

Abstract
Introduction
Thymic epithelial tumors (TETs) are rare tumors originating from the epithelia of the thymus, and therapeutic options beyond surgery are limited. Pathogenesis of TETs is poorly understood, and the scarcity of model systems for these rare tumors makes the study of their biology very challenging.
Methods
A new cell line (MP57) was established from a thymic carcinoma specimen and characterized using standard biomarker analysis, as well as next generation sequencing (NGS), and functional assays. Sanger sequencing was used to confirm the mutations identified by NGS.
Results
MP57 possesses all the tested thymic epithelial markers and is deemed to be a bona fide thymic carcinoma cell line. NGS analysis of MP57 identified a mutation in PIK3R2, a regulatory subunit of PI3K. Further analysis identified different mutations in multiple PI3K subunits in another cell line and several primary thymic carcinoma samples, including two catalytic subunits (PIK3CA and PIK3CG) and another regulatory subunit (PIK3R4). Inhibiting PI3K with GDC0941 resulted in in vitro antitumor activities in TET cells carrying mutant PI3K subunits.
Conclusions
Alterations of PI3K, due to mutations in its catalytic or regulatory subunits, is observed in a subgroup of TETs, in particular thymic carcinomas. Targeting PI3K may be an effective strategy to treat these tumors.
Keywords: Thymic epithelial tumor, PI3K, Mutation, PI3K inhibitor
Introduction
Thymic Epithelial Tumors (TETs) are rare tumors originating from epithelial cells of the thymus with an annual incidence rate of 0.13 per 100,000 persons in the United States. Based on the World Health Organization (WHO) classification system, TETs are histologically classified into thymomas types A, AB, B1, B2, and B3, and thymic carcinomas.1 Thymomas maintain the architectural structure of the thymus, whereas thymic carcinomas are not recognizable from carcinomas that arise in other sites. The WHO histological classification has prognostic value,2,3 and thymomas types A, AB, and B1 have a 10-year survival rate of more than 80%.4 Thymomas B2 ad B3 have an intermediate behavior and thymic carcinomas are aggressive tumors: they exhibit cytological atypia and high histological heterogeneity, lack typical structure of the thymus, and have higher metastatic potential, with 5-year survival rates at approximately 50%.5,6 Surgical resection is the main therapeutic intervention for TETs at early stages, while advanced and recurrent disease are treated with chemotherapy.7,8–13
Although targeted therapies have produced successful results in other cancer types in selected patient populations, only a few clinical trials have shown some efficacy with targeted treatments in TETs.2,11,14,15 This is largely due to lack of understanding of the biology of these tumors. However, recent genetic studies using comparative genomic hybridization, whole genome expression analysis and next generation sequencing have provided new insights in the biology of this rare cancer. Thymic carcinomas have a distinct molecular signature that segregates them from the thymoma subtypes.6,16–23 Such genetic differences, combined with the histology-based classification system, are clinically relevant for both prognosis and treatment of TETs and may contribute to identification of new molecular targets for therapeutic intervention.3,7,19,20
The rarity of TETs represents a great challenge to the study of the biology of these tumors as well as to the development of novel therapeutic strategies. The challenge is in large part due to the shortage of model systems and currently there are only six cell lines available for this tumor type.24–28 Here, we report the establishment of a new cell line (designated as MP57) derived from a patient with thymic carcinoma. Through characterization of MP57 along with other cell lines and primary tumors, we identified 5 actionable mutations in 4 different subunits of PI3K. These mutations were mostly found in thymic carcinomas. We further demonstrated that inhibiting PI3K has in vitro antitumor activity in the TET cells carrying mutant PI3K subunits.
Materials and Methods
Cell Lines
MP57 is a thymic carcinoma cell line newly established from a tumor specimen collected at autopsy. The patient history has been described elsewhere.9 Tumor cells were retrieved through autopsy from the primary mediastinal mass. The solid tumor was dissociated into single cell suspension using gentleMACs dissociator (Miltenyil Biotech, San Diego, CA, USA). After dissociation, the sample was cultured in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin (Gibco, Life technologies, Grand Island, NY, USA) in an incubator with 5% CO2 atmosphere at 37°C. Medium was replaced every 3–4 days. T1889 is a thymic carcinoma cell line kindly provided by Dr. Marco Breinig,29 and IU-TAB1 is a type AB thymoma cell line kindly provided by Dr. Sledge GW Jr. 27 All these cell lines are cultured in RPMI plus 10% FBS and 1% penicillin/streptomycin.
Immunofluorescence (IF) Staining
MP57 cells were seeded on 12 mm cover glass coated with Poly-L-Lysine (Corning, NY, USA). Sixteen hours later, cells were fixed in 4% formaldehyde for 15 minutes, and incubated with blocking buffer (1× PBS pH7.4 plus 8% BSA and 0.3% Triton™ X-100) for 60 minutes. Cells were then incubated for 1 hour at room temperature (RT) with different primary antibodies, followed by incubation with the secondary antibody AlexaFluor488 and DAPI for 90 minutes. After IF staining, the cover glasses were mounted with cover slips, and photos were taken on a Fluorescence Microscope. Primary antibodies for EpCAM, E-Cadherin, c-KIT, p63, Vimentin and N-cadherin were purchased from Cell Signaling Technology (Danvers, MA). Anti-Cytokeratin clone AE1/AE3 antibody was from Dako (Carpinteria, CA), and the AlexaFluor488 and DAPI were purchased from Invitrogen Life technologies (Grand Island, NY).
Flow Cytometric Analysis
Cells were trypsinized, washed in 1x PBS pH7.4, centrifuged at 350× g for 5 minutes, and resuspended in ice-cold staining buffer (1x PBS pH7.4, plus 1% BSA). Cells were then incubated with the fluorochrome-conjugated antibodies for 20 minutes on ice, and flow cytometric analysis of the stained cells was performed using FACS Calibur (BD, San Jose, CA, USA). APC anti-human c-Kit (clone 104D2), PE anti-human EpCAM (clone 9CD), and Pe/Cy7 anti-human E-Cadherin antibodies were purchased from BioLegend (San Diego, CA, USA). Anti-Fibroblast-PE (human; clone: REA165) was obtained from MACS Miltenyl Biotec, (San Diego, CA).
Cell Proliferation Assay
Cells were seeded at 5000 cells/well density in 96-well plates for CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI). The plates were incubated for 24, 48 or 72 hours at 37°C and 5% CO2 before adding 100 μl of Cell Titer-Glo reagent to lyse the cells. After 10 minutes incubation period at room temperature, the luminescent intensity was measured using GloMax Multidetection system (Promega, Madison, WI, USA).
The Trypan Blue dye exclusion assay was used as an alternative method to assess cell proliferation. Cells (5 × 104 cells/well) were seeded in 24-well plates. After 24, 48 and 72 hours, cells were detached from the well using TrypLe (Gibco, Life technologies, Grand Island, NY), and resuspended in medium. Cell viability was evaluated by adding Trypan Blue solution (0.4% in phosphate buffered saline PBS) (Gibco, Life technologies, Grand Island, NY) to the cell suspension. After 3 minutes incubation, the unstained cells (non-viable) and stained cells (viable) were counted in a hemacytometer.
Drug Sensitivity Testing
Cells were plated at 5,000 cells/well in 96-well plates in growth medium. After overnight culturing, cells were treated with/without GDC-0941 (Selleck Chemicals LLC, Houston, TX) for 72 hours. Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO) was used as solvent for GDC-0941 and as treatment control. Cell viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI), according to the manufacturer’s instructions. The assay was performed in triplicate for each experiment.
Cell Cycle Analysis
Cells were plated at a density of 200,000 cells per well in 24-well plates and incubated at 37°C overnight. Subsequently, cells were treated with DMSO or GDC-0941 for 24 hr. Cells were then washed, trypsinized, fixed with ethanol and analyzed using FACS Calibur, and the data were processed with FLOWJO (Tree Star).
Apoptosis Assay
Apoptosis was measured by Annexin V/propidium iodide (PI) staining (Life technologies, Grand Island, NY), followed by FACS analysis.
Western Blotting
Cell lysates were extracted in ice-cold RIPA lysis buffer (ThermoFisher, Waltham, MA) containing protease/phosphatase inhibitor cocktail (Roche). Equal amounts of total protein were separated on polyacrylamide gels and then transferred onto PVDF membranes (Life technologies, Grand Island, NY). The membranes were incubated with blocking buffer for 1 hour at RT and then with the primary antibodies for overnight at 4°C. The following day, the membranes were incubated with the appropriate peroxidase-conjugated secondary antibodies, and proteins were detected by an ECL detection system (Pierce, ThermoFisher, Waltham, MA). Anti-p-AKT (S473), anti-AKT, anti-p-ERK, and anti-ERK antibodies were purchased from Cell Signaling, and anti-ß-actin antibody was obtained from Sigma.
Transwell Migration Assay
Cells were seeded in polycarbonate membrane filter inserts (8.0 μm pore size) in 6-well Transwells (Corning Life Sciences, Acton, MA) at 5×105 per well with 2 ml medium in the upper insert chamber and 1 ml medium in the lower chamber. After 16 hours incubation with/without GDC-0941, cells at the lower chamber were stained with crystal violet and counted, according to the manufacturer’s instructions.
Xenograft Tumor Growth in Mice
Cells (1×106 cells/mouse) were subcutaneously inoculated into 4–6 weeks old female immunocompromised athymic nude mice (Charles River, Charles River, MA). Tumors were measured weekly by vernier caliper, and the volumes were calculated using the equation V= ½ × (L × W2). Tumor sections were stained by hematoxylin and eosin (H&E) for histology or by immunohistochemistry for detection of p63 (Ventana Medical Systems, Inc., Tucson, AZ) and cytokeratin-7 (CK7) (Santa Cruz Biotechnology, Inc., Dallas, TX).
Genomic DNA Extraction
Genomic DNA was extracted from cell line pellet, whole blood or formalin-fixed paraffin-embedded (FFPE) tissue using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). DNA was quantified using the Quantifluor ONE dsDNA kit with the K562 genomic DNA standard on the GloMax-Multi+ Microplate Reader (Promega, Valencia, CA), following the manufacturer’s protocol.
Next-Generation-Sequencing Library Construction
Briefly, each DNA sample was sheared to a target size of 150 to 200 base pairs (bp) using the Covaris M220 Focused-ultrasonicator (Covaris, Woburn, MA). MP57 samples and IU-TAB1 sheared DNA samples were constructed into individual libraries using the SureSelect XT reagent kit (Agilent Technologies, Santa Clara, CA) or the Personal Genome Diagnostics (PGDx) custom library kit (PGDx, Baltimore, MD), according to the manufacturers’ protocols. Each library was hybridized to RNA baits from the SureSelect Human Kinome v1 panel (Agilent Technologies, Santa Clara, CA, USA) or our PGDx custom gene panel (PGDx, Balitmore, MD) for 24 hours and captured using streptavidin-coated beads. A unique index tag was added to each library sample by a 12-cycle PCR amplification. Quality and quantity of the indexed libraries were assessed using the High Sensitivity DNA 1000 kit on the 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA). The indexed libraries were combined into 4 nM pools and sequenced on the MiSeq.
MiSeq Sequencing and Data Analysis
Paired end sequencing was performed on the MiSeq according to the manufacturer’s protocols (Illumina, San Diego, CA). The Human Kinome panel targets over 500 kinases and 612 genes in total, which equates to a 3.2 megabase pair (Mb) region size. The PGDx custom gene panel (1.3 Mb region size) targets the exons of 206 cancer genes and also evaluates specific genes for copy number variation and translocations. A complete list of the genes in the Human Kinome panel can be found on Agilent Technologies website, while the full list of the targeted genes in the PGDx custom panel can be found in the supplementary material (Supplementary Table 1). Alignment to the human reference genome 19 (GRCh37, UCSC hg19 assembly), quality trimming, and variant calling were executed by the MiSeq Reporter software or the PGDx custom server analysis pipeline.
Annotations and filtering of all variants were completed through Illumina’s VariantStudio software v2.2.1. The databases used to annotate variants included VEP v2.8 (Ensembl, SIFT, PolyPhen), 1000 Genomes, COSMIC version 65, ClinVar (2013), dbSNP v137, RefSeq, NHLBI Exome Variant Server, and UCSC (hg19). The following filters were applied to all variants called in the FFPE tumor: (1) PASS filter - variant not flagged for low coverage depth, low genotyping, low quality, low variant frequency, indel repeat greater than 8, and strand bias; (2) Mapping quality score greater than 30; (3) Read depth greater than 30; (4) Synonymous variants and non-coding (intron) variants found outside of splicing regions were filtered out; (5) removal of variants that also exist in the paired normal blood sample.
RESULTS
Establishment and characterization of MP57 thymic carcinoma cell line
A 45-year-old male with history of 30 packs/year smoking and 11 years of asbestos exposure initially presented with a two-week history of cough, upper respiratory symptoms and worsening left shoulder pain radiating to the forearm. A CT and PET-FDG scanning revealed an anterior mediastinal prevascular mass (7.4 cm), several positive mediastinal nodes and a lesion in the right upper lobe of the lung with no extra thoracic disease. Biopsy of the mediastinal mass was performed by anterior mediastinotomy, and pathology revealed a poorly differentiated carcinoma compatible with a diagnosis of thymic carcinoma stage IVa. Patient was enrolled on phase I trial of belinostat in association with PAC chemotherapy (cisplatin, doxorubicin, and cyclophosphamide).8 The patient received only two cycles of therapy due to disease progression with increased size of the mediastinal mass and initial pericardial invasion. Patient was then enrolled in a phase II trial of single agent sunitinib.9 Unfortunately, he progressed very rapidly, developed extreme fatigue, and died shortly thereafter of right ventricular heart failure.
An autopsy was performed, and tumor tissue was collected from the mediastinal mass and processed for the establishment of MP57 cell line as detailed in the Materials and Methods section. The morphology of MP57 cells at passage 16th and 30th are identical with polygonal shape (Fig. 1A). They grew adherently as a monolayer and have been cultured for more than 80 passages. Immunofluorescence analysis showed that MP57 cells retained the positivity of several thymic epithelial markers including cytokeratin clone AE1/AE3, EpCAM, E-Cadherin, c-KIT, and p63 (Fig. 1B). The expression of these epithelial markers on the cells was also confirmed by flow cytometric analysis (Fig. 1C). There was no sign of fibroblast component present in the population, as determined by the absence of human fibroblast marker (Fig. 1C) and the negative staining for Vimentin and N-Cadherin (Supplementary Fig. 1).
Figure 1.
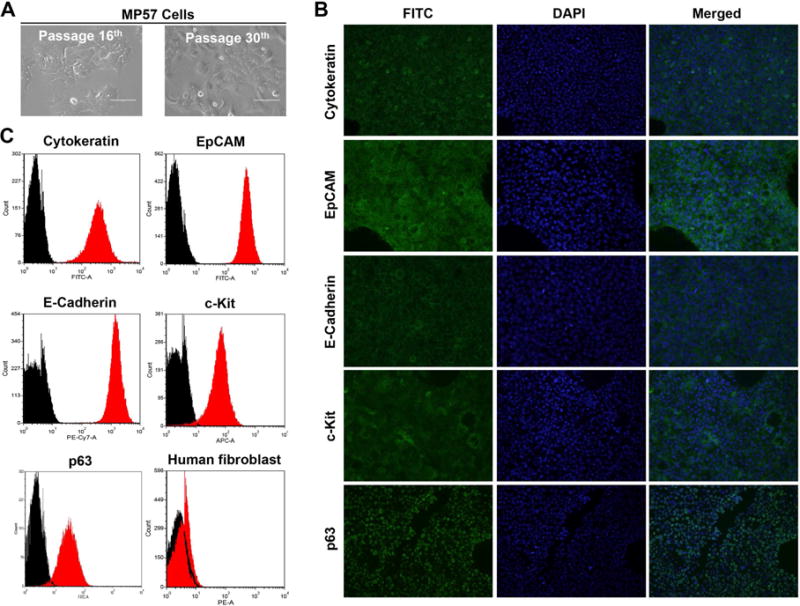
Morphology and characterization of MP57 primary cells. (A) Photographs of MP57 morphology at passage 16th and 30th, taken under a light inverted microscope (magnification 40×). (B) Immunofluorescence staining of thymic epithelial markers in MP57. The staining of cytokeratin clone AE1/AE3, EpCAM, E-Cadherin, c-Kit, and p63 are indicated by FITC. DAPI was used for staining of nuclei. (C) Cell surface marker analysis of MP57 by flow cytometry. Cytokeratin clone AE1/AE3, EpCAM, p63, c-Kit, and E-Cadherin, and human fibroblast antibody were analyzed. Black histogram represents control isotypes.
MP57 cells are tumorigenic in athymic nude mice
The doubling time of MP57 cells was approximately 24 hours, comparable to that of T1889 and IU-TAB1 cells as determined by CellTiter-Glo Luminescent Cell Viability Assay and Trypan Blue dye exclusion assay (Fig. 2A–B). We then evaluated tumorigenicity of MP57 cells in vivo. The cells (passage 5) were embedded in matrigel and inoculated subcutaneously into 10 athymic nude mice (1×106 cells/mouse). The xenograft tumors became apparent in about 2 weeks after inoculation, and in five weeks the average tumor size reached 200 mm3 (Fig. 2C–D), indicating that MP57 is tumorigenic.
Figure 2.
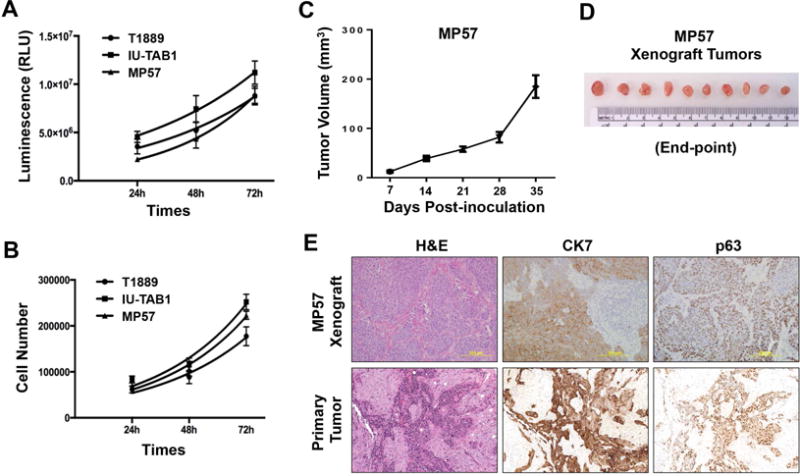
Proliferative and tumorigenic activities of MP57 cells. Cell proliferation was measured by (A) CellTiter Glo Viability assay, and (B) Trypan-Blue dye exclusion method. For comparison, T1889 and IU-TAB1 cells were also examined. (C) Tumorigenicity of MP57 cells in immunocompromised athymic nude mice (n=10). The curve represents the growth of subcutaneous tumors within the 5-week window. (D) MP57 xenograft tumors at the experimental end-point (day-35 post-inoculation). (E) Histological comparison between MP57 xenograft tumor and the primary tumor obtained from patient autopsy. Morphology of tumor cells was examined by H&E staining. The expression of cytokeratin 7 and p63 was determined by immunohistochemistry.
The xenograft tumors were then harvested and processed for histological analysis. The morphology of MP57 cells in the xenograft tumor resembled that of the primary tumor from the patient autopsy, and they both were positive for cytokeratin 7 (CK7) and p63, two thymic epithelial markers (Fig. 2E). These data confirmed that MP57 is a bona fide thymic carcinoma cell line.
Mutation analysis of MP57 primary tumor and cell line
We utilized a targeted exome sequencing approach using the MiSeq platform to profile genetic mutations in MP57. The MiSeq Kinome sequencing run that included the blood, cell line, and FFPE tumor samples yielded 5.5 gigabases (Gb) of data with 94.5% >Q30 (5.3 Gb). The median read depth of the targeted regions was 258, and in the tumor, 97.7%, 95.1%, and 61.7% of target regions were sequenced to at least 20, 100 and 500 reads, respectively. Each filtered variant location was viewed concurrently in IGV between the paired blood, cell line, and tumor sample. We focused on variants that had a >30% allelic frequency in the primary tumor, greater than 70% allelic frequency in the MP57 cell line, and zero frequency (100% wild type) in the blood of the same patient. After variant filtering, 6 single nucleotide variations (SNVs) and 1 deletion in the coding regions of the detected genes were revealed in both MP57 primary tumor and cell line (Supplementary Table 2). Among them are a TP53 frame shift mutation, which was identified in the thymic tumor biopsy from the same patient by an NGS study conducted previously,22 validating the power of the MiSeq approach. Other mutations were identified in the genes of PIK3R2, TAF1, CSNK2A3, SGK223, and TTN (Supplementary Table 2).
To further investigate the mutation profile of the MP57 cell line, we also sequenced it using our PGDx custom gene panel, which caters towards cancer-associated genes. The FFPE tumor was not sequenced with this panel due to insufficient input of DNA. The custom gene panel run yielded 6.17 Gb of data with 94% >Q30 (5.9 Gb), The MP57 cell line produced 525 Mb of data and 3.5 million reads with a 131 mean read depth. Additional mutations were discovered in the genes that were not included in the Kinome panel (Supplementary Table 3). MiSeq using PGDx custom panel also confirmed the TP53 frame shift mutation and detected a 4-fold amplification of the PIK3CA gene.
PI3K subunit mutations in thymic carcinomas
The PIK3R2 mutation identified in MP57 thymic carcinoma cell line using the Kinome panel is of particular interest. PIK3R2 is one of the regulatory subunits of PI3K, which consists of catalytic and regulatory subunits and requires both functional components for its kinase activity.30 Using Sanger sequencing, we confirmed that the mutation in PIK3R2 is a homozygous G>C missense mutation resulting in G373R amino acid substitution in the MP57 cell line (Fig. 3A and Supplementary Table 2). This mutation matches a COSMIC entry (COSM993028) and is considered deleterious and probably damaging, according to SIFT and PolyPhen analyses (Table 1).
Figure 3.
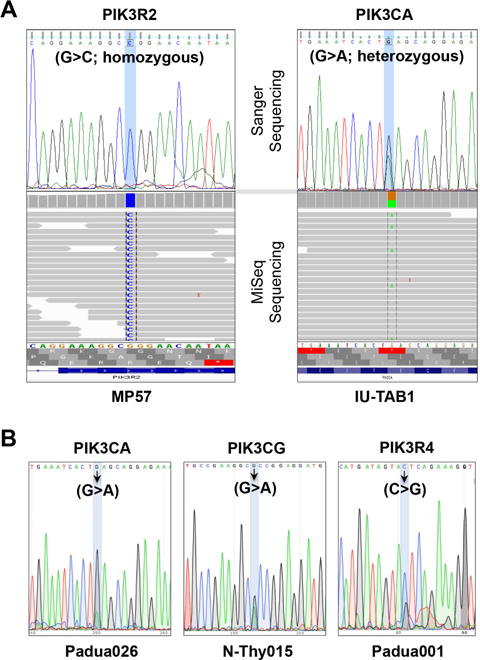
Identification of mutations in the genes encoding subunits of PI3K in TET samples. Sanger sequencing confirmation of mutations identified by MiSeq in (A) PIK3R2 in MP57 cells and PIK3CA in IU-TAB1 cells. (B) Sanger sequencing of mutations identified in PIK3CA, PIK3CG, and PIK3R4, in three TET primary tumor samples, respectively. Arrows indicate the mutated nucleotides.
Table 1.
Summary of PI3K subunit gene mutations identified in TET cell lines and primary tumors
| Sample ID | Sample Type | WHO Histotype | Mutation | Gene Symbol | PI3K Subunit | Effect | SIFT/PolyPhen | Sanger Seq |
|---|---|---|---|---|---|---|---|---|
| MP57 | Cell line & Tumor | Thymic Carcinoma | chr19:18273784G>C c.1117G>C p.G373R | PIK3R2 | p85β, Regulatory | Missense Mutation | deleterious/probably damaging | Homozygous |
| IU-TAB1 | Cell line | Thymoma Type AB | chr3:178936091G>A c.1633G>A p.E545K | PIK3CA | p110α, Catalytic | Missense Mutation | deleterious/probably damaging | Heterozygous |
| N-Thy015 | Tumor | Thymic Carcinoma | chr7:106508065G>A c.3309G>A p.R20H | PIK3CG | p110γ, Catalytic | Missense Mutation | deleterious/probably damaging | Confirmed |
| Padua001 | Tumor | Thymic Carcinoma | chr3:130427227C>G c.579C>G p.S173T | PIK3R4 | p150, Regulatory | Missense Mutation | benign/tolerated | Confirmed |
| Padua026 | Tumor | Thymic Carcinoma | chr3:178936091G>A c.1068G>A p.E545K | PIK3CA | p110α, Catalytic | Missense Mutation | deleterious/probably damaging | Confirmed |
Interestingly, a mutation in a different subunit of PI3K, along with other gene mutations, was also identified in the type AB thymoma cell line IU-TAB1 through MiSeq sequencing analysis using the PGDx custom gene panel (yielding 1.1 Gb of data and 7.3 million read with a 149 mean read depth) (Supplementary Table 4). The mutation is in the PIK3CA, a catalytic subunit of PI3K, and Sanger sequencing confirmed it as a heterozygous G>A mutation (Fig. 3A). This missense mutation causes an amino acid substitution of E545K in PIK3CA, which is also deleterious and possibly damaging with multiple COSMIC entries (Supplementary Table 4).
We then retrospectively analyzed the NGS data from a cohort of 54 human TET samples reported previously; among them, 28 samples was sequenced by whole exome sequencing, and the other 26 by targeted exome sequencing using a 197-gene panel in which only PIK3CA from the family was included.22 We found three different mutations in three different subunits of PI3K, and to our surprise, all these mutations were found in thymic carcinomas (Table 1). The three PI3K subunits found to have mutations in these primary tumors are PIK3CG catalytic subunit, PIK3R4 regulatory subunit, and PIK3CA. All three mutations were confirmed with Sanger sequencing (Fig. 3B). These gene mutation data suggest that alteration of PI3K activity may play a critical role in the pathogenesis of thymic carcinoma.
In vitro antitumor activities of PI3K inhibition in TET cells
We examined the effect of PI3K inhibition on the signaling and behavior of TET cells with/without mutations in the subunits of PI3K using GDC-0941, a pan-PI3K inhibitor. GDC-0941 treatment resulted in a dose-dependent decrease of AKT phosphorylation in MP57 and IU-TAB1, as well as in T1889 cells (Fig. 4A), indicating that GDC-0941 can inhibit the signaling downstream of both mutant and wild type PI3K. In parallel, GDC-0941 significantly inhibited the cell viability of MP57, IU-TAB1, and T1889 (Fig. 4B). IU-TAB1 cells, which harbor an activating mutation in the PIK3CA, displayed the best sensitivity to GDC-0941 (IC50 134 nM), whereas PI3K-wild-type T1889 cells were least sensitive (IC50 at 416 nM) among the three tested cell lines. In MP57 cells, it is possible that both PIK3R4 mutation and PIK3CA amplification contribute to aberrant activation of AKT.
Figure 4.
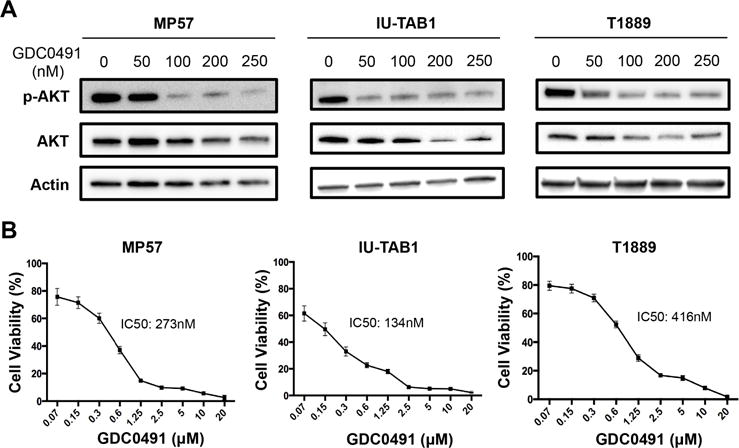
PI3K inhibition suppressed AKT activation and reduced viability of TET cells. (A) Western blot analysis of AKT phosphorylation in MP57, IU-TAB1, and T1889 cells treated with the indicated concentration of GDC-0941 for 6 hours. As controls, total AKT and β-actin were also probed. (B) The viability of the indicated cells treated with increasing concentrations of GDC-0941 (ranging from 70nM to 20 μM) for 72 hours, determined by CellTiter Glo Viability assay. The IC50 concentrations were calculated using GraphPad Prism program. The assay was performed in triplicate, and the error bars represent standard deviations.
To determine the effect of GDC-0941 on cell cycle progression, we treated both MP57 and IU-TAB1 cells with GDC-0941 at the indicated concentrations (0, 200nM, 500nM and 1 μM) and performed FACS analysis 24 hours after treatment (Fig. 5A). We observed a significant increase of cell numbers in the G0-G1 phase, particularly in the IU-TAB1 cells treated with GDC-0941, indicating that GDC-0941 induce G1 arrest in these cells. Interestingly, prolonged exposure of MP57 cells to the inhibitor resulted in significant increase of apoptotic cells as determined by Annexin V and PI staining, but such effect was less significant in the GDC-0941-treated IU-TAB-1 cells (Fig. 5B). In addition, oncogenic PI3K is also known to play a role in cancer cell invasion and metastasis.31 We then performed migration assays to assess the effect of PI3K inhibition by GDC-0941 on the motility of MP57 cells. Compared to DMSO control, GDC-0941 treatment resulted in a strong inhibition of MP57 cell migration in a dose-dependent fashion (Fig. 5C). These data suggest that blocking PI3K in TET cell lines has antitumor activity, especially in those with mutations in catalytic or regulatory subunits of PI3K.
Figure 5.
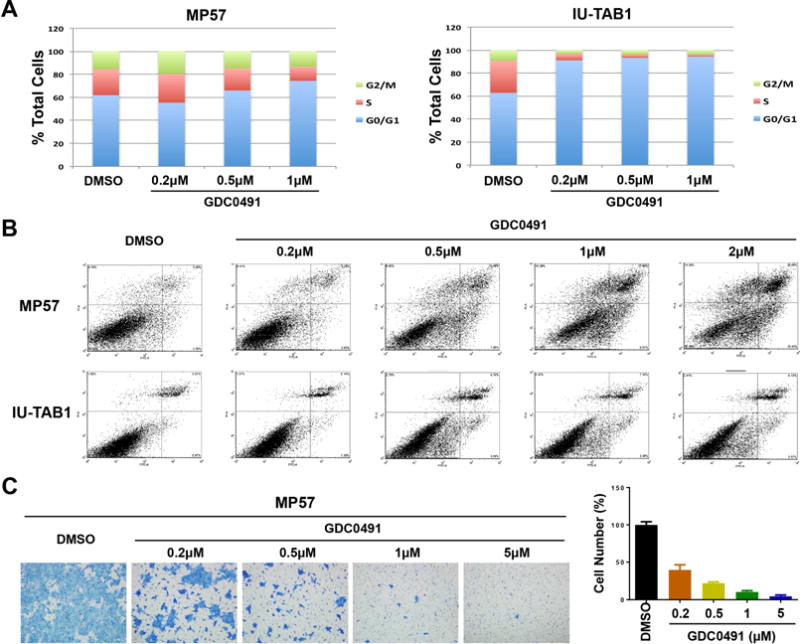
Effects of GDC-0941 on cell cycle, apoptosis, and motility of PI3K mutated TET cells. (A) Cell cycle analysis of MP57 and IU-TAB1 cells treated with the indicated concentrations of GDC-0941 for 72 hours, determined by flow cytometry of propidium iodide (PI) staining. DMSO treatment was used as the controls. (B) Analysis of apoptosis by flow cytometry using Annexin V and PI staining of the cells treated with the indicated concentrations of GDC-0941 for 72 hours. (C) Migration assay of MP57 cells treated with/without GDC-0941. Migrating cells were visualized by crystal violet staining (left), and the quantification was shown in the graph bars (right). Error bar represents standard deviation.
Discussion
Human cell lines derived from tumors represent important model systems for studying tumor biology and for evaluating drug efficacy. For TETs, such resources are very limited. In this study, we report a newly established thymic carcinoma cell line, MP57, which expresses all tested thymic epithelial markers and is tumorigenic in immune-compromised mice. Targeted exome sequencing of MP57 genomic DNAs revealed a homozygous loss-of-function mutation in the tumor suppressor TP53, causing frame-shift and truncation of the protein. We recently showed that mutations in TP53 are a frequent event in thymic carcinoma and are associated with poor prognosis.32
Importantly, in MP57 cells, we also identified a homozygous missense mutation in the PIK3R2 gene, which encodes one of the regulatory subunits of PI3K, p85β. This mutation results in a substitution of Gly373Arg in p85β subunit, and is considered to be deleterious/probably damaging by SIFT/PolyPhen prediction. This gene mutation has been previously reported in endometrial cancer, leading to enhanced PI3K activity and activation of its downstream AKT.33 Mutations of various genes in the PI3K pathway have been frequently detected in head and neck squamous cell carcinoma (HNSCC) and endometrial cancer.33,34 By extending our search for such alterations in TETs, we identified three additional mutations in four samples including three primary tumors of thymic carcinomas and one type AB thymoma cell line, IU-TAB1. These missense mutations are in the PIK3CG, PIK3R4 or PIK3CA gene, resulting in amino acid substitution of Arg20His in p110γ (catalytic subunit), Ser173Thr in p150 (regulatory subunit), or Glu545Lys in p110α (catalytic subunit), respectively. In HNSCC and endometrial cancer, PIK3CA was found to be the most frequently mutated gene among the genes encoding the subunits of PI3K, and its Glu545Lys mutation has been reported in both cancer types.33,34
Deregulation of the PI3K pathway plays a critical role in the development and progression of cancer, and has been frequently implicated in a wide spectrum of malignancies, including glioma, prostate, breast, ovarian, and endometrial cancer.6,35,36–38 PI3K has many important biological activities, especially cell proliferation and survival via activation of its downstream AKT and the mammalian target of rapamycin (mTOR). Components of the PI3K pathway are attractive molecular targets for cancer intervention, and a number of inhibitors targeting this pathway have been developed.39 GDC-0941 (Pictilisib) is an oral bioavailable, highly selective class I PI3K inhibitor.40 The antitumor activity of GDC-0941 has been assessed as a single agent and in combinations with various drugs including erlotinib, fulvestrant, and trastuzumab.41–45 Supported by preclinical studies, GDC-0941 is currently in early phase clinical trials in patients with advanced solid tumors.46,47 Patients with PIK3CA mutation showed relatively better response to PI3K/AKT/mTOR inhibitors in early phase trials, especially in those with PIK3CA H1047R mutation.48 Nonetheless, not all patients with PIK3CA mutations responded to those inhibitors, in part because such mutations often coexist with other genetic alterations such as KRAS and BRAF mutations.49–54 Thus, targeting PI3K alone might not be sufficient to achieve meaningful clinical response in those cases.
The identification of mutations in the subunits of PI3K in TETs has not been previously reported. Here, we reported not only the identification of PI3K subunit mutations, but also the antitumor activities of PI3K inhibitor in the TET cells. Using GDC-0941, we demonstrated that inhibiting PI3K resulted in a suppression of its downstream AKT and a dose-dependent inhibition of cell viability in the PI3K mutated MP57 and IU-TAB1 as well as in PI3K wild type T1889 cells. Although it is unclear whether different PI3K mutations might have different impact on its downstream signaling and biological activities, we found that GDC-0941 caused apoptosis in MP57 cells, but induced G1 cell cycle arrest in IU-TAB1 cells without clear indication of apoptosis. Noteworthy, MP57 is a thymic carcinoma cell line harboring a mutation in the PIK3R2 encoding a regulatory subunit, whereas IU-TAB1 is a type AB thymoma harboring a mutation in the PIK3CA encoding a catalytic subunit. Our data indicate that PI3K with mutations in different subunits might exert its oncogenic activities by preferentially engaging selective pathways, for instance, cell cycle arrest versus apoptosis.
Taken together, we have established a new thymic carcinoma cell line (MP57) and identified mutations in the genes encoding different subunits of PI3K in multiple TETs. We also demonstrated that PI3K inhibitor has antitumor activities in TETs in vitro. Given that most PI3K mutations were identified in thymic carcinomas, we speculate that targeting PI3K pathway might be a viable target for treatment in this rare malignancy. Further characterization of the oncogenic activities of PI3K and evaluation of its inhibitors in TETs, particularly thymic carcinomas, are needed.
Supplementary Material
Supplementary Fig. 1. MP57 cells do not express vimentin and N-cadherin. Immunofluorescence staining was used to detect vimentin and N-cadherin (FITC). DAPI was used to stain the nucleus of the cells.
Supplementary Table 1. Targeted genes in the PGDx custom gene panel.
Supplementary Table 2. Somatic gene mutations identified in MP57 primary tumor (FFPE) and its cell line using the Kinome panel.
Supplementary Table 3. Somatic gene mutations identified in MP57 cell line using the PGDx custom gene panel.
Supplementary Table 4. Somatic gene mutations identified in IU-TAB1 cell line using PGDx custom gene panel.
Acknowledgments
Funding support: Lombardi Comprehensive Cancer Center Core Grant
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors report no conflict of interest
References
- 1.Zucali PA, Di Tommaso L, Petrini I, et al. Reproducibility of the WHO classification of thymomas: practical implications. Lung Cancer. 2013;79:236–41. doi: 10.1016/j.lungcan.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strobel P, Hohenberger P, Marx A. Thymoma and thymic carcinoma: molecular pathology and targeted therapy. J Thorac Oncol. 2010;5:S286–90. doi: 10.1097/JTO.0b013e3181f209a8. [DOI] [PubMed] [Google Scholar]
- 3.Kelly RJ, Petrini I, Rajan A, et al. Thymic malignancies: from clinical management to targeted therapies. J Clin Oncol. 2011;29:4820–7. doi: 10.1200/JCO.2011.36.0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okumura M, Ohta M, Tateyama H, et al. The World Health Organization histologic classification system reflects the oncologic behavior of thymoma: a clinical study of 273 patients. Cancer. 2002;94:624–32. doi: 10.1002/cncr.10226. [DOI] [PubMed] [Google Scholar]
- 5.Eng TY, Fuller CD, Jagirdar J, et al. Thymic carcinoma: state of the art review. Int J Radiat Oncol Biol Phys. 2004;59:654–64. doi: 10.1016/j.ijrobp.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 6.Weissferdt A, Wistuba II, Moran CA. Molecular aspects of thymic carcinoma. Lung Cancer. 2012;78:127–32. doi: 10.1016/j.lungcan.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Kelly RJ. Systemic treatment of advanced thymic malignancies. Am Soc Clin Oncol Educ Book. 2014:e367–73. doi: 10.14694/EdBook_AM.2014.34.e367. [DOI] [PubMed] [Google Scholar]
- 8.Thomas A, Rajan A, Szabo E, et al. A Phase I/II Trial of Belinostat in Combination with Cisplatin, Doxorubicin, and Cyclophosphamide in Thymic Epithelial Tumors: A Clinical and Translational Study. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-14-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tiseo M, Rajan A, Thomas A, et al. “Pseudocavitation” in thymic carcinoma during treatment with sunitinib. J Thorac Oncol. 2013;8:511–2. doi: 10.1097/JTO.0b013e318284378f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strobel P, Bargou R, Wolff A, et al. Sunitinib in metastatic thymic carcinomas: laboratory findings and initial clinical experience. Br J Cancer. 2010;103:196–200. doi: 10.1038/sj.bjc.6605740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajan A, Carter CA, Berman A, et al. Cixutumumab for patients with recurrent or refractory advanced thymic epithelial tumours: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2014;15:191–200. doi: 10.1016/S1470-2045(13)70596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loehrer PJ, Sr, Kim K, Aisner SC, et al. Cisplatin plus doxorubicin plus cyclophosphamide in metastatic or recurrent thymoma: final results of an intergroup trial. The Eastern Cooperative Oncology Group, Southwest Oncology Group, and Southeastern Cancer Study Group. J Clin Oncol. 1994;12:1164–8. doi: 10.1200/JCO.1994.12.6.1164. [DOI] [PubMed] [Google Scholar]
- 13.Rajan A, Giaccone G. Chemotherapy for thymic tumors: induction, consolidation, palliation. Thorac Surg Clin. 2011;21:107–14. viii. doi: 10.1016/j.thorsurg.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Gower A, Wang Y, Giaccone G. Oncogenic drivers, targeted therapies, and acquired resistance in non-small-cell lung cancer. J Mol Med (Berl) 2014;92:697–707. doi: 10.1007/s00109-014-1165-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajan A, Giaccone G. Targeted therapy for advanced thymic tumors. J Thorac Oncol. 2010;5:S361–4. doi: 10.1097/JTO.0b013e3181f21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girard N, Shen R, Guo T, et al. Comprehensive genomic analysis reveals clinically relevant molecular distinctions between thymic carcinomas and thymomas. Clin Cancer Res. 2009;15:6790–9. doi: 10.1158/1078-0432.CCR-09-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badve S, Goswami C, Gokmen-Polar Y, et al. Molecular analysis of thymoma. PLoS One. 2012;7:e42669. doi: 10.1371/journal.pone.0042669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrini I, Meltzer PS, Zucali PA, et al. Copy number aberrations of BCL2 and CDKN2A/B identified by array-CGH in thymic epithelial tumors. Cell Death Dis. 2012;3:e351. doi: 10.1038/cddis.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly RJ. Thymoma versus thymic carcinoma: differences in biology impacting treatment. J Natl Compr Canc Netw. 2013;11:577–83. doi: 10.6004/jnccn.2013.0073. [DOI] [PubMed] [Google Scholar]
- 20.Belani R, Oliveira G, Erikson GA, et al. ASXL1 and DNMT3A mutation in a cytogenetically normal B3 thymoma. Oncogenesis. 2014;3:e111. doi: 10.1038/oncsis.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omatsu M, Kunimura T, Mikogami T, et al. Cyclin-dependent kinase inhibitors, p16 and p27, demonstrate different expression patterns in thymoma and thymic carcinoma. Gen Thorac Cardiovasc Surg. 2014 doi: 10.1007/s11748-014-0437-3. [DOI] [PubMed] [Google Scholar]
- 22.Petrini I, Meltzer PS, Kim IK, et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet. 2014;46:844–9. doi: 10.1038/ng.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YS, Thomas A, Lau C, et al. Mutations of epigenetic regulatory genes are common in thymic carcinomas. Scientific Reports. 2014;4 doi: 10.1038/srep07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuzume T, Kubonishi I, Takeuchi S, et al. Establishment and characterization of a thymic carcinoma cell line (Ty-82) carrying t(15;19)(q15;p13) chromosome abnormality. Int J Cancer. 1992;50:259–64. doi: 10.1002/ijc.2910500216. [DOI] [PubMed] [Google Scholar]
- 25.Patel DD, Whichard LP, Radcliff G, et al. Characterization of human thymic epithelial cell surface antigens: phenotypic similarity of thymic epithelial cells to epidermal keratinocytes. J Clin Immunol. 1995;15:80–92. doi: 10.1007/BF01541736. [DOI] [PubMed] [Google Scholar]
- 26.Inai K, Takagi K, Takimoto N, et al. Multiple inflammatory cytokine-productive ThyL-6 cell line established from a patient with thymic carcinoma. Cancer Sci. 2008;99:1778–84. doi: 10.1111/j.1349-7006.2008.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gokmen-Polar Y, Sanders KL, Goswami CP, et al. Establishment and characterization of a novel cell line derived from human thymoma AB tumor. Lab Invest. 2012;92:1564–73. doi: 10.1038/labinvest.2012.115. [DOI] [PubMed] [Google Scholar]
- 28.Wang GJ, Wang YG, Zhang P, et al. Establishment and characterization of a novel cell line derived from thymoma with myasthenia gravis patients. Thoracic Cancer. 2015;6:194–201. doi: 10.1111/1759-7714.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehemann V, Kern MA, Breinig M, et al. Establishment, characterization and drug sensitivity testing in primary cultures of human thymoma and thymic carcinoma. Int J Cancer. 2008;122:2719–25. doi: 10.1002/ijc.23335. [DOI] [PubMed] [Google Scholar]
- 30.Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol. 1999;11:219–25. doi: 10.1016/s0955-0674(99)80029-5. [DOI] [PubMed] [Google Scholar]
- 31.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 32.Moreira AL, Won HH, McMillan R, et al. Massively parallel sequencing identifies recurrent mutations in TP53 in thymic carcinoma associated with poor prognosis. J Thorac Oncol. 2015;10:373–80. doi: 10.1097/JTO.0000000000000397. [DOI] [PubMed] [Google Scholar]
- 33.Cheung LWT. High Frequency of PIK3R1 and PIK3R2 Mutations in Endometrial Cancer Elucidates a Novel Mechanism for Regulation of PTEN Protein Stability. Cancer Discovery. 2012;2:750–751. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–9. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Thomas A, Lau C, et al. Mutations of epigenetic regulatory genes are common in thymic carcinomas. Sci Rep. 2014;4:7336. doi: 10.1038/srep07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung LW, Hennessy BT, Li J, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–85. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riviere JB, Mirzaa GM, O’Roak BJ, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–40. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wirtz ED, Hoshino D, Maldonado AT, et al. Response of Head and Neck Squamous Cell Carcinoma Cells Carrying PIK3CA Mutations to Selected Targeted Therapies. Jama Otolaryngology-Head & Neck Surgery. 2015;141:543–549. doi: 10.1001/jamaoto.2015.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vadas O, Burke JE, Zhang X, et al. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2. doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- 40.Raynaud FI, Eccles SA, Patel S, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Molecular Cancer Therapeutics. 2009;8:1725–1738. doi: 10.1158/1535-7163.MCT-08-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang W, Hosford SR, Dillon LM, et al. Strategically timing inhibition of phosphatidylinositol 3-kinase to maximize therapeutic index in estrogen receptor alpha-positive, PIK3CA-mutant breast cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallin JJ, Guan J, Prior WW, et al. GDC-0941, a novel class I selective PI3K inhibitor, enhances the efficacy of docetaxel in human breast cancer models by increasing cell death in vitro and in vivo. Clin Cancer Res. 2012;18:3901–11. doi: 10.1158/1078-0432.CCR-11-2088. [DOI] [PubMed] [Google Scholar]
- 43.Floris G, Wozniak A, Sciot R, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19:620–30. doi: 10.1158/1078-0432.CCR-12-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munugalavadla V, Mariathasan S, Slaga D, et al. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene. 2014;33:316–25. doi: 10.1038/onc.2012.594. [DOI] [PubMed] [Google Scholar]
- 45.Wirtz ED, Hoshino D, Maldonado AT, et al. Response of head and neck squamous cell carcinoma cells carrying PIK3CA mutations to selected targeted therapies. JAMA Otolaryngol Head Neck Surg. 2015;141:543–9. doi: 10.1001/jamaoto.2015.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poovassery JS, Kang JC, Kim D, et al. Antibody targeting of HER2/HER3 signaling overcomes heregulin-induced resistance to PI3K inhibition in prostate cancer. Int J Cancer. 2015;137:267–77. doi: 10.1002/ijc.29378. [DOI] [PubMed] [Google Scholar]
- 47.Sarker D, Ang JE, Baird R, et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan-Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clinical Cancer Research. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janku F, Hong DS, Fu S, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377–87. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 50.Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858–66. doi: 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ihle NT, Lemos R, Jr, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–50. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janku F, Lee JJ, Tsimberidou AM, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One. 2011;6:e22769. doi: 10.1371/journal.pone.0022769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janku F, Wheler JJ, Westin SN, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–82. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. MP57 cells do not express vimentin and N-cadherin. Immunofluorescence staining was used to detect vimentin and N-cadherin (FITC). DAPI was used to stain the nucleus of the cells.
Supplementary Table 1. Targeted genes in the PGDx custom gene panel.
Supplementary Table 2. Somatic gene mutations identified in MP57 primary tumor (FFPE) and its cell line using the Kinome panel.
Supplementary Table 3. Somatic gene mutations identified in MP57 cell line using the PGDx custom gene panel.
Supplementary Table 4. Somatic gene mutations identified in IU-TAB1 cell line using PGDx custom gene panel.