

Abstract
Objective:
To identify genetic loci associated with plasma tau concentrations in healthy elders and individuals with Alzheimer disease.
Methods:
Four hundred sixty-three non-Hispanic white individuals exceeding quality control criteria were included from the Alzheimer's Disease Neuroimaging Initiative (ADNI-1) cohort. Association of plasma tau with genetic polymorphisms was performed with a linear regression model. Significant associations were validated in an independent replication cohort consisting of 431 healthy elders or individuals with mild cognitive impairment recruited from the University of California, San Francisco Memory and Aging Center.
Results:
The minor allele (A) of rs242557 in the microtubule-associated protein tau gene (MAPT) was associated with higher plasma tau levels at genome-wide significance (p = 4.85 × 10−9, empiric family-wise error corrected p = 0.0024) in a dose-dependent fashion. This association was also observed in the replication cohort (p = 1.0 × 10−5; joint analysis p = 1.2 × 10−12). Single nucleotide polymorphisms near PARK2 (rs2187213) (p = 6.15 × 10−6), IL2RA (rs7072793, rs7073236) (p = 7.89 × 10−6), and an intergenic locus on 9p21.3 (rs7047280) (p = 8.13 × 10−6) were identified as suggestive loci associated with plasma tau levels.
Conclusions:
MAPT H1c haplotype (rs242557) has previously been identified as a genetic risk factor for progressive supranuclear palsy and corticobasal degeneration. The current findings suggest that plasma tau concentration could be an endophenotype for identifying risk for 4-repeat tauopathies in older individuals.
Tau is a microtubule-associated protein that promotes assembly and stabilization of cytoskeletal microtubules.1 Accumulation of insoluble deposits of tau has been observed in a number of neurodegenerative disorders, including Alzheimer disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), some forms of frontotemporal lobar degeneration, and chronic traumatic encephalopathy, which are collectively classified as tauopathies.1
CSF tau concentrations are thought to reflect neuronal degeneration in AD, and CSF tau, either alone or in combination with β-amyloid peptide, has been confirmed as a useful biomarker for AD across the spectrum of disease severity.2 On the basis of this finding, CSF total tau (t-tau) and phosphorylated tau (p-tau) concentrations at residue 181 have been used as endophenotypes in genome-wide association studies (GWAS) to detect risk variants for AD, with the APOE locus showing the strongest association with elevated CSF tau.3,4 Enigmatically, despite strong genetic links to tau, CSF tau levels are normal or low in other tauopathies such as PSP and some tau gene (MAPT) mutation carriers.5,6
The recent development of an ultrasensitive assay for tau in peripheral blood makes it feasible to study the relationship between peripheral tau concentrations and tauopathies.7 In comparison to CSF, plasma tau levels in aging and neurodegenerative disease have not been well studied.
The use of quantitative traits in GWAS has been shown to increase statistical power over case-control designs.3,4 Here, we hypothesized that plasma tau, similar to CSF tau, may constitute a suitable endophenotype for identifying genetic risk factors for tauopathies. Within this context, we conducted a GWAS for plasma tau level and identified a single nucleotide polymorphism (SNP) (rs242557) within the MAPT gene that showed a genome-wide significant association with plasma, but not CSF, tau levels.
METHODS
Participants.
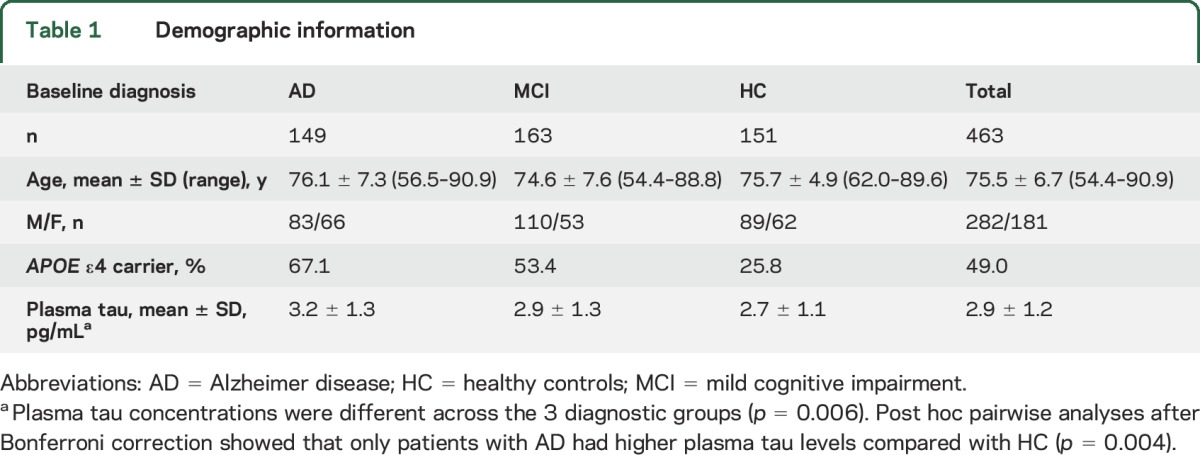
In this study, 463 (AD = 149, mild cognitive impairment [MCI] = 163, healthy controls [HC] = 151 at baseline) non-Hispanic white individuals whose data met all quality control (QC) criteria were included from the Alzheimer's Disease Neuroimaging Initiative 1 (ADNI-1) cohort. Table 1 shows the demographic data and description of the plasma tau levels in each group. Data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu).
Table 1.
Demographic information
The full cohort with both plasma tau and GWS data included 506 participants. The analysis was restricted to non-Hispanic white participants (n = 468) to reduce the potential bias of population stratification in the GWAS. Cryptic relatedness and population substructure, which can confound GWAS, were checked with genomic identity-by-descent and multidimensional scaling components using the PLINK v1.90b3.28 software8 (figure e-1 at Neurology.org). This step removed 2 participants who appeared cryptically related and clustering separately from the other samples (figure e-1a), resulting in 466 valid samples. Finally, with the use of the HapMap cohort, they showed tight clustering with individuals of European ancestry (figure e-1b).
Plasma and CSF tau measurements and QC.
Plasma tau concentrations were determined with the Human Total Tau kit from Quanterix (Lexington, MA), as described by the manufacturer. Assays were run at the University of Gothenburg. Intra-assay and interassay coefficients of variation were 10% to 15%. The lower limit of quantification was 1.22 pg/mL; samples with a reported plasma tau concentration below this value were removed from further analysis. The plasma tau concentrations could be downloaded from the ADNI1 database (http://adni.loni.usc.edu/). Detailed steps for measurements and QC using CSF t-tau and CSF p-tau have been previously reported.4 Further QC was performed to reduce the potential influence of extreme outliers on statistical results. Mean and SD of baseline plasma tau measures were calculated by investigators blinded to diagnostic information, and participants who had a value >4 or <4 SD from the mean value (7.7 pg/mL) were regarded as extreme outliers and removed from the analysis.4 This step removed 3 additional participants, resulting in 463 valid samples.
Standard protocol approvals, registrations, and patient consents.
The study was approved by institutional review boards of all participating institutions, and written informed consent was obtained from all participants or authorized representatives.
Genotyping and QC.
The ADNI-1 samples were genotyped with the Human 610-Quad BeadChip (Illumina, Inc, San Diego, CA). Stringent QC assessment was performed with the PLINK software with the following criteria: call rate for SNPs >95%, call rate for individuals >95%, minor allele frequencies >0.20, and Hardy-Weinberg equilibrium test p > 0.001. The restriction to SNPs with a minor allele frequency >20% served to reduce the likelihood of false-positive results from quantitative trait association in a relatively small sample size. The final, cleaned dataset included a total of 316,802 genotyped variants. The polymorphisms rs7412 and rs429358, which define the APOE alleles, were genotyped separately by an APOE genotyping kit.4
Statistical analyses.
The Spearman rank correlation coefficient was used to determine correlations of plasma tau concentrations with CSF tau concentrations. Values of p < 0.05 were considered statistically significant after adjustment for multiple comparisons with Bonferroni correction. A linear regression model was used to determine association of plasma tau concentrations with genetic polymorphisms using the PLINK software with an additive genetic model (i.e., dose-dependent effect of the minor allele). To correct for confounding by genetic ancestry that could lead to population stratification, the first 4 multidimensional scaling components of a representative, linkage disequilibrium (LD)–pruned genotype matrix were calculated in PLINK and used as covariates in the regression model. Age, sex, and diagnosis were also included as covariates. To account for multiple comparisons, thresholds of p < 5 × 10−8 and p < 1 × 10−5 were used for genome-wide significant and suggestive associations, respectively.9 As an additional alternative to exclude possible false-positive results, the PLINK max(T) permutation test with 5,000 permutations was used to generate empiric p values and for multiple-testing correction. Genome-wide associations were visualized with the R package qqman10; regional associations were visualized with the LocusZoom web tool.11
Replication of genome-wide significant associations.
An independent replication cohort of 431 healthy individuals free of neurologic diseases was identified from patients seen at the University of California, San Francisco and approved by the local Institutional Review Board. Subsequently, 29 of these participants were diagnosed with MCI. Plasma tau concentrations were determined with the Human Total Tau 2.0 kit from Quanterix at the University of Gothenburg. Samples were diluted 4-fold and run in singlicate. SNPs for the validation stage were chosen on the basis of the genome-wide significance threshold in the initial analysis (p < 5 × 10−8). Genotyping of these SNPs was performed with Taqman assays (rs242557: C_1016016_1_). In total, we acquired both plasma tau concentration measurements and genotyping of rs242557 in 387 participants. Data analysis was performed with linear regression implemented in R, accounting for age, sex, diagnosis, and cohort (discovery vs replication) as covariates.
RESULTS
Correlations between plasma tau and CSF tau levels.
There were 316 participants (AD = 83, MCI = 149, HC = 84) with both plasma and CSF tau levels. However, there was no correlation between tau levels in plasma and CSF in any diagnostic group (plasma tau vs CSF t-tau [figure 1A]: AD, r = 0.131, p = 0.234; MCI, r = 0.209, p = 0.066; HC, r = 0.002, p = 0.984; plasma tau vs CSF p-tau [figure 1B]: AD, r = 0.056, p = 0.611; MCI, r = 0.145, p = 0.077; HC, r = −0.027, p = 0.809), indicating that plasma tau levels do not reflect CSF tau levels.
Figure 1. Correlations between plasma tau and CSF tau levels.
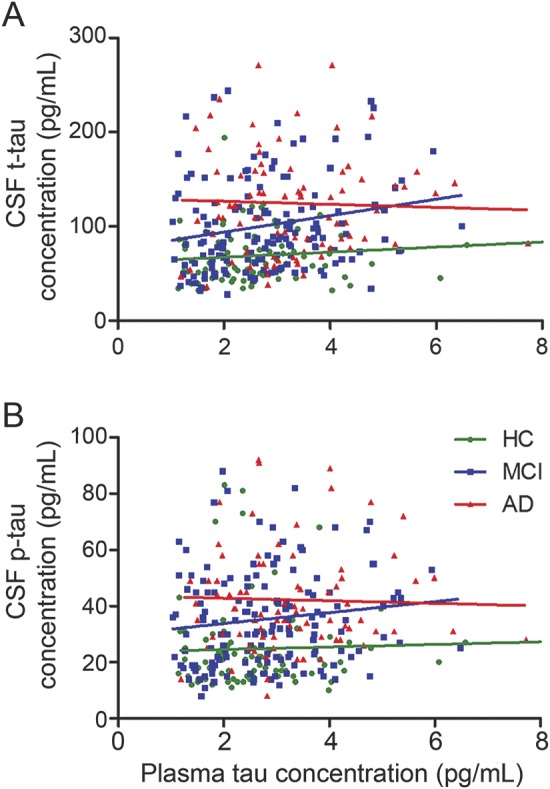
(A) There was no correlation between plasma tau levels and CSF total tau in any diagnostic group. (B) There was no correlation between plasma tau levels and CSF phosphorylated tau in any diagnostic group. AD = Alzheimer disease; HC = healthy controls; MCI = mild cognitive impairment.
The SNP rs242557, near MAPT, is associated with plasma tau levels.
A total of 463 individuals (AD = 149, MCI = 163, HC = 151 at baseline) were identified for GWAS (table 1). Plasma tau concentrations were different between the 3 diagnostic groups (p = 0.006) (table 1). Post hoc analysis after Bonferroni correction showed that only patients with AD had higher plasma tau levels compared with HC (p = 0.004). After adjustment for age, sex, and diagnosis, the association summary statistics appeared well calibrated with no evidence of population stratification (the genomic inflation factor = 1.00). A genome-wide significant association of rs242557 (in the MAPT region) with elevated plasma tau levels (p = 4.85 × 10−9) was detected (table 2 and figure 2A). Assuming an additive genetic model, the genotype of rs242557 explained 6.3% of the variance in plasma tau concentrations. This locus survived permutation-based empiric corrections for multiple testing (empiric p < 0.0002; permutation-based corrected empiric family-wise error rate controlled at 0.0024). The minor allele (A) of rs242557 was associated with higher plasma tau levels in a dose-dependent effect within both combined groups and each diagnostic group (normal group, p = 9.1 × 10−5; MCI group, p = 0.0013; and AD group, p = 3.1 × 10−4) (figure e-2a).
Table 2.
Top SNPs associated with plasma tau
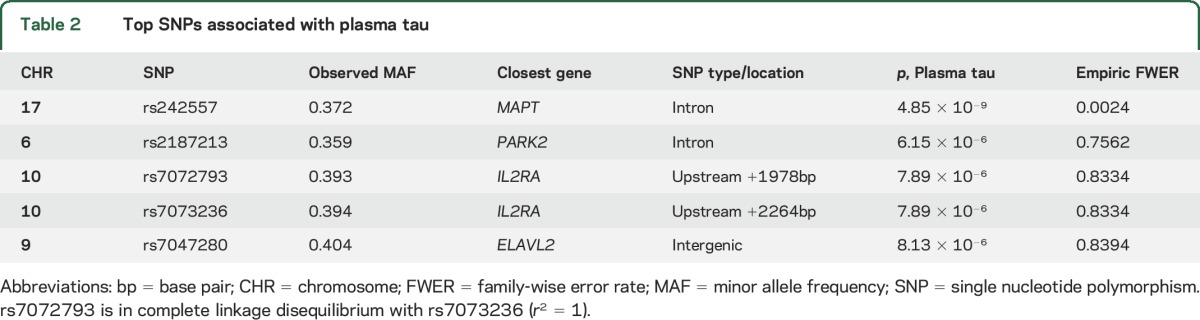
Figure 2. Manhattan and regional plots for associations with plasma tau.

(A) Genome-wide signal intensity (Manhattan) plots showing the −log10 (p value) for individual single nucleotide polymorphisms. (B) Regional association results for the MAPT region of chromosome 17. (C) Association results for 17q21.31 controlling for rs242557.
There were no other genome-wide significant associations with plasma tau outside the MAPT region. In the MAPT region, several SNPs in LD with rs242557 showed values of p < 0.001 for plasma tau levels (figure 2B). However, after controlling for rs242557 genotype (figure 2C), no SNPs in this region showed strong association with plasma tau levels (minimum uncorrected p = 0.007 at rs2239925), indicating that all of the association in this locus was driven by rs242557.
The LD pattern between rs242557 and nearby SNPs was nearly identical in the ADNI cohort compared with 1,000 Genomes European participants (figure e-3), suggesting that the SNP genotypes from this study were accurate. Moreover, LD between rs242557 and the H1/H2 haplotype-defining SNP, rs1560310, was calculated using the 1,000 Genomes European cohort (r2 = 0.197 and D' = 1), demonstrating that rs242557 is specific to the H1 clade.
The analysis identified 3 other suggestive loci near PARK2 (rs2187213) and IL2RA (rs7072793 and rs7073236) and within an intergenic locus on 9p21.3 (rs7047280) where p values reached the level of p < 10−5 (table 2, figure 2A). PARK2 and IL2RA minor alleles were associated with lower plasma tau levels (figure e-2b and e-2c) and the 9p21.3 minor allele was associated with higher plasma levels (figure e-2d) in a dose-dependent fashion within both combined groups and each diagnostic group. However, they did not survive after both permutation-based empiric corrections for multiple testing. Other plasma tau-associated SNPs that did not reach genome-wide significance and with p values that are greater than 10−4 are listed in table e-1.
APOE but not MAPT locus affects CSF but not plasma tau levels.
Among the 463 individuals analyzed in the plasma tau levels, 314 participants (AD = 82, MCI = 148, HC = 84) had CSF tau levels. We investigated whether the top SNPs identified in the plasma tau concentration GWAS (rs242557 within MAPT) and previous CSF tau concentration GWAS (APOE) were associated with CSF tau levels. After adjustment for age, sex, and diagnosis, APOE ε4 showed significant associations with both CSF t-tau (pc = 0.004) and CSF p-tau (pc = 0.028) after Bonferroni correction. However, rs242557 within MAPT was not associated with CSF t-tau (p = 0.988) or CSF p-tau (pc = 0.835). APOE ε4 was not associated with plasma tau levels (p = 0.312), nor were other SNPs in the APOE region.
Association with plasma tau at the MAPT locus replicates in an independent cohort.
To validate our finding of a locus near MAPT associated with plasma tau concentrations, we measured plasma tau concentrations and genotyped the rs242557 SNP in an independent cohort of 387 adult participants without any history of neurologic disease. After sample collection, 27 of the participants were diagnosed with MCI. In this cohort, the participants ranged from 30 to 99 years of age (mean 68.9 years, standard deviation 10.5 years) at the time of blood draw. Both sexes were well represented, including 161 male and 226 female participants. The mean plasma tau concentration was 2.2 pg/mL with an SD of 0.8 pg/mL. In the replication sample, the minor (A) allele of rs242557 was significantly associated with higher plasma tau levels (p = 1.0 × 10−5, figure 3) in a linear regression model accounting for age, sex, and diagnosis and explained 4.5% of the variation in plasma tau concentration. In a combined joint analysis incorporating the initial discovery cohort and the replication cohort, the association between plasma tau concentrations and rs242557 genotype was strengthened (p = 1.2 × 10−12). Fit into a joint regression model, each A allele of rs242557 increased plasma tau concentration by 0.38 pg/mL.
Figure 3. Plasma tau levels in a replication cohort as a function of rs242557 genotype.
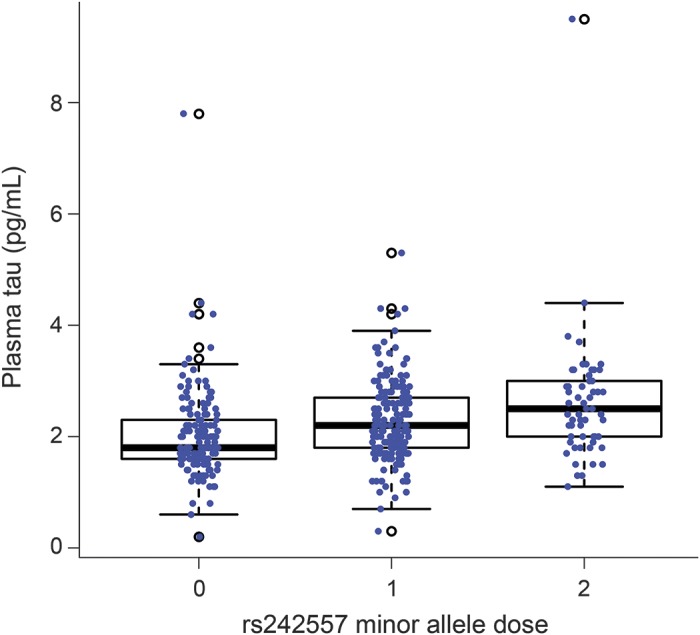
Plasma tau levels were compared across the GG, GA, and AA genotypes of rs242557 in an independent cohort of 387 participants to validate the initially observed association. A significant association of increasing plasma tau concentration with increasing minor allele (A) dose of rs242557 was observed (p = 1.0 × 10−5).
DISCUSSION
We present a GWAS of plasma tau levels in the ADNI cohort. We identified a genome-wide significant association of a SNP in the tau gene MAPT (rs242557) region with plasma tau levels and 3 additional suggestive association loci (in PARK2, IL2RA, and an intergenic region in 9p21.3). The minor allele of rs242557 (A) near MAPT and rs7047280 (C) in 9p21.3 was associated with higher plasma tau levels in a dose-dependent fashion, whereas minor alleles of SNPs near PARK2 (rs2187213-T) and IL2RA (rs7072793-C and rs7073236-C) were associated with lower plasma tau levels.
Several polymorphisms and mutations in and around MAPT confer risk for neurodegenerative tauopathies. The MAPT gene locus is located on chromosome 17q21.12,13 It exists as 2 major haplotype groups, H1 and H2, with the majority of individuals having the H1/H1 haplotype. Up to 25% of individuals in Western populations have a ≈970 kB sequence, including MAPT, oriented in the reverse orientation, precluding recombination and yielding a 1.3- to 1.6-MB region of linkage disequilibrium.13,14 Genetic studies, including GWAS, have identified both the inversion polymorphism and haplotype-specific polymorphisms influencing the risk of 4-repeat tauopathies (PSP and CBD).12,13,15–19 Moreover, the common subhaplotype H1c (tagged by rs242557) on the background of the H1 haplotype has been consistently found to be associated with both PSP and CBD.12,15,16,20 Previously published GWAS data with neuropathologically diagnosed cases showed that rs242557, tagging the MAPT H1c haplotype, was one of the most common SNPs associated with PSP (odds ratio = 1.96, p = 4.2 × 10−70) and CBD (odds ratio = 1.57, p = 7.91 × 10−6) (table 2).16,20 However, rs242557 was not associated with the risk of AD (p = 0.974),21 which is pathologically characterized by intracellular neurofibrillary tangles composed of roughly equal ratios of 3- and 4-repeat tau.1
The mechanism by which the MAPT H1c haplotype could increase plasma tau levels is not clear. The SNP rs242557 falls into a 182–base pair highly conserved region that was previously predicted to regulate MAPT expression (figure 2B).19 In cultured cells, the rs242557 A allele coupled with the H1 background promoter region had 2.7-fold greater transcription activity relative to the G allele on the H1 background and 4.2-fold greater activity relative to the H2 background.22 It has also been hypothesized that rs242557 could affect MAPT splicing,20,23 although this has not been supported by subsequent studies.24,25
Elevated plasma tau was not explained by elevated CSF tau. We did not identify a correlation between tau levels in plasma and CSF (figure 1), suggesting that different mechanisms are likely to regulate plasma tau concentrations. Previous GWAS have determined that the APOE locus is the strongest association for CSF tau and p-tau levels, although we have not observed an association between APOE genotypes and plasma tau concentrations.3,4 Although rs242557 was associated with higher CSF tau protein levels in one small sample of patients with AD (n = 89),26 both our data (n = 314) and another study with 313 individuals27 did not identify any association of this SNP with CSF tau protein levels. Multiple MAPT loci have been strongly associated with PSP and CBD, and some autosomal dominant MAPT mutations, particularly those in IVS10, are known to produce a PSP-like syndrome.28 Together, these data indicate that plasma tau levels might be a more useful endophenotype for identifying genetic risk for 4-repeat tauopathies (PSP and CBD) than for AD.
The alleles that we identified as suggestively associated with plasma tau might also be associated with risk for tauopathies. Mutations in the PARK2 gene, which encodes an E3 ubiquitin ligase, are the most common cause of early-onset parkinsonism.29,30 Interestingly, a PARK2 polymorphism (Val380Leu) is associated with lower risk of PSP, and PARK2 mutations produce clinical and pathologic features of PSP.31,32 IL2RA, a multiple sclerosis susceptibility gene, plays an important role in regulating immune response.33 Moreover, a SNP in IL2 (rs6852535, p = 1.3 × 10−7), which encodes the ligand for IL2RA, was identified as a suggestive locus for PSP risk in a previous GWAS.20 Recent data also suggest that microglia may play a role in tau-related neurodegeneration, which would be consistent with this association between an immunologic risk factor gene and plasma tau.34 Further studies are required to confirm these suggestive findings and to identify the potential roles of PARK2 and IL2RA in tauopathies.
A limitation of this report is the modest sample size, precluding stratified analyses for each diagnostic group. Furthermore, our study included data from HC and patients with AD spectrum disorders, raising the possibility that the identified associations result from confounding with AD pathology. Since rs242557 is a risk factor for non-AD tauopathies (CBD and PSP), this diminishes the possibility that an interaction between AD pathology and the H1c haplotype in our sample accounts for the association with plasma tau. Furthermore, the subgroup analysis showed that rs242557 (and the additional suggestive loci) was associated with plasma tau levels in a dose-dependent effect within each diagnostic group (HC, MCI, and AD, figure 2). This, along with replication of the rs242557 association in a cohort of participants largely without AD or other neurologic diseases, suggests that the presence of AD pathology is not necessary to observe the plasma tau association.
We detected a genome-wide significant SNP, rs242557, in MAPT and 3 suggestive loci (in PARK2, IL2RA, and an intergenic region in 9p21.3) associated with plasma tau levels measured in healthy elders and individuals with MCI or AD. Because rs242557 represents the MAPT H1c haplotype that has previously been identified as a major genetic risk factor for both PSP and CBD, our findings suggest that plasma tau concentration could be a useful endophenotype for identifying risk for 4-repeat tauopathies.
Supplementary Material
ACKNOWLEDGMENT
The authors thank contributors, including the staff at Alzheimer's Disease Centers who collected samples used in this study, patients, and their families whose help and participation made this work possible.
GLOSSARY
- AD
Alzheimer disease
- ADNI-1
Alzheimer's Disease Neuroimaging Initiative 1
- CBD
corticobasal degeneration
- GWAS
genome-wide association studies
- HC
healthy controls
- LD
linkage disequilibrium
- MCI
mild cognitive impairment
- p-tau
phosphorylated tau
- QC
quality control
- SNP
single nucleotide polymorphism
- t-tau
total tau
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Mr. Chen and Dr. Yu: design and conceptualization, analysis and interpretation of the data, drafting and revision of the manuscript. Mr. Wojta: experimental implementation, data collection and analysis. Dr. Wang: data analysis and interpretation of the data. Dr. Zetterberg and Dr. Blennow: experimental implementation, data collection and analysis, interpretation of the data, drafting and revision of the manuscript. Dr. Yokoyama: data analysis, drafting and revision of the manuscript. Dr. Weiner: experimental implementation, data collection, and interpretation of the data. Dr. Kramer, Dr. Rosen, and Dr. Miller: data collection and revision of the manuscript. Dr. Coppola: analysis and interpretation of the data, drafting and revision of the manuscript. Dr. Boxer: design and conceptualization, interpretation of the data, drafting and revision of the manuscript.
STUDY FUNDING
This study was supported by NIH grants U54NS092089, R01AG038791, and P01AG19724 and the Tau Research Consortium (Dr. Boxer). Mr. Chen is supported by an NIH–National Institute of Neurological Disorders and Stroke National Research Service Award (F31NS084556). Dr. Blennow receives research support from the Torsten Söderberg Foundation at the Royal Swedish Academy of Sciences and from the Research Council, Sweden. Dr. Yokoyama receives research support from the Association for Frontotemporal Degeneration Susan Marcus Memorial Fund Clinical Research Grant and the NIH–National Institute of Aging (K01AG049152). Data collection and sharing for this project were funded by the ADNI (NIH grant U01 AG024904) and Department of Defense ADNI (Department of Defense award W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, by the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc; Biogen; Bristol-Myers Squibb Co; CereSpir, Inc; Cogstate; Eisai Inc; Elan Pharmaceuticals, Inc; Eli Lilly and Co; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co, Inc; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corp; Pfizer Inc; Piramal Imaging; Servier; Takeda Pharmaceutical Co; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
DISCLOSURE
J. Chen founded and has an equity interest in Verge Genomics and serves as chief scientific officer. J. Yu serves as an associate editor-in-chief for Annals of Translational Medicine, is senior editor for Journal of Alzheimer's Disease, and has received research support from the National Natural Science Foundation of China (81471309). K. Wojta, H. Wang, and H. Zetterberg report no disclosures relevant to the manuscript. K. Blennow has served on advisory boards for IBL International, Roche Diagnostics, Eli Lilly, and Amgen and as a consultant for Novartis and Alzheon. J. Yokoyama, M. Weiner, J. Kramer, H. Rosen, B. Miller, and G. Coppola report no disclosures relevant to the manuscript. A. Boxer receives grant support from the NIH (U54NS092089, R01AG038791, and U01AG045390), the Tau Research Consortium, CBD Solutions, the Bluefield Project to Cure FTD, and the Alzheimer's Association; research support from Avid, BMS, Biogen, C2N, Cortice, Eli Lilly, Forum, Genentech, and TauRx for conducting clinical trials; personal compensation for consulting for Asceneuron, Ionis Pharmaceuticals, Janssen, and Merck and for serving on a DSMB for Neurogenetics; and stock/options for serving on a scientific advisory board for Alector and Delos. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001;24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 2.Jack CR, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron 2013;80:1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cruchaga C, Kauwe JS, Harari O, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron 2013;78:256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim S, Swaminathan S, Shen L, et al. Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology 2010;76:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagshal D, Sankaranarayanan S, Guss V, et al. Divergent CSF alterations in two common tauopathies: Alzheimer's disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2014;86:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosso SM, van Herpen E, Pijnenburg YA, et al. Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch Neurol 2003;60:1209–1213. [DOI] [PubMed] [Google Scholar]
- 7.Olivera A, Lejbman N, Jeromin A, et al. Peripheral total tau in military personnel who sustain traumatic brain injuries during deployment. JAMA Neurol 2015;72:1109–1116. [DOI] [PubMed] [Google Scholar]
- 8.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science 1996;273:1516–1517. [DOI] [PubMed] [Google Scholar]
- 10.Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. bioRxiv 2014. Available at: http://biorxiv.org/content/early/2014/05/14/005165. Accessed July 1, 2016. [Google Scholar]
- 11.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26:2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pittman AM, Myers AJ, Abou-Sleiman P, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet 2005;42:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pittman AM, Myers AJ, Duckworth J, et al. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet 2004;13:1267–1274. [DOI] [PubMed] [Google Scholar]
- 14.Zody MC, Jiang Z, Fung HC, et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 2008;40:1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams DR, Pittman AM, Revesz T, Lees AJ, de Silva R. Genetic variation at the tau locus and clinical syndromes associated with progressive supranuclear palsy. Mov Disord 2007;22:895–897. [DOI] [PubMed] [Google Scholar]
- 16.Kouri N, Ross OA, Dombroski B, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun 2015;6:7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conrad C, Andreadis A, Trojanowski JQ, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol 1997;41:277–281. [DOI] [PubMed] [Google Scholar]
- 18.Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 1999;8:711–715. [DOI] [PubMed] [Google Scholar]
- 19.Rademakers R, Melquist S, Cruts M, et al. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet 2005;14:3281–3292. [DOI] [PubMed] [Google Scholar]
- 20.Höglinger G, Melham N, Dickson D, Sleiman P, Müller U. V37 common variants affect risk for the tauopathy progressive supranuclear palsy. Basal Ganglia 2011;1:14. [Google Scholar]
- 21.Allen M, Kachadoorian M, Quicksall Z, et al. Association of MAPT haplotypes with Alzheimer's disease risk and MAPT brain gene expression levels. Alzheimers Res Ther 2014;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers AJ, Pittman AM, Zhao AS, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 2007;25:561–570. [DOI] [PubMed] [Google Scholar]
- 23.Caffrey TM, Joachim C, Paracchini S, Esiri MM, Wade-Martins R. Haplotype-specific expression of exon 10 at the human MAPT locus. Hum Mol Genet 2006;15:3529–3537. [DOI] [PubMed] [Google Scholar]
- 24.Trabzuni D, Wray S, Vandrovcova J, et al. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet 2012;21:4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majounie E, Cross W, Newsway V, et al. Variation in tau isoform expression in different brain regions and disease states. Neurobiol Aging 2013;34:1922.e7–1922.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laws SM, Friedrich P, Diehl-Schmid J, et al. Fine mapping of the MAPT locus using quantitative trait analysis identifies possible causal variants in Alzheimer's disease. Mol Psychiatry 2006;12:510–517. [DOI] [PubMed] [Google Scholar]
- 27.Kauwe JSK, Cruchaga C, Mayo K, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA 2008;105:8050–8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris HR, Osaki Y, Holton J, et al. Tau exon 10 +16 mutation FTDP-17 presenting clinically as sporadic young onset PSP. Neurology 2003;61:102–104. [DOI] [PubMed] [Google Scholar]
- 29.Schrag A, Schott JM. Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol 2006;5:355–363. [DOI] [PubMed] [Google Scholar]
- 30.Lücking CB, Dürr A, Bonifati V, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]
- 31.Ros R, Ampuero I, García de Yébenes J. Parkin polymorphisms in progressive supranuclear palsy. J Neurol Sci 2008;268:176–178. [DOI] [PubMed] [Google Scholar]
- 32.Morales B, Martínez A, Gonzalo I, et al. Steele-Richardson-Olszewski syndrome in a patient with a single C212Y mutation in the parkin protein. Mov Disord 2002;17:1374–1380. [DOI] [PubMed] [Google Scholar]
- 33.International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007;357:851–862. [DOI] [PubMed] [Google Scholar]
- 34.Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 2015;18:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.