

Abstract
In this review, the interactive mechanisms of mitochondria with the endoplasmic reticulum (ER) are discussed with emphasis on the potential protective role of the mitochondria derived peptide humanin (HN) in ER stress. The ER and mitochondria are dynamic organelles capable of modifying their structure and function in response to changing environmental conditions. The ER and mitochondria join together at multiple sites and form mitochondria-ER associated membranes that participate in signal transduction pathways that are under active investigation. Our laboratory previously showed that HN protects cells from oxidative stress induced cell death and more recently, described the beneficial role of HN on ER stress-induced apoptosis in retinal pigment epithelium cells and the involvement of ER-mitochondrial cross-talk in cellular protection. The protection was achieved, in part, by the restoration of mitochondrial glutathione that was depleted by ER stress. Thus, HN may be a promising candidate for therapy for diseases that involve both oxidative and ER stress. Developing novel approaches for retinal delivery of HN, its analogues as well as small molecular weight ER stress inhibitors would prove to be a valuable approach in the treatment of age-related macular degeneration.
Keywords: endoplasmic reticulum, mitochondria, mitochondrial-derived peptide, antioxidants, retinal pigment epithelium, age-related macular degeneration
Introduction
As one of the largest organelles in eukaryotic cells, the endoplasmic reticulum (ER) is a membrane bound network of branching tubules and flattened sacs that plays a major role in the synthesis, folding, and structural maturation of proteins, especially those destined for secretion or to the plasma membrane (Oakes and Papa, 2015). Multiple cellular stresses, such as oxidative stress, altered protein glycosylation, or protein folding defects, lead to accumulation of unfolded or misfolded proteins in the ER lumen and disturb ER function causing “ER-stress”. These ER-stress signals activate transcriptional and translational pathways that deal with unfolded and misfolded proteins, known as the unfolded protein response (UPR). Abnormally folded proteins in the ER can be flushed out through the ER-associated protein degradation (ERAD) pathway, in which misfolded proteins are translocated to the cytosol, where they undergo ubiquitylation and proteasome-mediated degradation. Further, UPR signaling promotes autophagy, which operates as an active mechanism to eliminate protein aggregates and damaged organelles via the lysosomal pathway (Oakes and Papa, 2015).
Recent studies have revealed the significance of ER-mitochondrial crosstalk in pathophysiological situations. The ER and mitochondria join together at several contact sites to form specific domains, termed mitochondria-ER associated membranes (MAMs) or mitochondria-associated ER membranes (MERCs) (Giacomello and Pellegrini, 2016). These contact sites help them to reciprocally transmit signals and communications with one another under stress conditions, triggering multiple, synergistic responses (Marchi et al., 2014). The roles played by the ER-mitochondria interface range from the coordination of calcium transfer to the regulation of mitochondrial fission, autophagy, accumulation of reactive oxygen species (ROS) and inflammasome formation. The relationship between the ER-mitochondria interface and inflammation was revealed with the observation that ROS promote the activation of NLRP3 inflammasomes which serve as a platform for caspase 1 activation (Bronner et al., 2015). Overall, it is revealed that the ER-mitochondria connection plays a fundamental role in the regulation of mitochondrial dynamics (Marchi et al., 2014).
ER Stress in Retinal Disease
A growing number of reports have suggested that accumulation of misfolded proteins plays an important role in the pathogenesis of several degenerative eye diseases such as retinitis pigmentosa, glaucoma, and age-related macular degeneration (AMD) (Salminen et al., 2010; Zhang et al., 2014). Even though there is no definitive evidence that ER stress is directly involved in AMD pathogenesis, oxidative stress, inflammation, cell death and angiogenesis are closely linked with ER stress and AMD (Salminen et al., 2010). For example, high concentrations of oxidized lipids that accumulate in the extracellular deposits found in AMD (drusen) could stimulate vascular endothelial growth factor (VEGF) via PERK/ATF4 signaling pathway and activate inflammatory stimuli which could trigger ER stress (Zhang et al., 2014). It has been proposed that misfolded protein-induced ER stress in retinal pigment epithelium (RPE) and/or choroid could lead to chronic oxidative stress, complement deregulation and AMD (reviewed in Zhang et al., 2014). Work from our laboratory demonstrated the involvement of ER stress in the regulation of cell death in RPE cells, the site of the early primary pathology in AMD (Dou et al., 2012). Induction of ER stress in the retina and RPE/choroid complex from mice exposed to cigarette smoke, a risk factor for AMD has been reported (Zhang et al., 2014). ER stress induced angiogenesis by up-regulating VEGF via UPR pathway and down regulating the antiangiogenic pigment epithelium-derived factor (PEDF) and promoting choroidal neovascularization (Salminen et al., 2010). Further, some ER inhibitors such as sterculic acid are reported to reduce inflammation and CNV formation (Zhang et al., 2014).
The Mitochondrial-Derived Peptide Humanin
Humanin is a mitochondria-derived 21–24 amino acid peptide encoded within the mitochondrial DNA that was first discovered through a search for neuroprotective factors from a cDNA library constructed from an unaffected brain fraction of an Alzheimer patient (Yen et al., 2013). Subsequently, multiple studies demonstrated its neuroprotective, anti-inflammatory, antiapoptotic, antiaging and antifibrilogenic properties in various cells and tissues (Yen et al., 2013). We have recently provided evidence that HN protected human RPE cells from oxidative cell death (Sreekumar et al., 2016). We found that HN exerted its protective function by utilizing intracellular and extracellular pathways involving mitochondria as well as STAT-3 signaling (Sreekumar et al., 2016).
Humanin Protection from ER Stress
The recent publication by Matsunaga et al. (2016) is the first study to evaluate the critical role of HN in protection from ER stress in any cell type. In this study, multiple ER stressors (tunicamycin, brefeldin A, and thapsigargin), induced RPE cell apoptosis, and HN pretreatment rendered dose-dependent protection from apoptosis. Tunicamycin (TM) treatment resulted in mitochondrial perturbations as evidenced by increased mitochondrial superoxide as well as decreased mitochondrial glutathione (GSH). HN co-treatment restored GSH synthesis by elevating the catalytic subunit of the rate-limiting glutamylcysteine ligase and inhibited superoxide production while other antioxidants such as catalase, Trx1, Grx1 and Grx2 and SOD II did not show appreciable change. The protective action of HN was not cell specific since HN also protected U-251 glioma cells exposed to TM (Matsunaga et al., 2016).
The study by Matsunaga et al. (2016) raises interesting possibilities to further our understanding of ER stress related mechanisms in the RPE and retina and potential roles of mitochondrial peptides in cellular protection. As alluded to earlier, chronic ER stress has been linked to an array of neurodegenerative diseases and inhibition of ER stress may provide a novel and effective therapeutic approach in the treatment of retinal diseases. In particular, small molecule inhibitors of the ER pathways could be potential drugs. However, targeting only one branch of the ER stress pathway might not be sufficient to prevent cell death given the close interaction via mitochondria-ER associated membranes (Giacomello and Pellegrini, 2016). Therefore, it is ideal to select drugs or agents that target both of these organelles for an effective therapy. Given the multipotent effects of HN in protecting against mitochondrial and ER stress, HN and its analogs could prove to be a valuable therapeutic approach for AMD especially since HN analogs have been reported to be several fold more potent than HN (Yen et al., 2013). HN being a short chain, low molecular peptide, has rapid tissue clearance resulting in less availability and hence may require frequent administrations. In order to overcome this problem, and to improve retention time, one approach would be to utilize thermally responsive elastin-like polypeptides (ELP) (Wang et al., 2014) recombinantly fused with HN.
Humanin and ER-Mitochondrial Crosstalk
With respect to mechanism of HN action in mitochondria, it would be of interest to study factors that affect ER-induced mitochondrial dysfunction and the pathways involved in this process. However, the localization of endogenous HN in subcellular compartments and sites of synthesis remain a topic of discussion. HN is expressed from an open reading frame (ORF) within the mitochondrial 16S ribosomal RNA. It is encoded from a 75 bp ORF sequence within the 1,567 bp cDNA, which yields either a 21 or 24 amino acid depending on the location of translation machinery. HN could also be encoded within one or more of the nuclear regions with 92% and 95% similarity to the original HN cDNA dispersed in multiple copies throughout the human genome. It is not clear whether HN is translated in the mitochondria (21-amino acid peptide) or the cytoplasm (24 amino acid peptide). Using fluorescein labeled HN and a mitotracker, we obtained evidence that there is rapid and abundant uptake of HN by RPE mitochondria using confocal microscopy (Figure 1). These experiments also showed the presence of HN in the cytoplasm under the experimental conditions (Figure 1). However, it is not known whether HN enters the ER compartment and whether it is found on the ER surface or is internalized. Thus, additional studies in unstressed and ER-stressed RPE will be needed to settle that question. Secondly, how ER stressors affect mitochondrial respiration and biogenesis in RPE and the nature of the salutary effects of HN will be an important issue to address. In this context, we showed recently that oxidative stress-induced decrease in mitochondrial bioenergetics and decrease in mitochondrial DNA copy number in human RPE cells was prevented by HN co-treatment (Sreekumar et al., 2016). Further, inhibition of the translocation of Bax from cytosol to mitochondria by HN was also observed (Sreekumar et al., 2016). The modulatory effect of ER stress on pro- and anti-apoptotic factors in RPE such as Bax, Bcl-2 and the effect of HN on these factors remains to be examined. Finally, the role of mitochondrial HN on mitochondrial-ER crosstalk mediated through MAMs (e.g. calcium signaling) should be evaluated.
Figure 1.
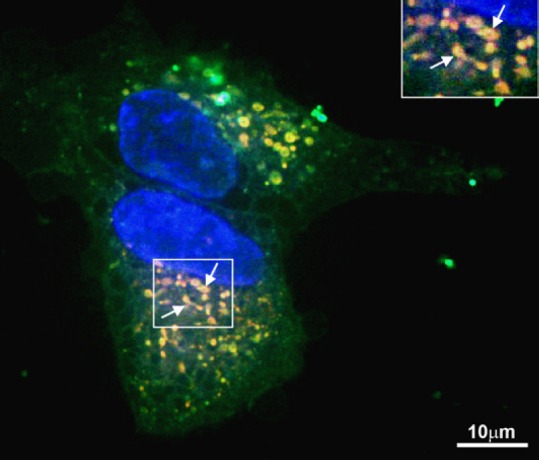
Uptake and translocation of FITC-labeled HN peptide in human RPE cells.
Serum-starved nonpolarized RPE cells were incubated with 10 μg FITC labeled HN for 2 hours. Fluorescent signal was evident in the cytosol and mitochondria (yellow orange). Arrow points to mitochondria colocalized with HN (inset). Red: Mitotracker. Green: Humanin. Yellow: Merge. Scale bar = 10 μm. Modified from Figure 4 in Sreekumar et al., 2016. FITC: Fluorescein isothiocyanate; HN: humanin; RPE: retinal pigment epithelium.
Based on the information available so far, a scheme for ER stress-induced mitochondrial dysfunction and protection by HN can be presented (Figure 2). According to this scheme, treatment of RPE by an ER stressor such as TM causes activation of caspase 3 resulting from the activation of ER-resident caspase 4 that eventually results in cell death. There is also a depletion of the antioxidant GSH in mitochondria with ER stress. In ER stress, exogenous HN rapidly enters mitochondria and protects RPE from apoptotic cell death by upregulating the rate-limiting enzyme of GSH biosynthesis and increasing mitochondrial GSH levels (Figure 2).
Figure 2.

Schematic representation of the ER stress induced by tunicamycin leading to apoptosis in RPE and the protective effect of exogenous Humanin on RPE apoptosis.
TM induced ER stress activates procaspase 4 to active caspase 4 which leads to increased apoptosis via activation of caspase 3. TM treatment caused increased ROS and decreased GSH in mitochondria. HN enters mitochondria in RPE co-treated with TM and HN and an inhibition of the formation of active caspase 4 and caspase 3 is found which blocks apoptosis. Attenuation of ROS generation and an increase in mitochondrial GSH occur with TM and HN co-treatment. The zones of close contact between ER and mitochondria called MAM is also shown. EM: Endoplasmic reticulum; HN: humanin; GSH: glutathione; MAM: mitochondria associated membranes; TM: tunicamycin; ROS: reactive oxygen species; RPE: retinal pigment epithelium.
Does the ER-induced oxidative stress involve mitochondrial inflammatory response such as the activation of inflammasome via NLRP3 and how does HN mediate this process? This question is important since stimulation of inflammatory cytokines with oxidative stress is well known and involvement of the inflammasomal component NLRP3 in AMD has been described. Further, a mechanism integrating cellular stress with innate immunity was reported in an infection model of ER stress sensor IRE1α (Bronner et al., 2015).
Humanin and Glutathione
As stated earlier, exposure of RPE to TM attenuated mitochondrial GSH and HN restored mitochondrial GSH levels suggesting that HN may be acting at least in part by upregulation of antioxidant enzymes. Glutathione (GSH) is a critical component of mitochondrial antioxidant defense system. GSH synthesis occurs exclusively in cytosol and mitochondria do not possess the synthetic machinery for GSH. However, GSH is found mostly in intracellular organelles including mitochondria and ER to maintain organelle-specific functions and cell survival. Specific carriers for import of GSH from cytosol to mitochondria are known (Ribas et al., 2014), whether HN activates the mitochondria specific GSH carriers in RPE cells remains to be determined.
Conclusions
A better understanding of the mechanisms that orchestrate the ER stress responses and its crosstalk with mitochondria may help to devise future strategies of safely modulating this process for therapeutic benefit in retinal neurodegenerative diseases such as AMD, and HN seems to be a promising candidate in this regard.
Acknowledgments
We thank Ernesto Barron for assistance in preparation of the figures.
Footnotes
Funding: This work was supported in part by Grants EY01545 (DRH), the Arnold and Mabel Beckman Foundation (DRH, RK) and an unrestricted grant to the Department of Ophthalmology from Research to Prevent Blindness, Inc.
Conflicts of interest: None declared.
References
- Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nuñez G, He Y, Yin XM, O’Riordan MX. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity. 2015;43:451–462. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou G, Sreekumar PG, Spee C, He S, Ryan SJ, Kannan R, Hinton DR. Deficiency of αB crystallin augments ER stress-induced apoptosis by enhancing mitochondrial dysfunction. Free Radic Biol Med. 2012;53:1111–1122. doi: 10.1016/j.freeradbiomed.2012.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M, Pellegrini The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Diff. 2016;23:1417–1427. doi: 10.1038/cdd.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim Biophys Acta. 2014;1837:461–469. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Matsunaga D, Sreekumar PG, Ishikawa K, Terasaki H, Barron E, Cohen P, Kannan R, Hinton DR. Humanin protects RPE cells from endoplasmic reticulum stress-induced apoptosis by upregulation of mitochondrial glutathione. PLoS One. 2016;11:e0165150. doi: 10.1371/journal.pone.0165150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Ann Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V, Garcia-Ruiz C, Fernandez-Checa JC. Glutathione and mitochondria. Front Pharmacol. 2014;5:151. doi: 10.3389/fphar.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kauppinen A, Hyttinen JM, Toropainen E, Kaarniranta K. Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol Med. 2010;16:535–542. doi: 10.2119/molmed.2010.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, Cohen P, Kannan R, Hinton DR. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci. 2016;57:1238–1253. doi: 10.1167/iovs.15-17053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sreekumar PG, Valluripalli V, Shi P, Wang J, Lin YA, Cui H, Kannan R, Hinton DR, MacKay JA. Protein polymer nanoparticles engineered as chaperones protect against apoptosis in human retinal pigment epithelial cells. J Control Release. 2014;191:4–14. doi: 10.1016/j.jconrel.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Lee C, Mehta H, Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol. 2013;50:R11–19. doi: 10.1530/JME-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SX, Sanders E, Fliesler SJ, Wang JJ. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp Eye Res. 2014;125:30–40. doi: 10.1016/j.exer.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]