Study participants were free of known dementia at enrollment, underwent annual clinical and neuropsychological evaluations, and agreed to annual blood draws and brain donation at the time of death.
Considering APOE/TOMM40 evolutionary history,18 we restricted our sample to participants of European ancestry. At the time of analyses, genotyping data were available on 2,072 (96.6%) of the 2,144 white participants who completed the baseline evaluation. We excluded 116 participants who were diagnosed with AD dementia at baseline and another 70 who had only one cognitive evaluation. Of the remaining 1,886 individuals, 417 (22.1%) had APOE ε4 (ε3/4 or ε4/4), 261 (13.9%) had ε2 (ε2/2 or ε2/3), and 38 (2.0%) had ε2/4. Our analyses focused on the 1,170 participants (62.0%) who were APOE ε3/3 homozygous. The mean age at baseline was 78.5 years (SD = 7.4 years, range 54.3–97.8 years); the majority were female (n = 825, 70.5%); and the mean years of education was 16.2 (SD = 3.5 years, range 5–30 years). These participants were followed up annually for up to 21 years, with a mean of 8.2 years (SD = 5.0).
Standard protocol approvals, registrations, and patient consent.
The studies were approved by the Institutional Review Board of Rush University Medical Center. Written informed consent was acquired from each participant, and all participants signed an Anatomic Gift Act for organ donation.
Cognitive assessments.
For each participant, comprehensive cognitive assessments were administered at baseline and each annual follow-up visit.19 The testing battery contains a total of 17 cognitive performance tests that assess 5 cognitive domains, including episodic memory, semantic memory, working memory, perceptual speed, and visuospatial ability. Composite measures were used to examine the longitudinal cognitive decline, as previously described.20 More details on the cognitive tests are provided in the e-Methods at Neurology.org. The Mini-Mental State Examination was used only for descriptive purpose.
Results from the cognitive testing were reviewed by a neuropsychologist for signs of cognitive impairment. Clinical judgment of AD dementia follows standard criteria and requires a history of cognitive decline and evidence of impairment in memory and at least one other cognitive domain.21
APOE and TOMM40′523 genotyping.
DNA was extracted from peripheral blood or frozen postmortem brain tissue. Genotyping was performed at Polymorphic DNA Technologies (Alameda, CA) by investigators blinded to all clinical and pathologic data. The APOE genotypes were determined by sequencing rs429358 (codon 112) and rs7412 (codon 158) at exon 4 of the APOE gene. The TOMM40′523 genotypes were determined by rs10524523 (chr19:44,899,792-44,899,826, human genome reference assembly GRCh38/hg38), a homopolymer-length polymorphism (poly-T), at intron 6 of the TOMM40 gene.7,18 On the basis of the length of the poly-T repeat, a short allele (′523-S) was defined by poly-T repeat ≤19, a long allele (′523-L) by 20 ≤ poly-T repeat ≤ 29, and a very long allele (′523-VL) by poly-T repeat ≥30.
Statistical analysis.
Analysis of variance, Kruskal-Wallis, and χ2 tests examined the bivariate relationship of the TOMM40′523 genotypes with baseline age, education, sex, length of follow-up, and baseline cognition. Linear mixed models tested the hypothesis that in APOE ε3/3 carriers the rate of linear decline in cognition differs by ′523 genotype. In the primary model, annual summary scores for global cognition are the longitudinal continuous outcome. The main predictors include a term for time in years since the baseline that estimates the mean rate of change in cognition (slope). The interaction terms of the ′523 genotypes and time examine the associations of the ′523 variant with the slope. We repeated the model for each of the 5 cognitive domains separately. The analyses were done with SAS/STAT software, version 9.3 for Linux (SAS Institute Inc, Cary, NC).
For older persons who died, the longitudinal cognitive trajectory features a precipitous drop a few years before death, commonly known as terminal decline.22 We reported previously that APOE ε4 was associated with both preterminal and terminal decline.23 Therefore, we explored the influence of the ′523 variant on terminal decline by using random change point models, a common modeling approach for nonlinear change.24,25 The model specification has been described previously.26 The model estimates different rates of decline before and after a change point that signals the acceleration of cognitive decline. The model examines the association of risk factors with the onset of accelerated decline (i.e., terminal decline) and the rate of decline before and after the onset. These analyses were done with OpenBUGS.27
All the models were controlled for age, sex, and education. Unless otherwise specified, statistical significance was determined a priori at an α level of 0.05.
RESULTS
Distribution of TOMM40′523 among APOE ε3/3 carriers.
The APOE ε3 allele was linked predominantly to either the ′523-S or ′523-VL allele (figure e-1). Of the 1,170 ε3/3 carriers in this study, 310 (26.5%) were ′523-S/S homozygous, 568 (48.5%) were ′523-S/VL heterozygous, and 281 (24.0%) were ′523-VL/VL homozygous. Fewer than 1% of the participants who had other ′523 genotypes (including 5 S/L and 6 L/VL cases) were excluded from the subsequent association analyses.
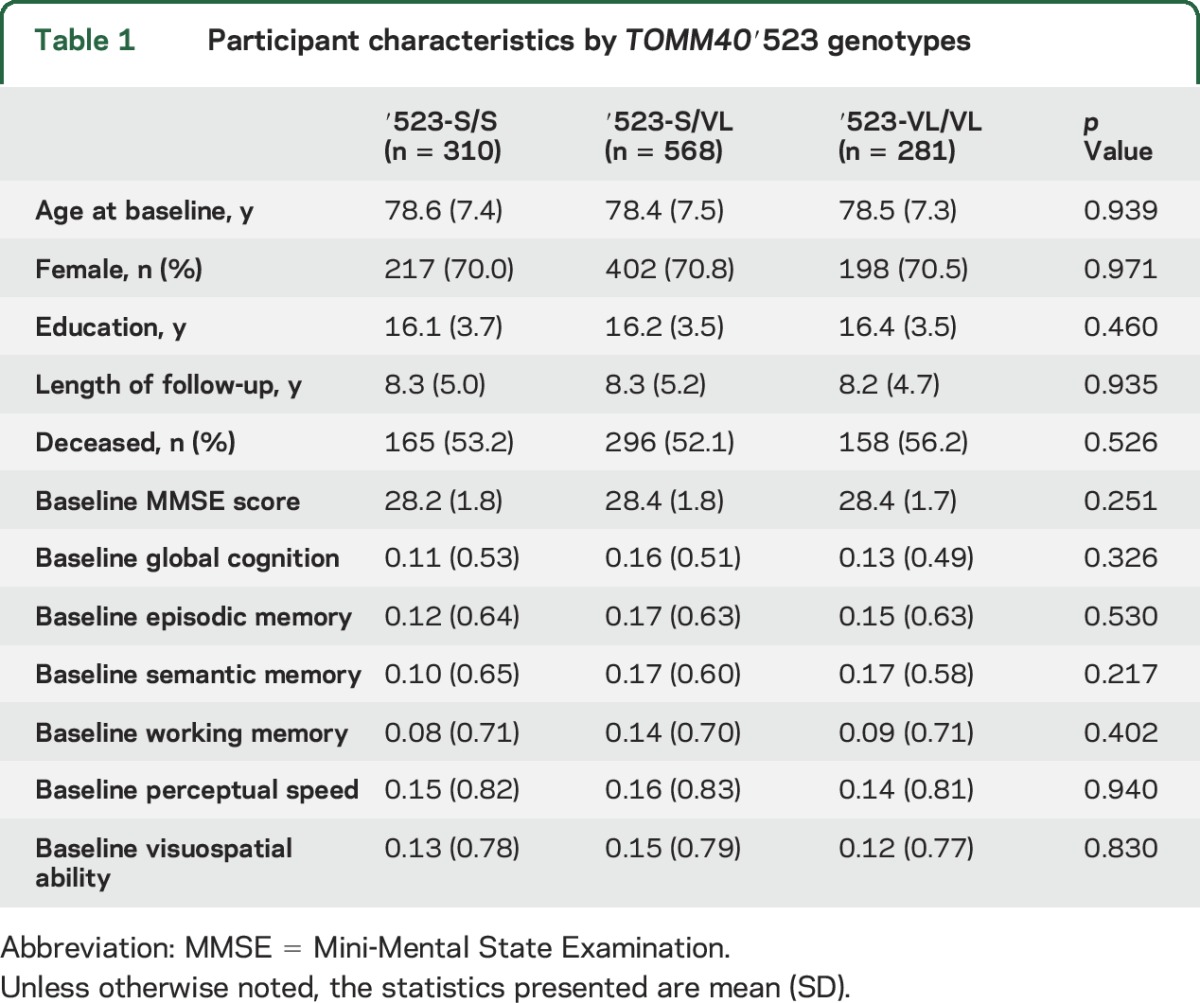
The baseline descriptives of study participants with the 3 major TOMM40′523 genotypes are summarized in table 1. We did not observe difference in demographics or length of follow-up by ′523 genotype. In addition, there was no difference in baseline cognition.
Table 1.
Participant characteristics by TOMM40′523 genotypes
TOMM40′523 and decline in global cognition.
We first examined the association of TOMM40′523 with decline in global cognition. In the model adjusted for baseline age, sex, and education, we observed that the mean annual rate of change in global cognition was −0.051 (standard error [SE] = 0.004, p < 0.001). We then augmented the model by including terms for the ′523 genotypes. Participants with ′523-S/S homozygosity had faster cognitive decline than those with ′523-S/VL (estimate = −0.021, SE = 0.007, p = 0.003) or ′523-VL/VL (estimate = −0.017, SE = 0.008, p = 0.030). Post hoc comparison did not reveal a significant difference between ′523-S/VL and VL/VL carriers in cognitive decline (p = 0.628), suggesting that the ′523-S/S genotype was associated with faster decline in a recessive manner. For model parsimony, we refitted the model by collapsing the S/VL and VL/VL into one group. As shown in figure 1, participants with ′523-S/S genotype had ≈40% faster decline in global cognition than the rest of ε3/3 carriers (p = 0.002). Depression might affect the association with cognitive decline; thus, we repeated the last model by controlling for depressive symptoms. Participants with more depressive symptoms had faster decline, but the association of ′523 genotype with cognitive decline was unchanged.
Figure 1. Linear decline in global cognition among APOE ε3/3 carriers by TOMM40′523-S/S genotype.

Spaghetti plots show the annual global cognition scores for 50 randomly selected participants without ′523-S/S genotype (light blue) vs 50 participants with ′523-S/S genotype (gray). Superimposed is model-derived cognitive decline in global cognition for the participants without ′523-S/S genotype (blue) vs with ′523-S/S genotype (black).
TOMM40′523 and decline in 5 cognitive domains.
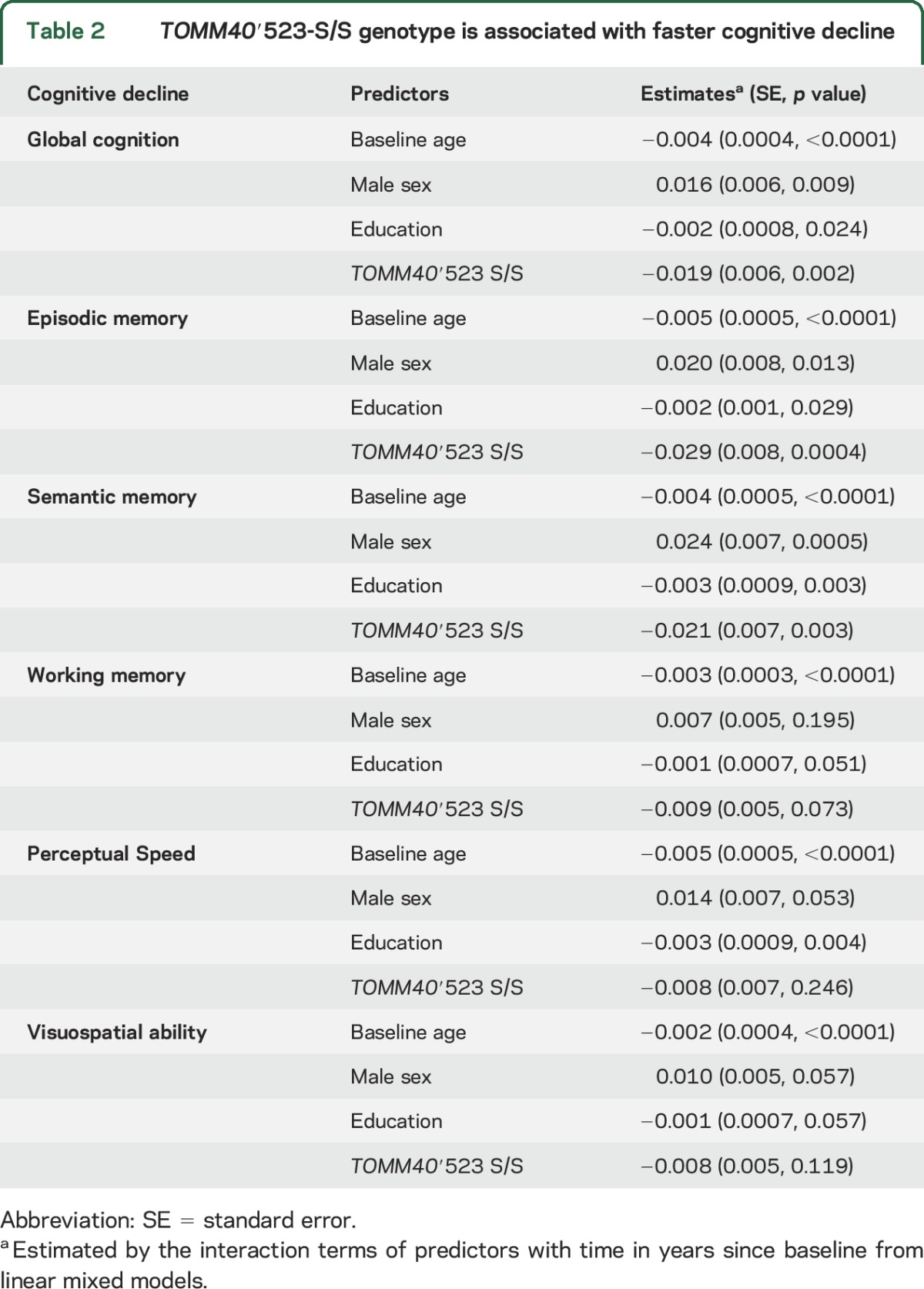
We repeated the linear mixed model and examined the effect of ′523-S/S genotype on decline in individual cognitive domains. As shown in table 2, among ε3/3 carriers, the presence of ′523-S/S genotype was associated with faster decline in episodic memory (p < 0.001) and semantic memory (p = 0.003). In contrast, we did not observe ′523 genotypic difference in decline in working memory, perceptual speed, or visuospatial ability (all p > 0.05). Notably, the results remained the same after adjustment for multiple testing (Bonferroni-corrected α = 0.01).
Table 2.
TOMM40′523-S/S genotype is associated with faster cognitive decline
TOMM40′523 and terminal decline.
Of the 1,170 ε3/3 homozygotes included in this study, slightly more than half died during the course of follow-up (n = 629, 53.8%). In linear mixed models stratified by death status, we observed that the deceased participants had faster cognitive decline on average (estimate = −0.084, SE = 0.007, p < 0.001) than those who were still alive (estimate = −0.023, SE = 0.004, p < 0.001). Furthermore, the association of ′523 S/S genotype with the rate of decline was stronger among participants who had died (estimate = −0.026, SE = 0.010, p = 0.008) than among the living (estimate = −0.010, SE = 0.007, p = 0.154). The results were similar for episodic memory and semantic memory.
Next, we focused on the subsample of the deceased participants and examined the associations of ′523-S/S with terminal decline in cognition. The average age at death was 89.2 years (SD = 6.4 years). Table 3 summarizes the results from the random change point models. Consistent with our previous report for the overall cohorts,28 in this sample, the terminal decline in global cognition occurred a mean of ≈3.4 years before death, and the mean decline in the terminal phase was 7 times faster than the preterminal phase. We found that the rate of terminal decline in global cognition was faster in participants with the ′523-S/S genotype compared to those without (figure 2). Similar results were observed in episodic memory and semantic memory. We did not find a significant difference in the onset of terminal decline by ′523-S/S status. The findings were inconclusive with respect to the effect of ′523-S/S on preterminal decline; the ′523-S/S genotype was associated with faster preterminal decline in semantic memory, but the associations with decline in global cognition and episodic memory were not significant.
Table 3.
TOMM40′523-S/S genotype is associated with faster terminal decline
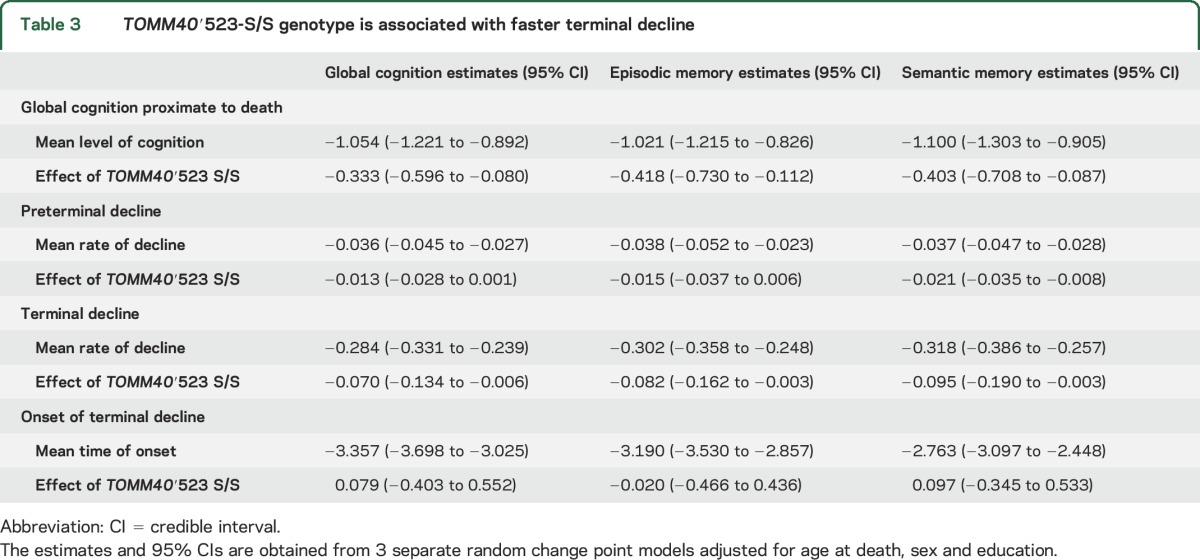
Figure 2. Terminal decline in global cognition among the deceased APOE ε3/3 carriers by TOMM40′523-S/S genotype.

Spaghetti plots show the annual global cognition scores for 50 randomly selected participants without ′523-S/S genotype (light blue) vs 50 participants with ′523-S/S genotype (gray). Superimposed is the predicted mean trajectory estimated from a random change point model for the participants without ′523-S/S genotype (blue) vs with ′523-S/S genotype (black).
DISCUSSION
The genetic role of TOMM40 in AD remains controversial because of its linkage disequilibrium with APOE.29 Studies targeting APOE 3/3 homozygosity are able to disentangle the effect of the TOMM40 variant independent of potential confounding due to ε4 or ε2. Recent phylogenetic analyses reveal that in whites the APOE ε3 allele is linked to the variable length at TOMM40′523 such that AD susceptibility differs depending on whether ε3 is linked to a ′523-S or -VL allele.6 Findings to date, however, are mixed. The ′523-VL allele has been shown to be related to earlier disease onset, poorer memory performance, or smaller gray matter volume, suggesting that ′523-VL increases the risk of AD.7,8,30,31 Null or even conflicting findings have also been reported.9–11,32,33 Using annual cognitive data from >1,000 community-based older white Americans who were ε3/3 homozygous, we examined the effect of the ′523 variant on late-life cognitive decline, We found that the ε3/3 in linkage to ′523-S/S is associated with faster decline in cognition compared with the linkage to either S/VL or VL/VL. The effect is driven primarily by episodic and semantic memory. This result supports that, although APOE and TOMM40′523 are in linkage disequilibrium, the APOE-TOMM40′523 haplotypes are more informative than the APOE genotypes alone in relation to cognitive decline. The direction of effect suggests that ′523-S rather than -VL is the potential risk allele. Our result is consistent with that from an earlier large-scale case-control study showing that, among individuals with ε3/3 genotype, the frequency of ′523-VL is lower in patients with AD than in controls.9 We observed that the ′523-S allele influences cognitive decline in a recessive manner such that, compared with S/S, ε3/3 in linkage to both S/VL and VL/VL shows slower decline, and we did not find a difference between S/VL and VL/VL. In addition, although prior literature suggests that the effect of TOMM40′523 is most evident at younger ages,14 in a subsample of deceased participants, we observed that the ′523-S/S genotype is associated with faster terminal decline. We previously reported a similar finding for APOE such that the ε4 allele not only contributes to the preterminal decline in cognition that occurs many years before death but also expedites the progression of dementia and influences cognition during the last few years of life.23
The underlying neurobiology of the effect of TOMM40′523 on cognitive decline is unclear, but the structural variants show effects in an array of neurologic areas. Findings from brain pathology of individuals with APOE ε3/3 show that the ′523 long allele (poly-T repeat ≥20) may increase the burden of AD pathology.34 Prior evidence also suggests that the ′523 variant may have a broader influence in other common neurodegenerative diseases, including Parkinson disease.35 The TOMM40 variant is implicated in affecting the level of neurofilament light proteins in CSF, a marker primarily for subcortical axonal injury and white matter disease.36 Older adults with the ′523-S allele also have lower white matter integrity.37 Beyond these studies specifically on TOMM40, the question of the extent to which results previously thought to relate purely to APOE ε4 may use a ′523-dependent mechanism is unanswered. For instance, although a few studies have found ′523 brain structure, far more extensive work in APOE ε4 has found that major connectivity tracts are affected at birth38 and that it ultimately disrupts the balance of specialized processing and information integration in normal brain networks.39 Moving beyond these disease associations toward some understanding of the role of ′523 in AD is difficult because the molecular pathways behind them are unclear.
To better define disease mechanisms in the future, the molecular effects of the APOE-TOMM40′523 haplotypes can be evaluated with a traditional focus on the sequence and chromatin state of that specific locus or from a global multi-omic perspective. For instance, methylation patterns at the locus coupling the 2 genes lead to an allele-specific effect on the expression levels of both genes.40 Examining effects beyond this locus may also be necessary, given the pleiotropy of APOE, the need to compare APOE and TOMM40 effects, and the broad role for molecular interactions in determining cellular state and AD susceptibility.39 Common examples of such broader approaches are coexpression or comethylation. These approaches can identify genome-wide sets of covarying genes, which can then be associated with disease state or genes with key disease variants. Indeed, the structure of these networks themselves may be altered in association with disease state or the presence of a given haplotype. Furthermore, directed causal networks leverage genetic variants to infer directed interactions and to help identify a sequence of signaling downstream of genetic variants. Novel omic assays can help to estimate these molecular effects. For instance, because ′523 is intronic, comparative chromatin conformation could be used to identify transcriptional regulatory changes due to higher-order chromatin state changes. Because APOE variants affect protein structure, the binding partners of APOE alleles may differ, and such knowledge would reduce the number of critical partners for disease effects. Overall, applying these omic approaches will assist in identifying the union, intersection, and interacting effects of TOMM40 and APOE in a manner that points to disease mechanisms.
The strength of the study is noted. A majority of previous reports on TOMM40 effects focus on age at onset or rely on case-control studies, and we elected to use the endophenotype of longitudinal cognitive decline. AD has been reconceptualized as a continuum from normality to mild cognitive impairment to AD dementia. By capitalizing on continuous repeated measures of cognitive performance, our approach avoids the potential confounding due to preclinical or subclinical contamination in the reference group, and the quantitative nature of the phenotype improves the statistical power to detect small effects. The study also has limitations. The ROS and MAP participants on average are older and have more education than the general population. Our findings need to be replicated in other studies, including studies that use alternative cognitive metrics.
The current study is restricted to the ε3/3 carriers in white Americans. The APOE-TOMM40′523 linkage patterns are different in blacks as a result of admixture. It is important to distinguish the APOE-TOMM40′523 haplotypes between older white and black populations. Finally, future studies are needed to examine whether the relationship between TOMM40′523 and cognition is driven by AD pathology. All the ROS and MAP participants are organ donors. However, only a subset of the study sample had died and undergone brain autopsy. As more postmortem samples are being collected, the 2 studies offer a unique opportunity to investigate this important question with the needed fidelity.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the investigators and staff at the Rush Alzheimer's Disease Center. They also thank all the participants of ROS and MAP. Information regarding obtaining data from ROS and MAP for research use can be found at the Rush Alzheimer's Disease Center Research Resource Sharing Hub (www.radc.rush.edu).
Glossary
- AD
Alzheimer disease
- MAP
Rush Memory and Aging Project
- ROS
Religious Orders Study
- SE
standard error
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Drafting or revising the manuscript for content: Drs. Yu, Lutz, Wilson, Burns, Roses, Saunders, Gaiteri, De Jager, Barnes, Bennett. Study concept or design: Drs. Yu, Lutz, Roses, Bennett. Analysis or interpretation of the data: Drs. Yu, Lutz, Wilson, Burns, Roses, Saunders, Gaiteri, De Jager, Barnes, Bennett. Acquisition of data: Drs. Lutz, Burns, Roses, Bennett. Statistical analysis: Dr. Yu. Study supervision or coordination: Drs. Burns, Bennett. Obtaining funding: Dr. Bennett.
STUDY FUNDING
This work was supported by the National Institute on Aging (R01AG17917, P30AG10161, RF1AG15819) and the Illinois Department of Public Health. The study was funded in part by an unrestricted educational grant from Zinfandel Pharmaceuticals, Inc. Genotyping was performed by investigators blinded to all clinical data, and statistical analyses were performed by investigators at Rush (Dr. Yu).
DISCLOSURE
L. Yu reports a relevant disclosure for this manuscript. This work was supported in part by an unrestricted educational grant from Zinfandel Pharmaceuticals, Inc. M. Lutz receives consulting fees from Zinfandel Pharmaceuticals. R. Wilson reports a relevant disclosure for this manuscript. This work was supported in part by an unrestricted educational grant from Zinfandel Pharmaceuticals, Inc. A. Roses is deceased; disclosures are not included for this author. A. Saunders receives consulting fees from Zinfandel Pharmaceuticals, Inc. Dr. Saunders is a member of the Biomarker Committee for the Zinfandel-Takeda Alliance and the TOMMORROW clinical trial. C. Gaiteri reports a relevant disclosure for this manuscript. This work was supported in part by an unrestricted educational grant from Zinfandel Pharmaceuticals, Inc. P. De Jager reports no disclosures relevant to the manuscript. L. Barnes reports a relevant disclosure for this manuscript. This work was supported in part by an unrestricted educational grant from Zinfandel Pharmaceuticals, Inc. D. Bennett reports relevant disclosures for this manuscript. The work was supported in part by an unrestricted educational grant from Zinfandel Pharmaceuticals, Inc. Dr. Bennett also serves on the adjudication committee for AD4833/TOMM40_301 study funded by Takeda Pharmaceuticals USA, Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 2.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–184. [DOI] [PubMed] [Google Scholar]
- 3.Schiepers OJ, Harris SE, Gow AJ, et al. APOE E4 status predicts age-related cognitive decline in the ninth decade: longitudinal follow-up of the Lothian birth cohort 1921. Mol Psychiatry 2012;17:315–324. [DOI] [PubMed] [Google Scholar]
- 4.Caselli RJ, Dueck AC, Osborne D, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med 2009;361:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deary IJ, Whiteman MC, Pattie A, et al. Cognitive change and the APOE epsilon 4 allele. Nature 2002;418:932. [DOI] [PubMed] [Google Scholar]
- 6.Roses AD, Lutz MW, Amrine-Madsen H, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J 2010;10:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lutz MW, Crenshaw DG, Saunders AM, Roses AD. Genetic variation at a single locus and age of onset for Alzheimer's disease. Alzheimers Dement 2010;6:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roses AD. An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Arch Neurol 2010;67:536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cruchaga C, Nowotny P, Kauwe JS, et al. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol 2011;68:1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crenshaw DG, Gottschalk WK, Lutz MW, et al. Using genetics to enable studies on the prevention of Alzheimer's disease. Clin Pharmacol Ther 2013;93:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jun G, Vardarajan BN, Buros J, et al. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Arch Neurol 2012;69:1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roses AD, Lutz MW, Crenshaw DG, Grossman I, Saunders AM, Gottschalk WK. TOMM40 and APOE: requirements for replication studies of association with age of disease onset and enrichment of a clinical trial. Alzheimers Dement 2013;9:132–136. [DOI] [PubMed] [Google Scholar]
- 13.Ertekin-Taner N, De Jager PL, Yu L, Bennett DA. Alternative approaches in gene discovery and characterization in Alzheimer's disease. Curr Genet Med Rep 2013;1:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caselli RJ, Dueck AC, Huentelman MJ, et al. Longitudinal modeling of cognitive aging and the TOMM40 effect. Alzheimers Dement 2012;8:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payton A, Sindrewicz P, Pessoa V, et al. A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in nonpathologic aging. Neurobiol Aging 2016;39:217 e1–217 e7. [DOI] [PubMed] [Google Scholar]
- 16.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the Religious Orders Study. Curr Alzheimer Res 2012;9:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory And Aging Project. Curr Alzheimer Res 2012;9:646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roses AD, Lutz MW, Saunders AM, et al. African-American TOMM40′523-APOE haplotypes are admixture of West African and Caucasian alleles. Alzheimers Dement 2014;10:592–601.e2. [DOI] [PubMed] [Google Scholar]
- 19.Wilson RS, Boyle PA, Yu L, Segawa E, Sytsma J, Bennett DA. Conscientiousness, dementia related pathology, and trajectories of cognitive aging. Psychol Aging 2015;30:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193. [PubMed] [Google Scholar]
- 21.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 22.Wilson RS, Leurgans SE, Boyle PA, Schneider JA, Bennett DA. Neurodegenerative basis of age-related cognitive decline. Neurology 2010;75:1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu L, Boyle P, Schneider JA, et al. APOE epsilon4, Alzheimer's disease pathology, cerebrovascular disease, and cognitive change over the years prior to death. Psychol Aging 2013;28:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall CB, Lipton RB, Sliwinski M, Stewart WF. A change point model for estimating the onset of cognitive decline in preclinical Alzheimer's disease. Stat Med 2000;19:1555–1566. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Dowling NM, Chappell R. Quantile regression with a change-point model for longitudinal data: an application to the study of cognitive changes in preclinical Alzheimer's disease. Biometrics 2015;71:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu L, Boyle P, Wilson RS, et al. A random change point model for cognitive decline in Alzheimer's disease and mild cognitive impairment. Neuroepidemiology 2012;39:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lunn DJ, Thomas A, Best N, Spiegelhalter D. WinBUGS–a bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000;10:325–337. [Google Scholar]
- 28.Boyle PA, Wilson RS, Yu L, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol 2013;74:478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerreiro RJ, Hardy J. TOMM40 association with Alzheimer disease: tales of APOE and linkage disequilibrium. Arch Neurol 2012;69:1243–1244. [DOI] [PubMed] [Google Scholar]
- 30.Johnson SC, La Rue A, Hermann BP, et al. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE epsilon3/epsilon3 genotype. Alzheimers Dement 2011;7:456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayden KM, McEvoy JM, Linnertz C, et al. A homopolymer polymorphism in the TOMM40 gene contributes to cognitive performance in aging. Alzheimers Dement 2012;8:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu SH, Roeder K, Ferrell RE, et al. TOMM40 poly-T repeat lengths, age of onset and psychosis risk in Alzheimer disease. Neurobiol Aging 2011;32:2328.e1–2328.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu CE, Seltman H, Peskind ER, et al. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer's disease: patterns of linkage disequilibrium and disease/marker association. Genomics 2007;89:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li G, Bekris LM, Leong L, et al. TOMM40 intron 6 poly-T length, age at onset, and neuropathology of AD in individuals with APOE epsilon3/epsilon3. Alzheimers Dement 2013;9:554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gottschalk WK, Lutz MW, He YT, et al. The broad impact of TOM40 on neurodegenerative diseases in aging. J Parkinsons Dis Alzheimers Dis 2014;1:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruno D, Pomara N, Nierenberg J, et al. Levels of cerebrospinal fluid neurofilament light protein in healthy elderly vary as a function of TOMM40 variants. Exp Gerontol 2012;47:347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyall DM, Harris SE, Bastin ME, et al. Alzheimer's disease susceptibility genes APOE and TOMM40, and brain white matter integrity in the Lothian birth cohort 1936. Neurobiol Aging 2014;35:1513 e1525–1513 e1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mc Donald J, Krainc D. Alzheimer gene APOE epsilon4 linked to brain development in infants. JAMA 2014;311:298–299. [DOI] [PubMed] [Google Scholar]
- 39.Gaiteri C, Mostafavi S, Honey CJ, De Jager PL, Bennett DA. Genetic variants in Alzheimer disease: molecular and brain network approaches. Nat Rev Neurol 2016;12:413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu CE, Cudaback E, Foraker J, et al. Epigenetic signature and enhancer activity of the human APOE gene. Hum Mol Genet 2013;22:5036–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.