

Background on chemotherapy-induced peripheral neuropathy (CIPN)
Incidence, prevalence, and consequences: Up to 90% of cancer patients experience CIPN at some point during or after anticancer treatment (Seretny et al., 2014). Although CIPN is second to hematologic toxicities in regards to the frequency of incidence, there are currently no approved treatments to prevent or treat CIPN, thus the neurotoxicity can be dose-limiting for some patients (Vasko et al., 2016). In addition, CIPN can persist following discontinuation of the drug: up to 40% of cancer patients continue to struggle with CIPN five years after treatment ends (Vasko et al., 2016) — and 10% remain symptomatic after more than 20 years. Thus, CIPN directly affects cancer survivorship, quality of life, and may limit future treatment options if cancer recurs (Vasko et al., 2016).
Symptoms and causative agents
CIPN's diverse toxicities usually manifest as diffuse, bilateral alterations in sensory neuronal function, which can include numbness, paresthesia, allodynia, thermal hyper- or hypoalgesia, cold intolerance, loss of proprioception, and reduced tendon reflexes (Vasko et al., 2016).
Many anticancer treatments cause CIPN, particularly:
Platinum compounds (cisplatin, carboplatin, oxaliplatin)
Vinca alkaloids (vincristine, vinblastine)
Taxanes (docetaxel, paclitaxel)
Epothilones (ixabepilone)
Immunomodulators (thalidomide, lenalidomide)
Bortezomib
Ionizing radiation
Each anticancer agent causes a slightly different complement of CIPN symptoms (Vasko et al., 2016). For example, platins accumulate in the dorsal root ganglia and reduce neurotransmitter release, causing parasthesias; while taxanes and epothilones disrupt axonal transport, causing sensorimotor and autonomic dysfunction, as well as neuropathic pain (Vasko et al., 2016).
Diagnostic challenges: The presentation of CIPN in patients is highly variable due to a number of factors, including the dose and schedule of chemotherapy administration, concurrent treatment with other neurotoxic agents, and the medical history of the patient; e.g., diabetes or alcoholism. The diagnosis of CIPN is further complicated by the extremely subjective nature of the symptoms of neuropathy. Consequently, the diagnosis of CIPN is usually made by a combination of the following: patient medical history, characterization of painful symptoms (location, frequency, spontaneous or induced, and intensity) and responsiveness to drugs that selectively block neuropathic vs. acute pain. Advanced clinical assessments of CIPN, via nerve conduction velocity measurements or quantitative sensory testing, do not necessarily correlate with patient self-assessments of neuropathy. As such, patients and caregivers may know the source of the neuropathy, but have a difficult time quantifying it, which is also an issue for clinical trials. Thus, no “gold standard” criteria for diagnosis or grading exist (Magge and DeAngelis, 2015). Moreover, effective treatments for CIPN are sorely lacking.
Unknown mechanisms of action = lack of effective treatments: Anticancer treatments affect sensory neurons with an increase (burning pain, hypersensitivity to touch) or decrease (numbness, loss of proprioception) in sensory function, and this variability in symptoms of CIPN increases the challenge to identify the mechanisms underlying CIPN. This is compounded by the numerous ways anticancer treatments affect the neurons directly or indirectly. For example, while platinum agents cause DNA damage through DNA cross-link adducts, some (like cisplatin and oxaliplatin) also produce significant reactive oxygen species (ROS) which creates oxidative DNA damage. Other agents, such as bortezomib are more involved in disruption of microtubular dynamics and axonal transport. This diverse list (Figure 1) would imply that equally diverse anti-CIPN therapeutics would be needed — in essence, to match the agent with the mechanism of each anticancer treatment (akin to precision medicine principles).
Figure 1.
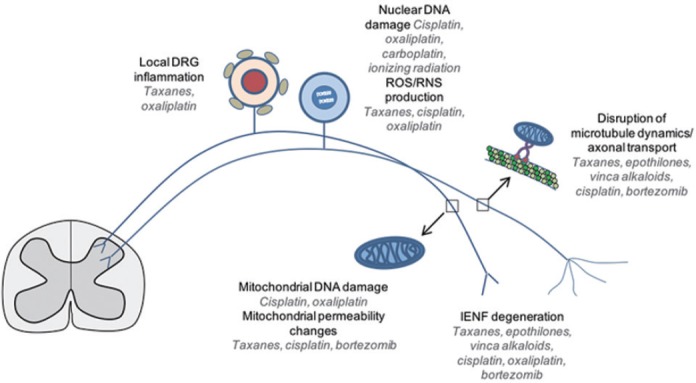
Putative sites of sensory neuronal dysfunction following specific anticancer drug treatments, indicated in italics.
DRG: Dorsal root ganglion; IENF: intraepidermal nerve fibers; RNS: reactive nitrogen species; ROS: reactive oxygen species.
Indeed, a plethora of treatment strategies have already been tried (Table 1). Both traditional and alternative/complementary treatments have been ineffective or inconsistent in suppressing the variety of CIPN symptoms (Pachman et al., 2014). Nevertheless, without definitive therapeutic targets or understanding the mechanism of action, developing preventive or therapeutic agents for CIPN is like operating blindly.
Table 1.
Current treatments for chemotherapy-induced peripheral neuropathy (CIPN)
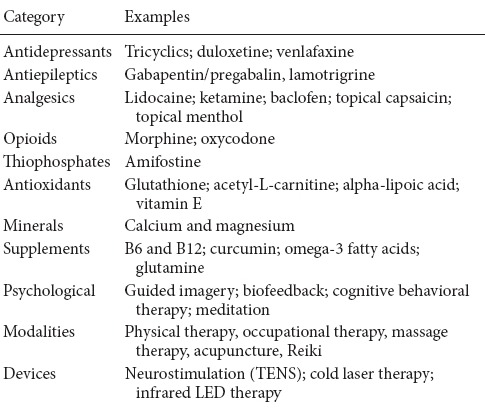
The latest ASCO guidelines (2014) mention pilot trials of several antidepressants as potentially having some efficacy for treating existing CIPN but notes the trials were not sufficiently powered. Notably, ASCO does not recommend any agents for CIPN prevention (Hershman et al., 2014). In similar fashion, the FDA has not yet approved any interventions or prevention strategies for CIPN (Hershman et al., 2014). This underscores the need to develop effective treatments to prevent or ameliorate this persistent problem.
A different approach: DNA damage and repair in CIPN: Although scientists are unsure of how anticancer therapies cause CIPN, the preponderance of sensory problems offers an important clue. The blood-brain barrier protects the central nervous system. In contrast, sensory nerves of the PNS are “exposed.” Their dorsal root ganglia exit the vertebral bodies; from there, individual nerves branch throughout the body, making both vulnerable to damage during cancer treatment (Vasko et al., 2016). Two therapeutic challenges exist regarding CIPN: healing after damage occurs, and neuroprotection before harm can happen. The latter is more desirable — but most likely a much tougher paradigm.
Nonetheless, most existing treatments for CIPN address the problem after the fact. They attempt to alleviate symptoms (such as pain) or correct for treatment sequelae (such as nutritional deficiencies). Similarly, modalities (such as physical therapy) compensate for, but do not cure deficits in sensory function (Pachman et al., 2014).
The goal of our laboratory is to provide neuroprotection against the deleterious effects of anticancer treatment, both before it can occur and after it is observed. Our work strongly supports the idea that any anticancer treatment capable of inducing oxidative stress or DNA damage can harm neurons, thus contributing to neuropathy. Several lines of evidence support this (Vasko et al., 2005; Vasko et al., 2011; Englander, 2013; Hershman et al., 2014; Kelley et al., 2014; Kim et al., 2015):
DNA damage in sensory neurons following ionizing radiation or chemotherapy correlates with CIPN symptoms.
The Base Excision Repair pathway (BER) repairs oxidative DNA damage, which is critical to long-lived, post-mitotic neurons.
BER is the primary means of repairing DNA damage in the nuclei and mitochondria of neurons.
APE1 is a pivotal enzyme in the BER pathway.
Reducing APE1 expression increases toxicity to sensory neurons exposed to anticancer therapies — but overexpression of APE1 prevents the damage.
APE1 is a multifunctional protein possessing both endonuclease and redox capabilities.
Inhibiting APE1's redox function partially unfolds the protein, which alters its ability to activate transcription factors. Disengaging APE1 from its redox activities appears to enhance its DNA repair capacity in neurons, but not tumors.
A targeted small-molecule therapeutic (E3330, now named APX3330) that inhibits redox signaling and enhances DNA repair in sensory neurons, provides neuroprotection in a manner analogous to genetic APE1 overexpression without diminishing the effectiveness of the anticancer treatment.
Thus, we have established a causal relationship between DNA damage and CIPN by modulating the DNA repair capacity of sensory neurons through the BER pathway and have developed a putative therapeutic that could prevent CIPN without compromising anticancer treatments.
Pipeline of molecules: More than a decade of testing APX3330 has confirmed its neuroprotective and tumor-killing properties, with no evidence of acute drug-related severe toxicity observed in vivo or in humans when used in a prior series of clinical studies by Eisai for chronic hepatitis C in Japan. APX3330 was recently given a Letter to Proceed from the FDA for Phase 1 clinical trials for safety and MTD determination (IND125360), which will start in 2017.
Additionally, we are utilizing a SAR (structure-activity relationship) approach for the development of second-generation compounds to target APE1 both for tumor killing and prevention or reversal of CIPN. We have modified APX3330 and have yielded a number of new compounds that have undergone or will undergo testing on sensory neuronal cultures. One example of a second-generation compound, APX2009, demonstrated neuroprotective activity against cisplatin and oxaliplatin similar to that of APX3330 — but at lower concentrations (Kelley et al., 2016). APX2009 has also demonstrated effective tumor killing properties and has a favorable half-life in human microsomes P450 studies, giving the compound a favorable pharmacokinetic profile suitable for treating patients. Collectively, these data suggest that APX2009 is effective in preventing or reversing platinum-induced CIPN without affecting the platinum's anticancer activity (Kelley et al., 2016). Further preclinical studies will ascertain not only APX2009's relative therapeutic indices, but determination of the other second-generation compounds anti-tumor and anti-CIPN effectiveness.
Conclusions and future directions
Effective prevention and management of CIPN hinges on understanding its pathophysiology. While more than one causal mechanism may be at work (Figure 2), we believe that the induction of oxidative DNA damage in sensory neurons is a major cause of CIPN. This drives our work with BER and APE1 — to find CIPN treatments that do not obstruct therapeutic anticancer regimens. Related to this, we are also examining whether inflammation contributes to peripheral sensitization by causing DNA damage which would dovetail with CIPN and mechanism of action. Initial findings in this field by our group has implications linking oxidative DNA damage either from chemotherapeutic agents or inflammation and the resulting peripheral neuropathy. Fully elucidating how anticancer drugs damage DNA and how the BER pathway reverses this damage can pave the way for identifying therapeutic targets and truly effective treatments for CIPN.
Figure 2.
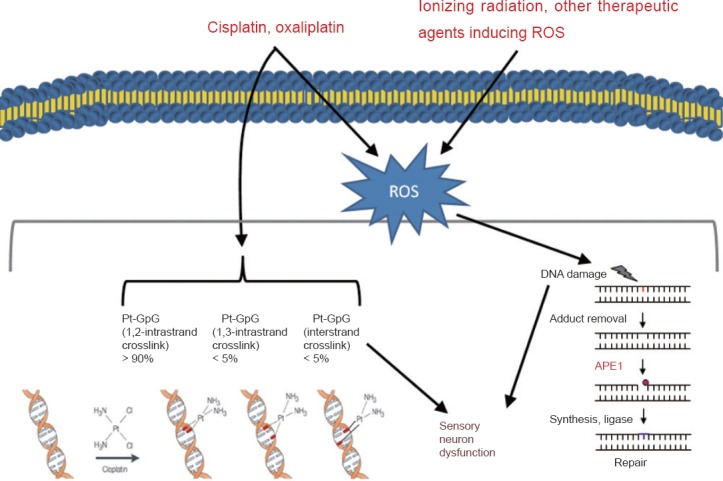
The role of oxidative DNA damage in altering sensory neuronal function following exposure to anticancer treatments inducing oxidative DNA damage such as cisplatin, oxaliplatin or ionizing radiation.
APE1: Apurinic/apyrimidinic endonuclease; Pt: platinum adduct; ROS: reactive oxygen species.
Financial support for this work was provided by the National Cancer Institute [CA122298 (MRK) and the National Institutes of Health, [R21NS091667 (MRK and JCF)]. Additional financial support was provided by, the Earl and Betty Herr Professor in Pediatric Oncology Research, Jeff Gordon Children's Foundation and the Riley Children's Foundation (MRK) and IU Simon Cancer Center Neurotoxicity Working Group (MRK and JCF).
Acknowledgments
Many thanks to Lana Christian of Create-Write Inc. for her expert writing and editing assistance.
References
- Englander EW. DNA damage response in peripheral nervous system: Coping with cancer therapy-induced DNA lesions. DNA Repair. 2013;12:685–690. doi: 10.1016/j.dnarep.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershman DL, Lacchetti C, Dworkin RH, Lavoie Smith EM, Bleeker J, Cavaletti G, Chauhan C, Gavin P, Lavino A, Lustberg MB, Paice J, Schneider B, Smith ML, Smith T, Terstriep S, Wagner-Johnston N, Bak K, Loprinzi CL. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American society of clinical oncology clinical practice guideline. J Clin Oncol. 2014;32:1941–1967. doi: 10.1200/JCO.2013.54.0914. [DOI] [PubMed] [Google Scholar]
- Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One. 2014;9:e106485. doi: 10.1371/journal.pone.0106485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, Fishel ML, Vasko MR. Identification and characterization of new chemical entities targeting apurinic/apyrimidinic endonuclease 1 for the prevention of chemotherapy-induced peripheral neuropathy (CIPN) J Pharmacol Exp Ther. 2016;359:300–309. doi: 10.1124/jpet.116.235283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Guo C, Thompson EL, Jiang Y, Kelley MR, Vasko MR, Lee SH. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat Res. 2015;779:96–104. doi: 10.1016/j.mrfmmm.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magge RS, DeAngelis LM. The double-edged sword: Neurotoxicity of chemotherapy. Blood Rev. 2015;29:93–100. doi: 10.1016/j.blre.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachman DR, Watson JC, Lustberg MB, Wagner-Johnston ND, Chan A, Broadfield L, Cheung YT, Steer C, Storey DJ, Chandwani KD, Paice J, Jean-Pierre P, Oh J, Kamath J, Fallon M, Strik H, Koeppen S, Loprinzi CL. Management options for established chemotherapy-induced peripheral neuropathy. Support Care Cancer. 2014;22:2281–2295. doi: 10.1007/s00520-014-2289-x. [DOI] [PubMed] [Google Scholar]
- Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, Colvin LA, Fallon M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain. 2014;155:2461–2470. doi: 10.1016/j.pain.2014.09.020. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Vasko MR, Shariati B, Zanville N. Kelly M, Fishel M. DNA Repair in Cancer Therapy (Second Edition) Second Edition. Boston: Academic Press; 2016. The role of DNA damage and repair in toxicity to postmitotic cells caused by cancer therapies A2; pp. 383–428. [Google Scholar]
- Vasko MR, Guo C, Thompson EL, Kelley MR. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair. 2011;10:942–952. doi: 10.1016/j.dnarep.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]