

Abstract
Demyelination alone has been considered sufficient for development of neurological deficits following central nervous system (CNS) disease. However, extensive demyelination is not always associated with clinical deficits in patients with multiple sclerosis (MS), the most common primary demyelinating disease in humans. We used the Theiler’s murine encephalomyelitis virus model of demyelination to investigate the role of major histocompatibility complex (MHC) class I and class II gene products in the development of functional and neurophysiological deficits following demyelination. We measured spontaneous clinical activity by two independent assays and recorded hind-limb motor-evoked potentials in infected class I-deficient and class II-deficient mice of an identical genetic background as well as in highly susceptible SJL/J mice. The results show that despite a similar distribution and extent of demyelinated lesions in all mice, only class I-deficient mice were functionally normal. We propose that the mechanism by which demyelinated class I-deficient mice maintain neurologic function results from increased sodium channel densities and the relative preservation of axons. These findings are the first to implicate a role for MHC class I in the development of neurological deficits following demyelination.
Theiler’s murine encephalomyelitis virus (TMEV) is a picornavirus that causes persistent demyelination in susceptible strains of mice. The virus replicates in astrocytes, oligodendrocytes and macrophages and, in susceptible strains of mice, induces neurologic deficits and pathological changes similar to those caused by multiple sclerosis1. In the Theiler’s model of MS, host genetics determines whether chronic demyelination will occur and virus will persist2–7. Mice of the H–2b genotype, namely, C57BL/6 × 129/J, β2-microglobulin (β2m)+/+, are resistant to virally induced demyelination and show no evidence of clinical disease. When mice of this resistant H–2b genotype (C57BL/6 × 129/J) have a genetic deficiency in expression of class I MHC products (β2-microglobulin-deficient mice)2–4 or class II MHC (C57BL/6 × 129/J Ab° mice)5, they develop extensive demyelination. Whereas class II-deficient mice showed severe neurological deficits and were frequently moribund by 120 days after infection, class I-deficient animals (otherwise of the same genotype) developed no clinical signs. From these observations, we hypothesized that functional deficits are the result of a class I MHC-restricted immune response.
Traditional assays for monitoring functional deficits in mice on the basis of appearance lack the sensitivity to detect subtle clinical deficits. Through the use of a computer-assisted activity monitor and an activity wheel, we were able to more accurately determine the functional status of experimental mice. In addition, we further characterized the electrophysiological function in mice by measuring motor-evoked potentials. This electrophysiological function was then compared with sodium channel densities to identify a potential mechanism for the preservation of function in demyelinated, class I-deficient mice.
Demyelination is extensive in class I-deficient mice
Myelin was normal in the spinal cords of infected nonmutant mice of the resistant H–2b genotype, which clear the TMEV infection (data not shown). In contrast there was extensive demyelination in the spinal cords of chronically infected class I-deficient and class II-deficient mice of the identical resistant H–2b genotype. This demyelination was similar to that observed in SJL/J mice of the susceptible H–2s genotype. Three-dimensional reconstruction of the lesions observed in the spinal cords of the four strains infected with TMEV (Fig. 1) illustrates that the extent and distribution of demyelination was similar in class I-deficient (Fig. 1, c and d), SJL/J (Fig. 1, e and f) and class II-deficient (Fig. 1, g and h) mice, in contrast to nonmutant H–2b mice (Fig. 1, a and b).
Fig. 1.
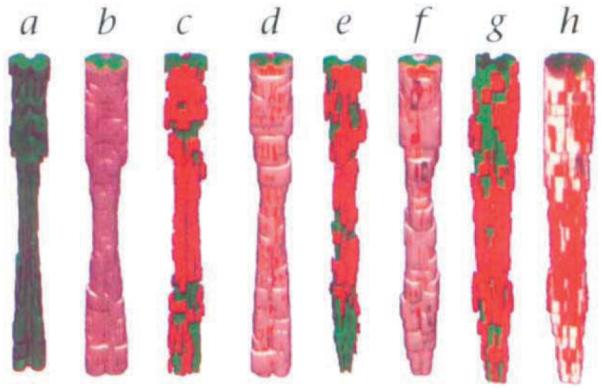
Three-dimensional reconstruction of demyelinated lesions from chronically infected nonmutant H–2b (β2m+/+), class I-deficient (β2m−/−), SJL/J and class II-deficient (Ab°) mice. The extent and distribution of demyelination is similar in β2m−/− (c and d), SJL/J (e and f) and class II-deficient (Ab°) (g and h) mice. β2m+/+ mice (a and b) show no demyelination. Green, gray matter; white, white matter; red, demyelinated lesions.
To further evaluate whether the distribution of demyelinated lesions was different in chronically infected class I-deficient, class II-deficient and SJL/J mice, the number of lesions located in the posterior, lateral, anterolateral and anterior columns were quantified from serial spinal cord sections. An identical distribution of lesions was observed for infected class I-deficient, class II-deficient and SJL/J mice (approximately 3% ± 0.5% in the posterior columns, 30% ± 1% in the lateral columns, 37% ± 1% in the anterolateral columns and 30% ± 0.1% in the ventral columns). These data emphasize that alterations in the distribution of demyelinated lesions are not contributing to the preservation of function observed in infected class I-deficient mice.
Spontaneous activity is normal in class I-deficient mice
In order to assess clinical changes in mice quantitatively, spontaneous activity was studied using a computerized monitoring system. Both horizontal and vertical movements were measured; however, vertical movement proved to be the best indicator of rear limb weakness and/or paralysis. The ratio of mean nocturnal activity (infected/uninfected) was calculated to allow for interstrain comparisons. Despite the extensive demyelination in their spinal cords, vertical activity in chronically infected class I-deficient mice was comparable to that of nonmutant H–2b mice, whereas class II-deficient mice showed a marked decrease in vertical activity (Fig. 2c). A time course of the mean nocturnal vertical activity ratios in susceptible SJL/J mice revealed decreased activity as early as 60 days and by 270 days after infection, it was completely abolished (Fig. 2, a and b). As a second functional assay, the ratio of mean revolutions per 24 hours (infected/uninfected) in an activity wheel were calculated over 3 to 5 days. Nonmutant H–2b and demyelinated class I-deficient mice displayed a similar number of revolutions, whereas significantly fewer revolutions per day were observed in susceptible SJL/J and class II-deficient mice (Fig. 2d).
Fig. 2.

Functional status of mice as measured by the Digiscan activity monitoring system, the activity wheel and conduction velocities in hind-limb motor-evoked potentials. Vertical activity was monitored hourly (over 3 days) for all groups of mice. a, The progressive decrease in nocturnal vertical activity from 0 to 180 days after infection is illustrated for susceptible SJL/J mice. b, A time course for SJL/J mice revealed a decrease in the ratio of nocturnal vertical activity as early as 60 days after infection when compared with nonmutant H–2b (β2m+/+) mice. This activity was completely abolished by 270 days after infection. c, At 180 days after infection there was no difference observed between the mean nocturnal vertical activity ratios for infected nonmutant H–2b (β2m+/+) and class I-deficient (α2m−/−) mice, indicating that hind-limb motor abnormalities were minimal in these mice. In contrast the ratios of mean nocturnal vertical activity for 180-day-infected SJL/J and 100-day-infected class II-deficient (Ab°) mice were significantly lower than those of nonmutant H–2b (β2m+/+) and class I-deficient β2m−/− mice. d, Spontaneous activity measured by the activity wheel. The ratios of mean revolutions per day revealed the same pattern of normal activity in class I-deficient (β2m−/−) mice when compared with infected H–2b (β2m+/+) mice, whereas infected SJL/J and class II-deficient (Ab°) mice displayed significantly decreased activities. e, Conduction velocities of 180-day-infected β2m−/− mice did not differ significantly from those of uninfected control β2m−/− mice or uninfected and 180-day-infected β2m+/+ mice. Conduction velocities for 180-day-infected SJL/J mice were slower when compared with uninfected SJL/J mice, 180-day-infected β2m+/+ mice and 180-day-infected β2m−/− mice. Conduction velocities for 100-day-infected, class II-deficient mice were also slower as compared with uninfected class II-deficient mice, 180-day-infected β2m+/+ and 180-day-infected β2m−/− mice. There were no differences observed between the mean conduction velocities for any of the uninfected controls. Uninfected, open symbols; chronically infected, shaded symbols. f, Representative tracings of recorded motor-evoked potentials from β2m+/+, β2m−/−, SJL/J and class II-deficient mice. Recordings from 180-day-infected β2m+/+ and β2m−/− mice are similar to their littermate uninfected controls. Recordings from 180-day-infected SJL/J and 100-day-infected class II-deficient mice showed prolonged latencies, dispersion of potentials and smaller amplitudes when compared with their uninfected controls. The arrow represents the point used to calculate latency to the beginning of the potential.
Conduction velocities are normal in class I-deficient mice
To examine electrophysiological disturbances in infected mice, we measured motor-evoked conduction velocities. Conduction velocities in chronically infected class I-deficient mice were not different from those of controls (Fig. 2e). In contrast there was a conduction delay in infected SJL/J and class II-deficient mice when compared with their uninfected littermates (Fig. 2e). Because class II-deficient mice were moribund by 100 days after infection, recordings were made earlier in these mice. This explains the slightly faster conduction velocities in class II-deficient mice when compared with infected SJL/J mice. No changes in the shape or amplitude of the potentials were detected in infected class I-deficient or nonmutant H–2b mice, whereas there was temporal dispersion as well as smaller amplitudes of the response in infected SJL/J and class II-deficient mice (Fig. 2f).
Sodium channel densities are increased in class I-deficient mice
One possible reason for the unexpected lack of electrophysiological deficits in class I-deficient mice is a compensatory increase in or redistribution of sodium channels. In extensively demyelinated areas, saltatory conduction would be affected by the decrease in sodium channels in the nodes of Ranvier, potentially leading to conduction block. However, reorganization of axonal membranes has been observed in peripheral nerve fibers where demyelination was induced after a crush injury. Following the injury, sodium channels were observed throughout the demyelinated segment of the axon8. Furthermore, it has also been shown that saltatory conduction and the formation of nodes can precede remyelination in demyelinated axons9,10. The redistribution of preexisting channels from the nodes or their increase within the internode may provide a mechanism whereby continuous, less efficient conduction could be maintained11.
To address this possibility, sodium channel densities in experimental mice were measured using two independent assays. Mice were assessed at the later stages of chronic infection in order to maximize the potential for the disruption of spinal cord white matter. In chronically infected class I-deficient mice, there were increased sodium channel fluorescent intensities when compared with those of uninfected controls (Fig. 3a), whereas there were markedly decreased sodium channel intensities in chronically infected SJL/J mice when compared with uninfected controls (Fig. 3b). Disrupted sodium channel staining was also observed in the lesions of chronically infected class II-deficient mice (data not shown). These results were supported by [3H]saxitoxin (STX) autoradiography (Fig. 3c), which showed a 1.7-fold increase in the [3H]STX grain density of chronically infected class I-deficient mice relative to that of uninfected class I-deficient controls. This contrasted with the chronically infected SJL/J mice, which showed a 50% decrease in [3H]STX-labeled grain density when compared with that of uninfected controls. No difference was observed between the grain densities for uninfected and infected nonmutant H–2b mice, as was expected for resistant mice (data not shown). When sodium channel intensities were plotted against myelin proteolipid protein (PLP) intensities, a positive linear correlation was seen for all groups except the chronically infected class I-deficient mice, indicating that some areas with the lowest PLP signal contained elevated levels of sodium channels (Fig. 3a). To illustrate this point, areas of low PLP signal in chronically infected class I-deficient mice were compared with similar areas in chronically infected SJL/J mice. These areas revealed an elevated and more homogenous sodium channel staining pattern in the chronically infected class I-deficient mice (Fig. 3, e and h), which contrasted with the markedly lower and disrupted sodium channel pattern in chronically infected SJL/J mice (Fig. 3, f and i).
Fig. 3.

Sodium channel distributions in uninfected (filled triangle) and chronically infected (open circle with dot) class I-deficient (β2m−/−) and SJL/J mice. a, A significant increase was observed in the sodium channel intensities of 323-day-infected β2m−/− mice when compared with their uninfected counterparts. b, In contrast, there was a significant decrease in sodium channel intensities of 270-day-infected SJL/J mice as compared with uninfected controls. a and b, A positive linear correlation (thickened gray lines, chronically infected groups; thin black lines, uninfected groups) exists between sodium channel and corresponding myelin proteolipid protein (PLP) intensities for uninfected SJL/J (r = 0.50), infected SJL/J (r = 0.20) and uninfected β2m−/− mice (r = 0.47). However, there is no positive linear correlation between sodium channel and PLP intensities for chronically infected β2m−/− mice (a), which can be explained by regions of high sodium channel intensities in areas of low PLP intensities. c, [3H]Saxitoxin autoradiography showed a 1.7-fold increase in the sodium channel grain density of 323-day-infected β2m−/− mice when compared with that of uninfected controls. In contrast, there was a 50% decrease in the sodium channel grain density of 270-day-infected SJL/J mice when compared with their uninfected controls. White bars, uninfected; black bars, chronically infected. Preincubation with 200-fold excess of unlabeled tetrodotoxin blocked approximately 50% of the [3H]saxitoxin labeling in an uninfected β2m+/+ mouse (data not shown). Representative examples of sodium channel staining (d, e and f) and PLP-myelin staining (g, h and I) in an uninfected β2m−/− mouse (d and g), a 323-day-infected β2m−/− mouse (e and h) and a 270-day-infected SJL/J mouse (f and i). Images for the sodium channel stain and their respective PLP stained counterparts were taken from the same field. A normal distribution of sodium channel (d) and myelin (g) is shown for an uninfected β2m−/− mouse. (Uninfected nonmutant mice had a comparable staining pattern.) Note the elevated and homogenous distribution of sodium channel staining (e) in an area of relative myelin loss (h) from an uninfected β2m−/− mouse. Note the severe loss of sodium channel intensity (f) in a similar area of low myelin (i) in a chronically infected SJL/J mouse. Even though the degree of myelin loss is similar in infected β2m−/− (h) and SJL/J (i) mice, there is more sodium channel expression in β2m−/− (e) compared with the SJL/J (f) mouse. Regions of negative staining were determined from the corresponding ultraviolet images to be cellular nuclei. Green, sodium channels; red, PLP.
It is possible that astrocytes may be the source of sodium channel upregulation following demyelination. To determine the cellular localization of sodium channels in the spinal cord white matter of chronically infected class I-deficient mice, increased sodium channel densities were colocalized with glial fibrillary acidic protein (GFAP) staining (data not shown). Although some sodium channels were present on the surface of astrocytes, the majority of the sodium channel foci did not localize to astrocytes or their processes. These results support the fact that the increased sodium channel densities are present on axons.
Axons are relatively well preserved in class I-deficient mice
To assess the integrity of axons in chronically infected class I-deficient mice, both Bielschowski stain and immunofluorescence anti-neurofilament stain were used independently to reveal axon fibers in the spinal cord (Fig. 4). The axons in chronically infected class I-deficient mice were relatively preserved even in regions of parenchymal cellular infiltration and demyelination (Fig. 4, b and e). In contrast, the axons in chronically infected SJL/J mice were markedly disrupted and degenerating (Fig. 4, c and f). The disruption of axons observed in SJL/J mice provides one explanation for the decline in detectable sodium channels and the presence of severe clinical deficits, whereas the relative preservation of axons in class I-deficient mice provides an additional factor that contributes to the absence of clinical deficits. The lack of axonal degeneration may also provide the framework for compensatory mechanisms, such as sodium channel redistribution and remyelination, to engage.
Fig. 4.

Representative examples of axons in the spinal cord white matter of chronically infected class I-deficient (β2m−/−) and SJL/J mice and uninfected class I-deficient and nonmutant H–2b (β2m+/+) mice. Representative examples of antineurofilament (a-c) and Bielschowski (d-f) staining are shown for an uninfected β2m+/+ mouse (a), an uninfected β2m−/− mouse (d), a chronically infected (323 days postinfection) class I-deficient mouse (b and e) and a chronically infected (270 days) SJL/J mouse (c and f). A normal distribution of axons is shown for an uninfected β2m+/+ mouse (a) and an uninfected β2m−/− mouse (d). The staining pattern in uninfected animals of all groups is comparable. Axons are relatively well preserved in chronically infected β2m−/− mice despite the presence of inflammatory cells and demyelination. Both the neurofilament (b) and the Bielschowski (e) stains show that the axons in β2m−/− mice are neither significantly disrupted nor degenerating. In contrast the axons in chronically infected SJL/J mice are severely disrupted. The neurofilament stain (c) shows a significant loss of immunostainable material, whereas the Bielschowski stain (f) shows swelling axons (red arrows) which are actively undergoing degeneration.
Discussion
This is the first study to investigate the contribution of a class I MHC-restricted immune response to the development of clinical deficits and electrophysiologic abnormalities in an inflammatory demyelinating disease. Because class I-deficient and class II-deficient mice were of an identical genetic background and only differed in the expression of the MHC class I or class lI gene products, this study strongly indicates that the development of neurological and electrophysiological deficits following virus-induced demyelination requires class I MHC. Class II MHC is not required for demyelination or development of functional deficits, since deletion of this gene was associated with severe paralytic white matter disease5.
T cells have been implicated in the development of demyelination and neurologic deficit following virus infection12. The distribution of T cells in TMEV-infected class I-deficient, class II-deficient and SJL/J mice has been previously described. For example, there were no CD8+ T cells found in spinal cord sections of class I-deficient mice at 45, 180 and 540 days after infection13. Alternatively, demyelinating lesions in class II-deficient mice were shown to have CD8+ T cells but no CD4+ T cells5. These both contrast to chronically infected SJL/J mice, which have been characterized as having both CD4+ and CD8+ T cells14.
Alterations in the cytokines secreted and the distributions of T cells present in the lesions of class I-deficient mice may contribute to the absence of neurologic deficit following demyelination. A persistent inflammatory response as a consequence of chronic virus replication1,5 may be detrimental to the functioning of sodium channels and axons. Cytokines have been shown to inhibit sodium channel currents15. Characterization of cytokines in the lesions of chronically infected animals revealed intense immunoperoxidase staining for interleukin-4 (IL-4) in SJL/J and class II-deficient mice, but not in class I-deficient mice (data not shown). Furthermore, interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) were detected in the lesions of SJL/J mice, but were minimal or undetectable in class I- and class II-deficient mice (data not shown).
Remyelination as described by Miller et al.13 alone probably does not account for preserved function in infected class I-deficient mice. Areas with elevated and homogeneously distributed sodium channel intensities were observed in infected class I-deficient mice without corresponding increases in myelin intensities (Fig. 3, a, e and h). In addition, a relative preservation of axons was also observed in infected class I-deficient mice (Fig., 4b and e). The pattern of sodium channel staining suggests that in infected class I-deficient mice with normal function, conduction could occur in demyelinated axons either through the upregulation or the redistribution of sodium channels16. Moreover, the localization of the increased sodium channel intensities was determined not to be primarily the result of an upregulation on astrocytes. Thus, the lack of a persistent class I-mediated immune response following demyelination may allow for the preservation of axons, the engagement of compensatory mechanisms and the subsequent absence of neurologic deficit.
Conduction velocity depends primarily on electrophysiological properties of a small percentage of the fastest conducting fibers17. We did not evaluate conduction of these central fibers with stimulus trains or their refractory period17, which may have unmasked subtle changes consistent with a decreased safety factor of conduction. Swimming stress tests indicated fatigability for infected class I-deficient mice, but they still performed better than SJL/J mice with similar demyelination (data not shown). Activity monitoring and multifiber recordings are appropriate for our focus on clinically relevant function of central axons.
This study has important implications to the pathogenesis of neurological deficits in MS. Magnetic resonance imaging studies18 and autopsy cases19,20 indicate that severe demyelination can occur in MS patients who are neurologically normal. Moll et al.21 described increased sodium channel densities in demyelinated lesions of MS when compared with those in normal white matter. Even though MS is hypothesized to be a class II-mediated CD4+ autoimmune disease, CD8+ T cells are found frequently in the parenchyma, intimately associated with demyelinated lesions22. These cells may be triggered to release cytokines and to function as cytotoxic cells in response to an endogenous or exogenous CNS antigen. Virus infection has been correlated with attacks of MS (ref. 23) and interferon-β, a cytokine elevated in virus infection24, decreases exacerbation rate. Therefore, activation of class I-restricted CD8+ T cells (potentially by a virus) in the vicinity of demyelinated vulnerable axons may result in functional neurologic impairment. These data indicate that manipulating the function of class I-restricted CD8+ T cells in the microenvironment of a demyelinated lesion may hold promise as a therapeutic strategy in MS.
Methods
Mice
The following mouse strains were used: SJL/J from the Jackson Laboratories (Bar Harbor, ME)(n = 24); C57BL/6 × 129/J (H–22, β2m+/+) mice from the Mayo Clinic lmmunogenetic Mouse Colony, C. David, Director (n = 20), C57BL/6F × 129/J (H–2b) β2m−/− mice, from R. Jaenisch, Whitehead Institute (Cambridge, MA) (n = 15); and C57BL/6 × 129/J (H–2b) Ab° (class II-deficient mice), from Chris Benoist (Strasburg, France) (n = 14). Mice were inoculated intracerebrally with 2 × 105 plaque-forming units of the Daniel’s strain of TMEV in 10 μl volume at 4 to 8 weeks of age.
Activity monitoring
Two independent assays of activity were used: (1) The Digiscan activity monitor system (Omnitech Electronics, Columbus, OH) consists of an acrylic cage (40 × 40 × 30.5 cm) supported by a metal frame that holds two sets of photocells. When the projected infrared beams are interrupted, they give a measure of both horizontal and vertical spontaneous activities of animals. For all groups both horizontal and vertical activities were monitored. However, vertical activity, which measures the rearing of mice and is related to the degree of rear limb paralysis, proved to be the most sensitive indicator. The ratios of nocturnal vertical activity (infected/uninfected) were calculated by dividing mean nocturnal vertical activities (from 6 p.m. to 5 a.m. over 3 days) for infected animals by mean nocturnal vertical activities for their uninfected, age-matched counterparts. No difference between the nocturnal vertical activities of infected and uninfected counterparts would result in a ratio of approximately equal to one. (2) The activity wheel (Lab Products Inc., Maywood, NJ) is a stainless steel standard exercise wheel with housing attached. Activity wheel data for each group were collected as the number of revolutions per day over 3 to 5 days. Ratios of revolutions per day (infected/uninfected) were calculated by dividing mean revolutions per day (over 3 to 5 days) for the infected animals by mean revolutions per day for their uninfected counterparts. Three mice were placed in the activity box and the activity wheel for at least 72 consecutive hours. Food and water were freely accessible; a normal 12-hour light/dark cycle and 70 °F ambient temperature were maintained. All uninfected controls were age-matched to their infected counterparts. Comparisons between hourly nocturnal vertical activity ratio data were calculated using the Kruskal-Wallis ANOVA on ranks. Comparisons between activity wheel data were calculated using an ANOVA. Pairwise comparisons for both were made with the Student-Newman-Keuls method (P < 0.05).
Motor evoked potentials (MEPs)
Mice were anesthetized with sodium pentobarbital, 0.08 mg/g mouse weight. Temperature was maintained between 35.7 °C and 36 °C by a heating lamp and monitored by a rectal probe. MEPs were elicited and recorded as described with a few minor changes25. Stimulus intensity was 8–10 mV, which corresponded to 10–20% over the voltage at which the maximum amplitude of the initial peak of the evoked potential was observed. Each recorded response was an average of 5–10 sweeps. Interelectrode distances were measured by placing the limbs at 90° angle to the body and adding the distance between the stimulating cathode to the spinous process of Sl plus the distance from the spinous process of Sl to the recording electrode in the hind-limb. For each group of mice, velocities of conduction were calculated by measuring the distance between the cathodes and by dividing the latency to the first observed peak of an evoked response by this number. Each data point represents the average of two recordings from one side of each mouse. To verify that responses represented spinal MEPs, rigorous specificity control experiments were performed. Intraspinal stimulation with platinum-iridium electrodes at the C8–T1 interspace and recording at the L6–S1 interspace gave MEP shapes similar to those observed in Fig. 2f. To exclude that responses were not motor, we recorded sensory-evoked potentials by stimulating at the lumbar and recording at the cervical segment. To exclude that responses were peripheral, we measured peripheral nerve sensory conduction in the digital nerve of the hind-limb and motor conduction in the hind-limb (tibialis anterior muscle). The shape, latency and amplitude of MEPs were different from peripheral motor and sensory potentials. Injection of 0.1 ml of 1% lidocaine in the spinal canal obliterated MEPs. All uninfected animals were age-matched to their infected counterparts. Velocities were compared with control values using an ANOVA. All pairwise comparisons were made with the Student-Newman-Keuls method (P < 0.05).
Fluorescence immunocytochemistry
To localize sodium channels, myelin and astrocytes, an affinity-purified rabbit anti-sodium channel (Upstate Biotechnology, Lake Placid, NY) IgG directed against the highly conserved intracellular III-IV loop, an AB3 monoclonal rat anti-myelin proteolipid protein (PLP; Agmed Inc., Bedford, MA) IgG and a monoclonal mouse anti-glial fibrillary acidic protein (GFAP; Sigma Biosciences, St. Louis, MO) IgG were used, respectively. Cellular nuclei were localized using 4,6-diamidino-2-phenylindole (DAPI). Frozen sections(l0 μm) were cut and fixed for 3 min in 95% ethanol/5% glacial acetic acid at 5 °C. To label sodium channels, all sections were stained as described26. This was followed by a myelin stain, which consisted of a 1-h incubation with primary antibody (1:10, RT), a rinse and a 1-h incubation with Texas Red (TR) affinity-pure F(ab’)2 fragment donkey anti-rat IgG (1:50, RT), or an astrocyte stain, which consisted of a 1-h incubation with primary antibody (1:400, RT), a rinse and a 1-h incubation with TR affinity-pure goat anti-mouse IgG (1:200, RT). All antibody dilutions for the myelin stain were done in PBS containing 0.1% saponin. Finally, sections were stained for 5 min with DAPI, rinsed and mounted with Mowiol/glycerol containing 2.5% 1,4-diazobicyclo-[2.2.2]-octane. To localize axons, two monoclonal mouse anti-neurofilament (NF; Boehringer Mannheim, Indianapolis, IN) IgG antibodies directed against the 68-kDa and 160-kDa neurofilament polypeptides were used in combination. Frozen sections (10 μm) were fixed for 10 min in 4% paraformaldehyde, rinsed, incubated for 1 h with the two primary antibodies (2.5 mg/ml of each, RT), rinsed and incubated for 1 h with FITC affinity-pure F(ab’)2 fragment goat anti-mouse IgG (1:100, RT). Nuclei were localized with DAPI as described above. Photography for sodium channels/myelin or sodium channel/astrocytes was done after quantitative fluorescence analyses by simultaneous digitization of both the red (myelin) and green (sodium channels) channels using a Zeiss laser scanning confocal instrument (Zeiss, Jena, Germany) with a ×63 oil objective and under identical amplified settings. Photography for neurofilaments involved an identical setup; however, only the green channel was digitized.
Bielschowski stain
A Bielschowski stain was performed on 10-μm frozen spinal cord sections. Briefly, sections were placed in a 20% silver nitrate solution for 15 min at 37 °C followed by a rinse. Sections were then placed in 50 ml of 20% silver nitrate containing l0–13 ml of ammonium hydroxide for 10 min at 37 °C and rinsed with distilled water (dH2O) containing 1% ammonium hydroxide. Afterward, slides were developed, washed in ammonia water, toned in 0.2% gold chloride solution for 30 s, washed in dH2O, incubated in a 5% sodium thiosulfate solution for 1 min and washed for 10 min in tap water. Finally, sections were dehydrated in alcohols, cleared in xylene and mounted. Photography was done using a Zeiss Axiophot microscope and a ×63 oil objective.
Cytokines
To localize IL-4, IFN-γ and TNF-α, a purified monoclonal rat antimouse IL-4 IgG, a purified monoclonal rat anti-mouse IFN-γ IgG and a purified monoclonal rat anti-mouse TNF-α IgG (all from PharMingen, San Diego, CA) were used, respectively. Frozen sections (10 μm) were fixed for 10 min with acetone at 5 °C and rinsed. This was followed by a 24-h incubation with primary antibody (IL-4, 1:50; IFN-γ, 1 :25; TNF-α, 1:50) at RT, a rinse and an incubation for 1 h with a biotinylated rabbit anti-rat IgG (1:100, RT). Detection of the antibodies involved the use of the previously described avidin–biotin immunoperoxidase technique (Vector Laboratories, Burlingame, CA) using the Hanker-Yates reagent (Polysciences, Warrington, PA)4.
Quantitative multispectral fluorescence analysis
Images for quantitative fluorescence were digitized with identical amplified settings in a single session using an ×40 objective, a Zeiss Axiophot microscope, a silicon-intensified target camera and 768 × 512 pixel resolution with the appropriate optics for each fluorochrome, yielding a red (myelin), green (sodium channel) and ultraviolet (nuclear) image for each field. Fluorescent intensity within the images was digitally corrected for gain by using a low-pass filtered fluorescent standard. Mean fluorescent intensities were quantified using the KS400 software (Kontron Elektronik Gmbh, Munich) on a Pentium platform in registered red and green images. An average total area of 149,666 mm2 was quantified for each group. Using the ultraviolet image as a template, tissue overlying cell nuclei was excluded from the analysis (n = 95 or 96 samples per group of three animals). Fluorescent intensities were normalized against the minimum and maximum values for all samples in the experiment and were plotted as percentile rank. Statistical differences between fluorescent intensities for uninfected and infected animals were detected using a Kruskal-Wallis ANOVA on ranks. Pairwise comparisons were made using the Student-Newman-Keuls method (P < 0.05). Correlation coefficients for the fluorescent intensities were calculated using the Pearson product moment correlation.
[3H]Saxitoxin binding and quantification
To determine the distribution of saxitoxin (STX) binding sites associated with voltage-sensitive sodium channels, 10-μm frozen mouse spinal cord sections were exposed to 5 nM [3H]STX (1.85 Mbq, Amersham, Arlington Heights, IL) for 1 h at 4 °c as described27. Nonspecific binding was evaluated by preincubating with 1 μM tetrodotoxin. After application of STX, slides were fixed for 30 min in 4% paraformaldehyde, rinsed and dried. Slides were exposed for 5 weeks with Kodak NTB2 emulsion (Kodak, Rochester, NY), developed and then counterstained with hematoxylin. Densities of [3H]STX silver grains in spinal cord white matter were obtained from images digitized with identical amplified settings using a ×63 oil objective and a Zeiss Axiophot microscope fitted with a Newvicon camera. The KS400 image analysis software was used to calculate [3H]STX grain densities (expressed as the number of grains per unit area) from the grain image using the tissue image as a template to avoid perivascular cellular infiltrates (n = 60 samples per group of 2 or 3 animals). The emulsion background grain density was determined from fields without tissue and was subtracted from the mean tissue grain densities. Statistical differences between [3H]STX grain densities for uninfected and infected animals were detected using a Kruskal-Wallis ANOVA on ranks. Pairwise comparisons were made using the Student-Newman-Keuls method (P < 0.05).
Light microscopy, three-dimensional analysis and lesion distribution
Mice were perfused via intracardiac puncture with Trump’s fixative (phosphate-buffered 4% formaldehyde with 1% glutaraldehyde, pH 7.4). The spinal cord was removed, sectioned coronally into 1-mm blocks, postfixed with osmium tetroxide and embedded in Araldite (Polysciences). Cross sections (1 mm thick) were cut from each block and stained with 4% paraphenyldiamine. For three-dimensional analysis, cross sections from 30 to 40 consecutive spinal cord blocks from a representative animal from each group were digitized. Images were combined into a 3-D volume after correcting for slice-to-slice misalignments. White matter, gray matter and demyelinated lesions were traced manually and used to render a three-dimensional image of the entire spinal cord. Image processing, registration and rendering were performed using ANALYZE, a 3-D medical image display and analysis software system developed by the Biomedical Imaging Resource, Mayo Clinic, Rochester, Minnesota28. To determine the distribution of demyelinated lesions in chronically infected class I-deficient (235 days; n = 4), class II-deficient (135 days; n = 5) and SJL/J (200 days; n = 7) mice, 10 spinal cord blocks described above were quantified from each animal for the presence of lesions in the posterior, lateral, anterolateral and anterior columns. This yielded 113 total lesions for class I-deficient mice, 71 total lesions for class II-deficient mice and 132 total lesions for SJL/J mice. Percentages were based on the number of lesions in a given area of white matter divided by the total number of lesions. Statistical differences between the number of lesions in previously described areas were assessed using a Kruskal-Wallis ANOVA on ranks.
Acknowledgments
The authors gratefully acknowledge the expert technical assistance of K.D. Pavelko and J. J. Camp. This work was supported by National Institutes of Health grants R01 NS32129, R01 NS24180 & N01-A1-4-5197.
References
- 1.Rodriguez M, Oleszak E, Leibowitz J. Theiler’s murine encephalomyelitis: A model of demyelination and persistence of virus. Crit. Rev. Immunol. 1987;7:325–365. [PubMed] [Google Scholar]
- 2.Fiette L, Aubert C, Brahic M, Rossi CP. Theiler’s virus infection of β2-microglobulin-deficient mice. J. Virol. 1993;67:589–592. doi: 10.1128/jvi.67.1.589-592.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pullen LC, Miller SD, Dal Canto MC, Kim BS. Class I-deficient resistant mice intracerebrally inoculated with Theiler’s virus show an increased T cell response to viral antigens and susceptibility to demyelination. Eur. J. Immunol. 1993;23:2287–2293. doi: 10.1002/eji.1830230935. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez M, et al. Abrogation of resistance to Theiler’s-induced demyelination in H–2b mice deficient in β2-microglobulin. J. Immunol. 1993;151:266–276. [PubMed] [Google Scholar]
- 5.Njenga MK, et al. Theiler’s virus persistence and demyelination in major histocompatibility complex class II-deficient mice. J. Virol. 1996;70:1729–1737. doi: 10.1128/jvi.70.3.1729-1737.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez M, Leibowitz J, David CS. Susceptibility to Theiler’s virus-induced demyelination: Mapping of the gene within the H–2D region. J. Exp. Med. 1986;163:620–631. doi: 10.1084/jem.163.3.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez M, David CS. Demyelination induced by Theiler’s virus: Influence of the H-2 haplotype. J. Immunol. 1985;135:2145–2148. [PubMed] [Google Scholar]
- 8.Foster RE, Whalen CC, Waxman SG. Reorganization of the axon membrane in demyelinated peripheral nerve fibers: Morphological evidence. Science. 1980;210:661–663. doi: 10.1126/science.6159685. [DOI] [PubMed] [Google Scholar]
- 9.Bostock H, Hall SM, Smith KJ. Demyelinated axons can form ’nodes’ prior to remyelination. J. Physiol. (Lond.) 1980;308:21P–23P. [Google Scholar]
- 10.Smith KJ, Bostock H, Hall SM. Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J. Neurol. Sci. 1982;54:13–31. doi: 10.1016/0022-510x(82)90215-5. [DOI] [PubMed] [Google Scholar]
- 11.Waxman SG. Demyelination in spinal cord injury and multiple sclerosis: What can we do to enhance functional recovery? J. Neurotrauma. 1992;9:S105–S117. [PubMed] [Google Scholar]
- 12.Rodriguez M, Sriram S. Successful therapy of Theiler’s virus-induced demyelination (DA strain) with monoclonal anti-Lyt-2 antibody. J. Immunol. 1988;140:2950–2955. [PubMed] [Google Scholar]
- 13.Miller DJ, Rivera-Quinones C, Njenga MK, Leibowitz J, Rodriguez M. Spontaneous CNS remyelination in γ2 microglobulin-deficient mice following virus-induced demyelination. J. Neurosci. 1995;15:8345–8352. doi: 10.1523/JNEUROSCI.15-12-08345.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller DI, Njenga MK, Murray PD, Leibowitz J, Rodriguez M. A monoclonal natural autoantibody that promotes remyelination suppresses central nervous system inflammation and increases virus expression after Theiler’s virus-induced demyelination. Int. Immunol. 1996;8:131–141. doi: 10.1093/intimm/8.1.131. [DOI] [PubMed] [Google Scholar]
- 15.Brinkmeier H, Kaspar A, Wietholter H, Rudel R. Interleukin-2 inhibits sodium currents in human muscle cells. Pfluegers Arch. 420:621–623. doi: 10.1007/BF00374643. 19?? [DOI] [PubMed] [Google Scholar]
- 16.Black JA, Felts P, Smith KJ, Kocksis JD, Waxman SG. Distribution of sodium channels in chronically demyelinated spinal cord axons: Immuno-structural localization and electrophysiological observations. Brain Res. 1991;544:59–70. doi: 10.1016/0006-8993(91)90885-y. [DOI] [PubMed] [Google Scholar]
- 17.McDonald WI, Sears TA. Effect of demyelination on conduction in the central nervous system. Brain. 1970;93:583–598. doi: 10.1093/brain/93.3.583. [DOI] [PubMed] [Google Scholar]
- 18.Filippi M, et al. Correlations between changes in disability and T2-weighted brain MRI activity in multiple sclerosis: A follow-up study. Neurology. 1995;45:255–260. doi: 10.1212/wnl.45.2.255. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert JJ, Sadler M. Unsuspected multiple sclerosis. Arch. Neurol. 1983;40:533–536. doi: 10.1001/archneur.1983.04050080033003. [DOI] [PubMed] [Google Scholar]
- 20.Mackay RP, Hirano A. Forms of benign multiple sclerosis: Report of two "clinically silent" cases discovered at autopsy. Arch. Neurol. 1967;17:588–600. doi: 10.1001/archneur.1967.00470300030007. [DOI] [PubMed] [Google Scholar]
- 21.Moll C, Mourre C, Lazdunsky M, Ulrich J. Increase of sodium channels in demyelinated lesions of multiple sclerosis. Brain Res. 1991;556:311–316. doi: 10.1016/0006-8993(91)90321-l. [DOI] [PubMed] [Google Scholar]
- 22.Hauser SL, et al. lmmunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann. Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- 23.Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–1315. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The IFNβ Multiple Sclerosis Group Interleron beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double blind, placebo-controlled trial. Neurology. 1993;43:655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 25.Iuliano BA, Schmelzer JD, Thiemann RL, Low PA, Rodriguez M. Motor and somatosensory-evoked potentials in mice infected with Theiler’s murine encephalomyelitis virus. J. Neurol. Sci. 1994;123:186–194. doi: 10.1016/0022-510x(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 26.Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J. Neurosci. 1995;15:492–503. doi: 10.1523/JNEUROSCI.15-01-00492.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mourre C, Widmann C, Lazdunski M. Saxitoxin-sensitive Na+ channels: Presynaptic localization in cerebellum and hippocampus of neurological mutant mice. Brain Res. 1990;533:196–202. doi: 10.1016/0006-8993(90)91340-m. [DOI] [PubMed] [Google Scholar]
- 28.Robb RA, et al. ANALYZE: A comprehensive,operator-interactive software package for multidimensional medical image display and analysis. Comput. Med. lmag. Graphics. 1989;13:433–454. doi: 10.1016/0895-6111(89)90285-1. [DOI] [PubMed] [Google Scholar]