

Different outcomes of astrocyte inflammatory signalling in injury and neurodegeneration: It is emerging that astrocytes have a significant impact on the neuronal network by modulating synaptic connections and neuronal viability in both normal and pathological states. This provides a novel insight into pathomechanistic discoveries and therapeutics. It has been proposed that activation of the innate immune system and concomitant astrocytic and microglial inflammatory responses are contributors to synapse and neuronal dysfunction in neurodegenerative disorders (Heneka et al., 2014). On the contrary, current studies (Anderson et al., 2016) and our own work (Tyzack et al., 2014) indicate that astrocytic response in moderate traumatic injury induced inflammation promotes neuronal viability and synaptic recovery. This poses the question how beneficial and harmful aspects of inflammatory astrocyte signaling are altered in neurodegenerative diseases. Another central issue is how this effects synaptic connections then, which is an emerging theme in early pathogenesis in neurodegenerative conditions. Do astrocytes exacerbate synaptic dysfunction or impair plasticity? Discovery in this field had been hampered by the lack of cell-type specific experimental methods and human disease models suitable for exploring precise molecular events. Recent advances now provide an unprecedented opportunity to single out astrocyte mediated mechanisms relevant to degeneration or recovery of neuronal networks.
Astrocyte mediated synaptic plasticity in traumatic injury-induced inflammation: The recently developed “RiboTag” mouse system has brought a promising new prospective in providing a molecular profile of individual cell-types in animal models. This allows the pull-down of mRNAs by cell-type specific ribosomal tags, helping to define translational changes in communicating cells from whole tissue samples. Using this approach in combination with astrocyte-specific gene manipulation, a breakthrough study by the Sofroniew group has reversed the long-standing negative view that astrocytes activated by inflammatory cues impede repair in injury (Anderson et al., 2016). They have also elegantly proven that astrocytes require inflammatory pathway activation through signal transducer and activator of transcription-3 (STAT3) to promote the regrowth of axonal terminals in moderate spinal cord damage. Our own studies (Tyzack et al., 2014) have revealed that this same pathway is required for the recovery of central axon terminals onto motor neurons (MNs) as part of the re-arrangement of viable neuronal networks. In particular, we have shown a novel mechanism of structural synaptic plasticity, which is governed by STAT3 dependent re-expression and release of a synaptogenic molecule, thrombospondin-1 by astrocytes (Tyzack et al., 2014). Increasing evidence suggest that not only structural, but functional plasticity is also dependent on the astrocytic inflammatory response. Under normal conditions, astrocytes in accord with microglia balance synaptic and neuronal excitation via glutamate, adenosine, D-serine release, glutamate and extracellular potassium concentration and the postsynaptic glutamate receptor subunit composition (Haydon and Nedergaard, 2014). One of the most studied inflammatory synapse modulation via astrocytes is mediated by tumor necrosis factor (TNF)-α. This facilitates excitatory synaptic transmission by inducing the traffic of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-receptors (AMPAR) to the postsynaptic cell membrane and also by astrocytic release of glutamate that potentiates presynaptic activity via their N-methyl-D-aspartate receptors (NMDAR) (Habbas et al., 2015). While it is now clear that balanced synaptic excitation facilitates neuronal survival, it is less understood how astrocyte mediated responses limit detrimental neuronal overactivation in the inflammatory processes of regeneration. Functional compensatory mechanisms dependent on increased astrocytic glutamate uptake or initial structural synapse removal (stripping) have been long proposed as protective events following neuronal injury. Recent advances show that synapse stripping involves pro-inflammatory astrocyte and microglial activation, involving a complement (C1q) mediated process (Stevens et al., 2007). It transpires that synaptic recovery involves different glial inflammatory pathways, though this still not adequately explored. Our recent study indicates that STAT3 signaling during inflammation is not responsible for synapse removal but instead shifts the balance towards a recovery process (Tyzack et al., 2014). Understanding the triggers and master regulators in this injury-related astrocyte response may therefore bring us closer to understand how to modify glial inflammation so as to restore the neuronal network in neurotrauma and other neurological diseases (Figure 1).
Figure 1.
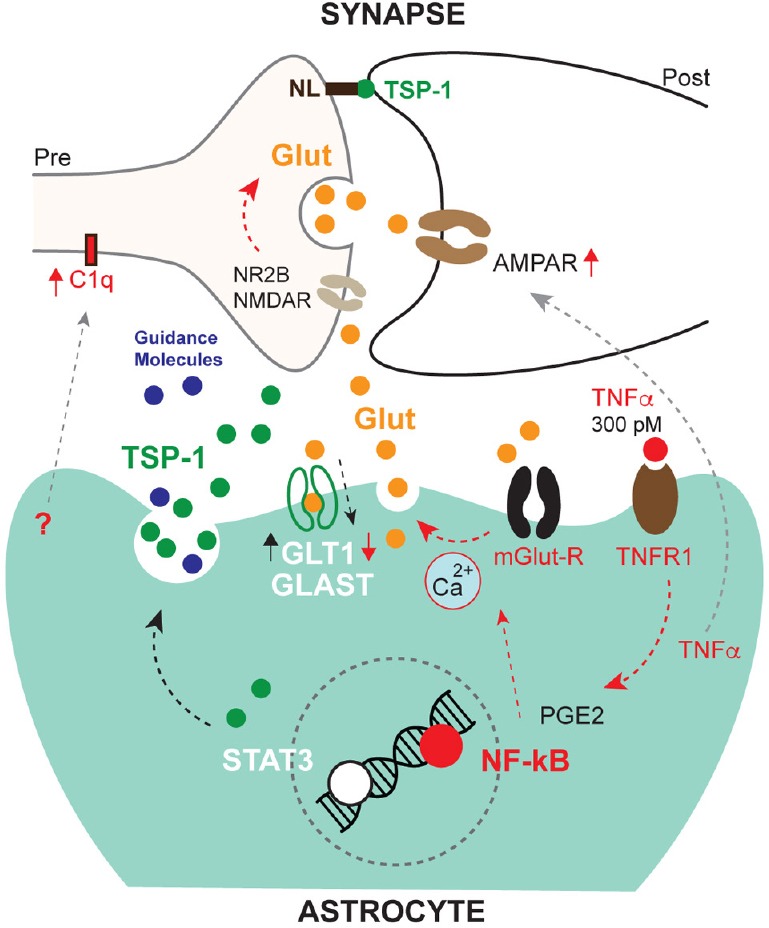
Effects of astrocyte mediated inflammatory responses on synapses.
TNFα signals via TNFR1/NF-κB and facilitates glutamatergic synaptic excitation by several mechanisms. This is detrimentally overactivated (red arrows) by TNFα at higher concentrations than 300 pM, which also impedes compensatory glutamate uptake by downregulating glial glutamate transporter-1 (GLT1). This inflammatory response is also conducive to a response characterised by synapse removal, involving the complement system (C1q). On the contrary, STAT3 signalling induces a synapse recovery process and may help excess glutamate uptake by GLAST (black arrows). Less characterized mechanisms are indicated by gray arrows. GLAST: Glutamate aspartate transporter; Glut: glutamate; mGlut-R: metabotropic glutamate receptor; NL: neuroligin; NR2B: NMDAR subunit; PGE2: prostaglandin E2; TNFR1: tumor necrosis factor-receptor1; TNFα: tumor necrosis factor-α.
Inflammatory mediators in amyotrophic lateral sclerosis (ALS) and perturbed astrocyte-synapse interactions: ALS is a rapidly progressive and fatal neurological disease with essentially no effective treatment, which affects motor and other neuronal populations and their synapses, leading to muscle weakness and to a variable degree of cognitive dysfunction. It appears that apart from intrinsic damage to MNs, non-cell autonomous processes, such as inflammation are conducive to neuronal degeneration. But it is less clear how these processes affect synaptic input onto MNs, which is known to be affected from the early stages in ALS. This is of immense relevance as the balance of neuronal excitation influences neuronal viability in neurodegeneration. Limited evidence suggests that there is an overall loss of synaptic input with some degree of potential plasticity and overactivation of surviving neurons (Matsumoto et al., 1994). The latter may have a synergistic effect on motor neuron (MN) activation along with the increased extrasynaptic excitation and intrinsic hyperexcitability (Wainger et al., 2014). Apart from microglia, astrocytes are most likely to contribute to this process in light of their intimate communication with synapses and their involvement in inflammatory activation. An elegant study using cell-specific translational profiling in a mouse ALS model has directly demonstrated increased pro-inflammatory signaling in astrocytes from an early disease stage (Sun et al., 2015). In particular, nuclear factor (NF)-kappa B (κB) appears to be a master regulator in ALS related inflammation, featuring in neuronal pathology (Ikiz et al., 2015), which points to the importance of interleukin (IL)-6 and TNF-α. These mediators signal through the NF-κB pathway and are increased in the brains and spinal cords of patients. Experimental examples from traumatic CNS injuries imply that they would lead to astrocyte mediated synapse overactivation and progressive stripping in ALS. Indeed, there is now evidence that pro-inflammatory mediators through NF-κB exacerbate MN overactivation. Then hyperexcitability may lead to ER stress (Wainger et al., 2014) and to other degeneration-promoting pathways. This raises an important and therapeutically relevant question: why is inflammatory signaling is over-represented in astrocytes in ALS? Is it predominantly an extrinsic influence on astrocytes or do they lose their compensatory or anti-inflammatory properties observed in the recovery phase of traumatic MN injuries? The answers may certainly guide us towards identifying more sophisticated targets for anti-inflammatory therapies that have been promising in animal models but have so far failed in human clinical trials. Thus there is an urgent need to integrate data from in vivo mouse models and in vitro human patient derived induced pluripotent stem cell-based disease systems to allow more precise elucidations for the role of astrocytes in synaptic and neuronal dysfunction.
Conclusion and future perspective: Restorative neuroscience has been for long borrowing ideas from injury systems characterized by regenerative events. Now with the development of state-of-art tools providing high-throughput cell-type specific molecular data, master regulators of glial inflammatory response may be more precisely dissected in this paradigm. Embarking on this path, it should be further addressed what aspects of detrimental or beneficial inflammatory signalling are dysregulated in ALS, which could affect the neuronal network. The use of cell-type specific screening approaches in both mouse and human models now may help pathway specific revisions of anti-inflammatory strategies in ALS.
References
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew M V. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habbas S, Santello M, Becker D, Stubbe H, Zappia G, Liaudet N, Klaus FR, Kollias G, Fontana A, Pryce CR, Suter T, Volterra A. Neuroinflammatory TNFα impairs memory via astrocyte signaling. Cell. 2015;163:1730–1741. doi: 10.1016/j.cell.2015.11.023. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Nedergaard M. How do astrocytes participate in neural plasticity? Cold Spring Harb Perspect Biol. 2014;7:a020438. doi: 10.1101/cshperspect.a020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–477. doi: 10.1038/nri3705. [DOI] [PubMed] [Google Scholar]
- Ikiz B, Alvarez MJ, Ré DB, Le Verche V, Politi K, Lotti F, Phani S, Pradhan R, Yu C, Croft GF, Jacquier A, Henderson CE, Califano A, Przedborski S. The regulatory machinery of neurodegeneration in in vitro models of amyotrophic lateral sclerosis. Cell Rep. 2015;12:1–11. doi: 10.1016/j.celrep.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Goto S, Kusaka H, Ito H, Imai T. Synaptic pathology of spinal anterior horn cells in amyotrophic lateral sclerosis: an immunohistochemical study. J Neurol Sci. 1994;125:180–185. doi: 10.1016/0022-510x(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S, Kopelevich A, Kaspar BK, Lagier-Tourenne C, Cleveland DW. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A. 2015;112:E6993–7002. doi: 10.1073/pnas.1520639112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzack GE, Sitnikov S, Barson D, Adams-Carr KL, Lau NK, Kwok JC, Zhao C, Franklin RJM, Karadottir RT, Fawcett JW, Lakatos A. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat Commun. 2014;5:4294. doi: 10.1038/ncomms5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SSW, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]