

Abstract
Circulating autoantibodies to beta cell antigens are present in the majority of patients with Type 1 diabetes. These autoantibodies can be detected before and at time of clinical diagnosis of disease. Although the role of autoantibodies in the pathogenesis of the disease is debated, their presence indicates a dysregulation of the humoral immune response. Mechanisms regulating autoantibodies in Type 1 diabetes are not well understood. In contrast, in other autoimmune diseases there is acceptance that autoantibodies are regulated not only by antigen but also by other antibodies that bind to the antigen-binding site of these autoantibodies (anti-idiotypic antibodies). The proposed purpose of this network is to maintain an equilibrium between autoantibodies and their anti-idiotypic antibodies, preventing autoimmunity, while allowing a robust response to exogenous antigen. Anti-idiotypic antibodies regulate both autoantibody binding and their levels by a) neutralizing autoantibodies, and b) inhibiting the secretion of autoantibodies. Because it has been proposed that the B lymphocytes that produce autoantibodies function as autoantigen presenting cells, inhibiting their binding to autoantigen by anti-idiotypic antibodies may prevent development of autoimmune disease. This hypothesis is supported by the presence of anti-idiotypic antibodies in healthy individuals and in patients in remission from autoimmune diseases, and by the lack of anti-idiotypic antibodies during active disease. We recently reported the presence of autoantibodies to glutamate decarboxylase in the majority of healthy individuals, where their binding to autoantigen is prevented by anti-idiotypic antibodies. These anti-idiotypic antibodies are absent at clinical diagnosis of Type 1 diabetes, revealing the presence of autoantibodies. Type 1 diabetes (T1D) is an autoimmune disease characterized by the dysfunction and destruction of insulin-producing beta cells by autoreactive T cells. Although much progress has been made towards understanding the respective roles of effector and regulatory T cells in this beta cell destruction, the development of autoantibodies to beta cell proteins is widely considered simply a by-product of the autoimmune destruction of the beta cells, rather than having an active role in the pathogenesis. This view is starting to change based on increasing recognition that autoantibodies can have defined roles in other autoimmune diseases, and the emergence of new data on their role in T1D. This exploration of the role of autoantibodies in autoimmune disease has been spurred, in part, by increasing recognition that development of autoimmune diseases is influenced by regulatory antibodies (anti-idiotypic antibodies) directed against the unique binding site of autoantibodies. This review provides an overview of the development and function of these anti-idiotypic antibodies, and present evidence supporting their role in the development of autoimmune diseases. Finally, we conclude this review with a model of the events that may cause loss of anti-idiotypic antibodies and the implications for the development of T1D.
Keywords: autoimmune disease, autoantibodies, type 1 diabetes, glutamate decarboxylase, Idiotypic Network Hypothesis
The specific antigen binding sites of an antibody are located in the three-dimensional structure created by the variable regions of the antibody’s two light and two heavy chains. This part of the antibody is referred to as the idiotypic determinant [1]. This antigen-specific “idiotype” of each antibody determines its unique recognition of its antigen. However, the idiotype itself can serve as an antigen and is recognized by anti-idiotypic antibodies (anti-Id), which can function as a critical part of a regulatory network.
The concept of anti-Id as regulatory factors was first formulated by Niels Jerne in the Network Hypothesis nearly 40 years ago [2–4]. He postulated that this unique ability of antibodies both to recognize an antigen and be recognized by other antibodies as an antigen creates a balanced network that acts to regulate the humoral arm of the immune system. Anti-Id are proposed to maintain the homeostasis of the adaptive humoral immune responses by neutralizing idiotypic antibodies and regulating idiotypic antibody secretion (for reviews see [5,6]).
Although this Network Hypothesis dominated the immunological field for over a decade, interest subsequently waned in part because of its seeming contradiction with aspects of the clonal selection theory, especially the concept of self-tolerance. Today, however, there is increasing recognition that low levels of autoimmunity are a common phenomenon in healthy individuals. These low levels of autoantibodies and autoreactive T cells have been found in many healthy cohorts used as controls for patients with clinical autoimmune disease. This natural autoimmunity is held in check by regulatory mechanisms [6].
Moreover, the apparent conflict of the Network Hypothesis with the clonal selection theory has largely been resolved with the recognition that the process of affinity maturation introduces significant amino acid changes in the idiotype – thus creating an antigen to which self-tolerance does not apply (see below). For these reasons, interest in the Idiotypic Network has re-emerged during the last decade, especially in the field of autoimmune disease [7–9].
Development of an immune response to antibodies
(A) Diversity of the variable region of antibodies develops during affinity maturation. When B lymphocytes encounter an antigen, they begin rapidly to proliferate and enter the proliferative compartment – the dark zone – of the germinal center (GC) of the lymph nodes. It is during this rapid proliferation phase that B lymphocytes undergo somatic mutation at a very high rate (somatic hypermutation), a process critical for the generation of antibodies with high affinity towards a specific antigen. As the B lymphocytes move towards the light zone of the GC, only those B lymphocytes that express antibodies with increased affinity are selected to survive and exit the GC as antibody producing plasma cells or memory cells (for details see [10]) (Figure 1).
Figure 1.

Affinity maturation of antigen-stimulated B lymphocytes. Upon binding to antigen the B lymphocytes enter germinal centers (GC) and undergo rapid proliferation and somatic hypermutation (in the dark zone). The somatic hypermutations cause random changes in the affinity of the antibody to the original antigen. B lymphocytes with higher affinity to the antigen, can interact successfully with GC T cells and follicular dendritic cells (FDCs) and receive survival signals from them (selection in the light zone). These B lymphocytes eventually differentiate into antibody secreting plasma B cells or memory B cells (Figure adapted from [120]).
The somatic hypermutations occur predominately in the hypervariable region of the antibody and on average 15 amino acid substitutions occur per antibody during this process [11], causing substantial differences in the antigenic determinants of the idiotype (Id). The antibodies generated during affinity maturation not only have improved antigen-binding abilities, but also differ significantly from the germline sequences. While the immune response in general is tolerant towards the germline sequences presented on naïve antibodies (for review see [12]), the immune system is not tolerant to the amino acid changes in the idiotypic region. Thus Id-specific antibodies (anti-Id) are stimulated during this change of idiotype [13–15]. The new idiotype of the antibody is indeed “foreign” to the body and will therefore not be recognized as “self” by the immune system. Therefore B cells reactive to the idiotype of this antibody will not be eliminated through the usual mechanisms applying to peripheral tolerance.
Anti-Id (also termed Ab2) can be directed to different epitopes of an antibody (Ab1). Depending on the binding sites they are classified as Ab2-α or Ab2-β. Ab2-α bind outside the antigen-binding site of Ab1, while Ab2-β bind to the complementarity determining region (CDR) of Ab1. Ab2-β can represent the internal image of antigen and compete with the binding of Ab1 to antigen, thus participating in the regulatory network [16] (Figure 2). These characteristics of anti-Id have important implications in their therapeutic use, as are discussed later.
(B) T cell responses to antibodies after affinity maturation. The original Idiotypic Network Hypothesis considered predominantly the humoral aspects of the Network. However, the observation that T cell-deficient animals do not develop anti-Id when immunized with antibody [17,18], demonstrated that T cells are an essential part of the Network. Therefore, below we provide an updated view of the Idiotypic Network by discussing the role of T lymphocytes within the Network.
Figure 2.

Anti-Id presenting the internal image of the antigen. The epitope of an antigen is bound by Ab1. Ab2-β (anti-Id) has an idiotype that is structurally similar to the epitope presented by the antigen. It therefore binds to the antigen-binding site of Ab1 and competes with the antigen for binding to Ab1, thus regulating antibody function.
The first step in the development of anti-Id is the presentation of the idiotypic antibody. Idiotypes are endocytosed by antigen-presenting cells (APC), such as macrophages, dendritic cells and B lymphocytes, and subsequently presented to T cells on MHC class II molecules [19]. B lymphocytes continuously present their endogenous Id-peptides on their MHC class II molecules [19,20] (Figure 3). Presentation of these Id peptides is essential for the generation of anti-Id. The presentation of idiotypic peptides on MHC class II stimulates T cells, which in turn provide signals for the B lymphocytes to differentiate into antibody-producing plasma cells and secrete anti-Id [15,21,22].
Figure 3.

Presentation of idiotypic peptides by B lymphocytes. B lymphocytes present peptides derived from their endogenous antibodies on MHC class II molecules (1). They also endocytose anti-Id that bind to the cells’ B cell receptor and subsequently present the anti-Id-peptides on MHC class II molecules.
Furthermore, B lymphocytes internalize anti-Id that bind to their B cell receptor (BCR). Consequently anti-Id peptides are presented on MHC class II molecules (conventional T-B collaboration) [23] (Figure 3). These interactions between T and B cells create an antigen-independent cooperation between T cells and B cells called Id-driven T-B cell collaboration, permitting continuous activation of both Tand B cells even in the absence of antigen [19,24–27]. This antigen-independent cooperation is an essential part of the “Relay Hypothesis” a mechanism proposed to account for the maintenance of the immunological memory in the absence of antigen [28,29].
Anti-Id in the Idiotypic Network
As discussed previously, B lymphocytes present peptides derived from anti-Id. Low concentrations of anti-Id stimulate T cells, which in turn stimulate idiotypic antibody secretion, resulting in a equilibrium between complementary B and T cells – the Idiotypic Network.
Antigen stimulation transiently disturbs this network. Antigen is taken up, presented to antigen-specific T cells, which stimulate B lymphocyte to secrete antigen-specific antibodies. This increase of idiotypic antibody titer induces the secretion of anti-Id, which firstly neutralizes the antibody, and secondly down-regulates the secretion of the antibody as discussed below (Figure 4). This series of events was demonstrated in individuals receiving tetanus toxoid vaccination [30].
Figure 4.
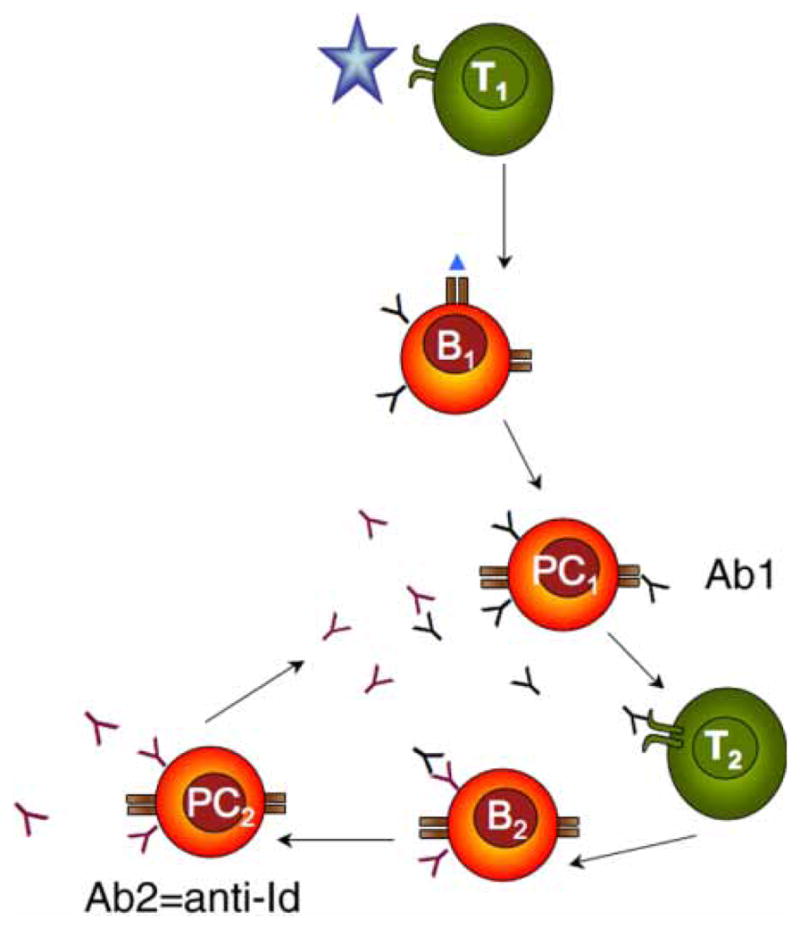
Re-establishment of the homeostasis after antigen stimulation. Antigen is presented to T cells (T1) that stimulate B lymphocytes (B1) to differentiate to plasma cells (PC1). PC1 secrete antibody (Ab1). Ab1 is recognized by T cells (T2), that stimulate B lymphocytes (B2) capable of recognizing the idiotypic determinant of Ab1. B2 lymphocytes differentiate to plasma cells (PC2) secreting Ab2, a.k.a. anti-Id, which neutralize Ab1.
Serum samples from vaccinated individuals were evaluated for tetanus toxoid specific antibodies and their anti-Id. The initial injection of vaccine antigen triggered an increase in tetanus toxoid antibody titer. This increase of idiotypic antibody titer was followed by an increase in anti-Id and a subsequent decrease in the titer of free tetanus toxoid antibody. This apparent decrease of tetanus toxoid antibody titer was caused by competition of anti-Id for the tetanus toxoid binding site on the idiotypic antibody. There is also a real decrease of tetanus toxoid antibody, which was caused by the anti-Id-induced inhibition of secretion of idiotypic antibody (for mechanism see below).
In addition to neutralizing secreted antibody, anti-Id can also down-regulate the secretion of the idiotypic antibody [31,32]. This effect is only observed at high concentrations of anti-Id. It has been demonstrated in vitro where anti-Id suppressed the synthesis of antibodies by human B lymphocytes [33–35]. This down-regulation of antibody secretion by anti-Id is due to the simultaneous binding of anti-Id to both the BCR and the Fc receptor (FcR) on B lymphocytes (Figure 5).
Figure 5.
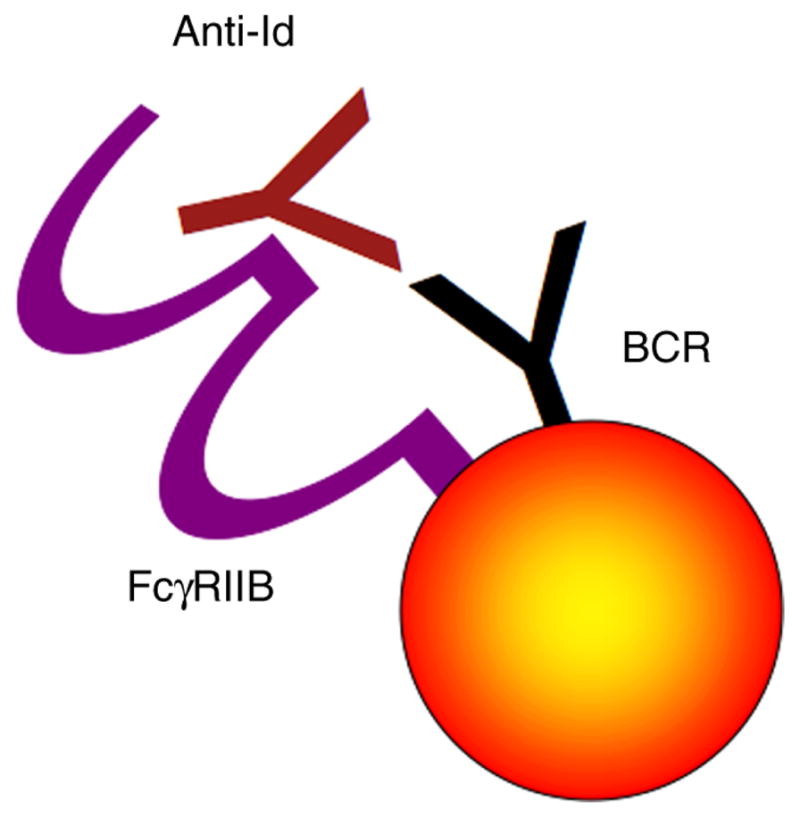
Co-ligation of FcγRIIB with the BCR. The Fc portion of anti-Id binds to the B lymphocyte’s FcγRIIB. Simultaneously the Fab portion of anti-Id binds to the BCR, thereby co-ligating these two membrane proteins. Co-ligation of the FcγRIIB to the BCR abolishes the BCR-induced stimulation of antibody secretion.
B lymphocytes express only one subtype of FcR: the FcγRIIB. This FcR is an inhibitory receptor and if in close proximity to the BCR (e.g., through anti-Id mediated co-ligation), the activated FcγRIIB abrogates the BCR initiated signal [36,37] and – if maintained long enough - results in B cell apoptosis [38,39]. Because FcγRIIB is a low affinity receptor, this inhibitory pathway is activated only at high anti-Id levels. As the threshold for the FcγRIIB is reached, anti-Id will inhibit secretion of the antibody, causing antibody levels to decrease.
The importance of the FcγRIIB-mediated regulatory mechanism in the maintenance of a balanced immune response was first proven in FcγRIIB-deficient mice. These animals developed uncontrolled antibody secretion and exacerbated autoimmunity [40–42]. Autoantibody levels are influenced by FcγRIIB also in human autoimmune diseases since reduced levels of FcγRIIB are found in patients with active systemic lupus erythematosus and those with untreated multiple sclerosis [43,44], two autoimmune diseases characterized by increased levels of autoantibodies.
Function and uses of anti-Id
The ability of anti-Id to neutralize potentially pathogenic antibodies is thought to be one of the major mechanisms by which administration of Intravenous Immunoglobulin (IVIg) exerts its therapeutic benefit in the treatment of several autoimmune diseases, as outlined below [45,46].
IVIg preparations consist of pooled IgG fractions from over 10,000 donors and exert their therapeutic effects through different modes of action [47]. Among these is the binding of pathogenic autoantibodies by anti-Id present in the IVIg, which include anti-Id that neutralize or bind to autoantibodies directed to anti-Factor VIII, anti-thyroglobulin, anti-DNA, anti-intrinsic factor, anti-platelet GPIIb/IIIa, and antigens in the cytoplasm of neutrophil granulocytes [48–52]. That anti-Id are the mediator of the therapeutic effect of IVIg is demonstrated by the elimination of the therapeutic action of IVIg after removal of anti-Id and confirmed by the demonstration that the isolated anti-Id fractions restore the therapeutic activity of IVIg [53].
The role of anti-Id in IVIg treatment has been best studied in hemophilia patients. Replacement therapy of hemophilia A patients with coagulation factor VIII (FVIII) can elicit the development of antibodies that neutralize FVIII. These anti-FVIII antibodies are a serious obstacle to the chronic treatment of hemophilia patients. Anti-FVIII autoantibodies can also develop spontaneously in acquired hemophilia [54]. Hemophilia patients are treated successfully with IVIg, which contain anti-Id to anti-FVIII antibodies [55], and anti-Id have been demonstrated to completely neutralize the inhibiting activity of polyclonal anti-FVIII antibodies from hemophilia A patients [56].
Other examples of anti-Id-mediated neutralization of autoantibodies occur in Myasthenia Gravis, Lambert-Eaton syndrome, Immune thrombocytopenic purpura, patients with primary and secondary Sjögren’s syndrome and scleroderma [57–59] and antibody-mediated neuropathies [60–63]. The observation that IVIg contains anti-Id to so many different autoantibodies illustrates how common they are in healthy individuals. The wide prevalence of anti-Id also explains the efficacy of IVIg in the treatment of different autoimmune diseases [47,64].
Anti-Id in autoimmune diseases
Support for the proposed role of anti-Id as regulators in the immune response comes from studies of auto-immune diseases. A protective role of anti-Id against autoimmune diseases is based on two major observations:
anti-Id specific to autoantibodies are present in patients during remission and/or in healthy individuals,
anti-Id are absent during periods of active disease.
This negative correlation between anti-Id and autoimmune disease has been studied in great detail in different forms of systemic lupus erythematosus and autoimmune thyroid diseases. Similar, but less extensive associations have been found in other autoimmune diseases (Table I).
Table I.
Autoantibodies and anti-Id in autoimmune diseases other than those discussed in this review.
| Disease | Target of autoantibodies | Anti-Id | Anti-Id absent during acute disease | Anti-Id present during remission | Possible mechanism | Reference |
|---|---|---|---|---|---|---|
| Membranoproliferative glomerulonephritis | Alternative pathway C3/C5 convertase | Yes | Yes | Yes | Suppression of autoantibody secretion | [107–109] |
| Idiopathic thrombo-cytopenic purpura | Platelets | Yes | Yes | Yes | Suppression of autoantibody secretion | [110–112] |
| ANCA-associated vasculitides | Anti-neutrophil cytoplasmic antibodies (ANCA) | Yes | Yes | Yes | Neutralization of autoantibody | [48,113–116] |
| Sjogren’s Syndrome | Ribonucleoproteins = La/Ro RNP | Yes | Not tested | Not tested | Neutralization of autoantibody | [72] |
| Primary biliary cirrhosis | E2 subunit of pyruvate dehydrogenase complex | Yes | Not tested | Not tested | Neutralization of autoantibody | [117] |
| Myasthenia Gravis | Acetylcholine Receptor | Yes | Not tested | Not tested | Neutralization of autoantibody | [118,119] |
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the presence of autoantibodies directed against nuclear antigens (nucleosomes, DNA, histones) and phospholipids. Available evidence strongly suggests a protective role for anti-Id directed against these autoantibodies.
The most frequently detected autoantibodies in SLE are those against double-stranded DNA (dsDNA). Naturally occurring anti-Id to anti-DNA antibodies can be detected in relatives of patients with SLE [65], individuals who were in contact with such patients [66], and even in healthy controls [67–69]. In marked contrast, these anti-Id are not present in most patients with active SLE [70,71]. However, patients in remission from SLE do show a resurgence of anti-Id. Thus in SLE anti-Id levels are inversely correlated with disease activity [69,72].
Immunization of a mouse model of SLE with the mouse anti-dsDNA monoclonal antibody 3E10 induced the development of anti-Id and was associated with inhibition of anti-dsDNA antibodies and suppression of lupus nephritis [73]. Vaccination of nine humans with SLE with the same antibody in a phase 1 trial induced the development of anti-Id in five patients, who remained disease free for the 2-year follow-up period [74].
The human anti-ssDNA monoclonal antibody 16/6 Id was found to correlate with levels of SLE disease activity, being elevated during active disease [75,76]. Passive administration of anti-Id to 16/6 Id suppressed production of the anti-ssDNA antibody in mice and temporarily attenuated SLE [77].
Another major autoantibody found in patients with SLE is directed against ribosomal P phosphoproteins. These autoantibodies are highly specific for SLE and correlate with disease activity. Stafford et al. discovered concealed autoantibodies to ribosomal P phosphoproteins in healthy individuals and in those SLE patients who had tested negative for autoantibodies in conventional antibody assays [78–80]. These autoantibodies were in complexes with specific anti-Id and were detectable only after the removal of the latter [78].
These findings further support the notion that autoantibodies are present in healthy individuals but concealed by the presence of anti-Id. Thus, although most healthy controls appear to be autoantibody-negative using conventional antibody assays, more sophisticated assays show that they in fact have autoantibodies that are masked by anti-Id (discussed in more detail later).
Neonatal lupus syndrome
Neonatal lupus syndrome (NLS) is a rare systemic autoimmune disease caused by the transplacental passage of maternal autoantibodies to the fetus. These maternal autoantibodies are directed against Ro/SSA, La/SSB, and/or U1-ribonucleoprotein. The mothers often have no symptoms at the time of birth. The affected child shows cardiac and dermatologic symptoms, which have been attributed to Ro/SSA and La/SSB autoantibodies, respectively. Sera from mothers of healthy children exhibit higher anti-Id levels towards anti-La/SSB autoantibodies compared to mothers giving birth to a child with NLS, suggesting that the anti-Id to La/SSB autoantibodies may protect the fetus by blocking the pathogenic maternal autoantibody [81].
Autoimmune thyroid diseases
Autoantibodies present in sera of patients suffering from autoimmune thyroid diseases, such as Graves’ disease and Hashimoto’s thyroditis, are directed to three major autoantigens; thyroid-stimulating hormone receptor (TSHR), thyroid peroxidase, and thyroglobulin. Naturally occurring anti-Id to these autoantibodies have been characterized [82,83] and anti-Id to anti-TSHR are associated with remission in Graves’ disease [84]. Anti-Id to anti-TSHR are also present in 8–30% of untreated patients [85,86] and presence of anti-Id has been linked to a better response of patients with Graves’ disease to anti-thyroid drugs [86]. These findings suggest that an idiotypic network also exists in autoimmune thyroid diseases.
Technical considerations in the detection of anti-Id in sera
The intrinsic capacity of anti-Id to bind to autoantibodies often prevents the detection of the anti-Id, particularly in healthy individuals where both antibodies are present in immune complexes. Thus, in these sera, the bound anti-Id need to be “unmasked” before they can be detected. Likewise, the autoantibodies present in the sera of healthy individuals are usually missed when measured by conventional assays. The reason autoantibodies are routinely detected in patients with autoimmune disease is precisely due to the paucity of anti-Id in these individuals. Several methods are used to “unmask” anti-Id: absorption of autoantibodies to a large excess of immobilized antigen [79], blocking of anti-Id using peptides that present the antigenic determinants of the autoantibody (complementary peptides) [72,81,87], or absorption of anti-Id to immobilized autoantibodies [88]. The removal of inhibitory anti-Id from the serum reveals the presence of autoantibodies in seemingly autoantibody-negative samples [81,87,88]. The presence of anti-Id can be confirmed by the re-addition of the isolated anti-Id to the autoantibody, resulting in the inhibition of autoantibody binding to the antigen [66,78].
The function of these concealed autoantibodies is currently unclear. However, what is becoming increasingly clear is that when a serum is found to be autoantibody negative in conventional autoantibody ligand binding assays, this can be due to the absence of autoantibodies or the presence of anti-Id in complex with autoantibodies.
Anti-Id in Type 1 diabetes
Work suggesting a protective role of anti-Id in Type 1 Diabetes (T1D) is beginning to emerge, consistent with the more extensive data on the role for anti-Id in SLE and the inverse correlation of anti-Id with clinical disease in other autoimmune diseases, detailed above and in Table I. Progression to T1D is characterized by the presence of circulating autoantibodies directed against several beta cell autoantigens: insulin, the smaller isoform of glutamate decarboxylase (GAD65), Insulinoma 2-associated protein, and the Zinc Transporter 8 protein (for review see [89]). Although the pathological role of these autoantibodies has not been established, the notion that autoantibodies may be involved in the pathogenesis of T1D has received recent support from the findings that GAD65 autoantibodies (GAD65Ab) enhance presentation of GAD65-derived peptides and can promote cytotoxic T cell responses [90–92].
IVIg treatment of T1D was attempted with mixed results depending on the duration of disease and duration of IVIg treatment [93–97]. However, all studies had at least one subgroup of patients who showed some benefit from IVIg treatment, with higher rates and longer durations of remission and preserved c-peptide release, indicative of improved beta cell function [98].
In T1D, anti-Id against insulin autoantibodies were first described in both humans [99] and the BB rat model [100]. The presence of GAD65Ab-specific anti-Id was initially suggested by the observation of an immunoglobulin inhibiting the binding of GAD65Ab to GAD65 in human sera. However, the lack of appropriate monoclonal antibodies prevented further identification of this inhibitor [101]. We subsequently observed that NOD mice injected with a human GAD65Ab monoclonal antibody develop high titers of anti-Id [102]. Since these animals did not develop diabetes and had reduced levels of insulitis, the induction of anti-Id was linked to prevention of T1D in this animal model.
To determine whether a GAD65Ab specific Idiotypic Network is present in humans, we tested a large cohort of healthy individuals and found that their sera contained both anti-Id and GAD65Ab in complexes [88], although dissociation of the complexes was necessary for the detection of both. This finding suggests that GAD65Ab are present in the majority of healthy individuals, rather than being restricted to T1D, and that anti-Id normally bind GAD65Ab and, as discussed above, mask their detection. By extension, these findings suggest that loss of anti-Id may be associated with the development of T1D. This latter hypothesis has received further support from our finding that anti-Id specific to GAD65Ab are low or missing in T1D patients [88] and that anti-Id levels increase in T1D patients who experienced a temporary remission after diagnosis of disease (honeymoon period). In contrast, those patients who did not undergo a remission phase did not show an increase of anti-Id levels [103]. This correlation of increased anti-Id with improved beta cell function has also been found in Latent Autoimmune Diabetes in Adults (LADA) patients who received alum-GAD vaccination [103]. In this instance, patients who experienced an improvement in beta-cell function also showed an increase in anti-Id levels, while patients in the placebo groups or patients who did not respond to the vaccination, did not demonstrate such an increase. Although these findings do not establish causality, they support the hypothesis that anti-Id protect against the development and progression of T1D. These studies are critically needed to test this hypothesis to identify the mechanism by which such protection occurs.
Moreover, it is likely that the by us described GAD65-specific Idiotypic Network is not the only network present in T1D. Because of the scarcity of human monoclonal autoantibodies to other major autoantigens in T1D – insulin, Insulinoma 2-associated protein, and the Zinc Transporter 8 protein – we have not yet tested whether also these autoantibodies and their anti-Id are present in healthy individuals. Until these reagents become available, we propose GAD65 and its Idiotypic Network as a model system for the study of the mechanisms by which anti-Id may be involved in the immune system.
As briefly mentioned here and reviewed in detail elsewhere [104], prevention and intervention therapy of T1D in humans still faces difficulties. Existing therapies are either only marginally successful, or cause side effects that may outweigh the benefits. Further work establishing a possible protective function of anti-Id in T1D is therefore warranted.
Model for the failure of the Idiotypic Network in T1D
Our model of a GAD65Ab-specific Idiotypic Network in T1D incorporates our hypothesis of how loss of anti-Id occurs and its possible consequences. In the healthy immune system GAD65Ab, anti-Id, and T cells are in equilibrium. According to the Network Hypothesis, introduction of antigen, GAD65, stimulates the production of GAD65Ab. GAD65Ab in turn trigger an increase in anti-Id, thus restoring the equilibrium. (Figure 6 left panel). Indeed, vaccination with GAD65 has been shown to induce a temporary increase in GAD65Ab [105,106]. Whether the subsequent decrease in GAD65Ab titer is caused by an increase in anti-Id is an important, but unanswered, question. Prolonged release of GAD65, e.g., during destruction of pancreatic beta cells and release of islet cell autoantigens, is predicted to continuously stimulate GAD65Ab secretion (Figure 6 right panel).
Figure 6.

Model for the Failure of the Idiotypic Network in T1D during prolonged GAD65 exposure. Left panel: In the healthy individual, transient introduction of GAD65 (1) will cause a temporary disturbance of the Network’s equilibrium. GAD65 will stimulate the secretion of GAD65Ab, which will in turn stimulate the secretion of anti-Id from specific B lymphocytes (2), restoring the equilibrium (3). Right panel: The continuous presence of GAD65 (beta cell injury/death) will destroy the Network. As before, GAD65 stimulates secretion of GAD65Ab (1), which initially will trigger the production of anti-Id (2). However, GAD65 itself binds to the BCR on GAD65Ab-specific B lymphocytes, both stimulating these specific B lymphocytes and preventing the anti-Id from co-ligating the BCR with the FcγRIIB (3). With increasing GAD65Ab levels, co-ligation of the BCR with the FcγRIIB on anti-Id specific B lymphocytes (4) will generate a net inhibitory signal, resulting first in the inhibition of anti-Id secretion and eventually in the apoptosis of anti-Id specific B lymphocytes.
In addition, GAD65 itself competes with anti-Id for binding to membrane-bound GAD65Ab, thus preventing both co-ligation of the BCR with FcγRIIB by anti-Id and downregulation of GAD65Ab secretion. Therefore, when the autoantigen is present continuously, the GAD65Ab-specific B lymphocytes will receive a net stimulatory signal. At the same time anti-Id specific B lymphocytes receive a net inhibitory signal as detailed below, resulting in down-regulated anti-Id secretion. Based on observations in other systems, continuous suppression of anti-Id secretion will eventually lead to the apoptosis of anti-Id specific B lymphocytes [38,39]. Loss of anti-Id will then permanently remove the inhibition of GAD65 binding to GAD65Ab [88] and allow GAD65Ab-mediated antigen presentation and prolonged stimulation of autoreactive T cells.
Future directions
Further studies are needed to prove that GAD65Ab-specific anti-Id protect individuals from developing T1D.
In addition, to determine the specific steps leading to the development of GAD65Ab-specific anti-Id, it will be necessary to analyze GAD65Ab and anti-Id levels in longitudinal samples obtained from individuals who go on to develop T1D. We expect that such an analysis will reveal that during progression to T1D GAD65Ab levels increase as anti-Id levels decrease.
To support our hypothesis that anti-Id develop as a consequence of increased GAD65Ab levels, it will be necessary to investigate anti-Id levels in individuals who were vaccinated with GAD65 and show a transient rise in GAD65Ab levels. We predict that this temporary rise in GAD65Ab levels will be followed by an increase in anti-Id levels and is responsible for the later masking of GAD65Ab.
Finally, it is essential to demonstrate that anti-Id protect against the development of T1D. Injection of NOD mice with GAD65Ab-specific anti-Id before and after development of T1D followed by a careful analysis of the different components of the immune response, such as T cells and infiltrating lymphocytes should establish whether anti-Id can prevent T1D or cause its remission in this animal model. Such studies evaluating the efficacy of monoclonal anti-Id specific to GAD65Ab to prevent T1D in animals are currently underway. Positive outcomes of these experiments may lead to the development of a new therapeutic tool for the prevention of human T1D.
Acknowledgments
The author gratefully acknowledges the dedicated and critical help in writing this review from Drs. Gerald J. Taborsky Jr. and Jerry P. Palmer (University of Washington, Seattle).
Nomenclature
- Idiotype (Id)
Antigenic determinants of an antibody directed to a particular antigen; found only in the variable region
- Anti-Idiotypic antibody (anti-Id)
Antibody directed towards the idiotype of an antibody.
- Hypervariable regions:
Portions of the light and heavy immunoglobulin chains with highly variable amino acid sequences, that constitute the antigen-binding site of the antibody.
- MHC class II
Complex expressed on antigen presenting cells, that present antigen to CD4 + T cells.
- Fragment of antibody (Fab)
Fragment of antibody that contains the antigen-binding site.
- Intravenous immunoglobulin (IVIg)
Pooled IgG from > 1000 blood donors, used for the treatment for autoimmune diseases.
- Affinity maturation:
Affinity-selected differentiation of activated B cells, which increases the affinity of the antibody for the antigen.
- Plasma cell
B lymphocyte specialized in producing antibodies.
- Memory cells
B lymphocytes with prolonged life span that enable the immune system to respond faster to a second exposure.
- B-cell antigen receptor (BCR)
Membrane bound antibody expressed on B lymphocytes.
- FcγRIIB
Inhibitory receptor. Only IgG receptor expressed on B-lymphocytes.
- Internal image
The part of an anti-Id that is structurally complementary to the idiotype.
Footnotes
Declaration of Interest statement: The author reports no conflicts of interest. This work was performed as independent research sponsored by the National Institutes of Health (DK26190, and DK17047), and a Basic Science Award from the American Diabetes Association. The author is responsible for the content and writing of this paper.
References
- 1.Kunkel HG, Mannik M, Williams RC. Individual Antigenic Specificity of Isolated Antibodies. Science. 1963;140:1218–1219. doi: 10.1126/science.140.3572.1218. [DOI] [PubMed] [Google Scholar]
- 2.Jerne NK. Towards a network theory of the immune system. Ann Immunol (Paris) 1974;125C:373–389. [PubMed] [Google Scholar]
- 3.Jerne NK. Clonal selection in a lymphocyte network. Soc Gen Physiol Ser. 1974;29:39–48. [PubMed] [Google Scholar]
- 4.Jerne NK. The immune system. Sci Am. 1973;229:52–60. doi: 10.1038/scientificamerican0773-52. [DOI] [PubMed] [Google Scholar]
- 5.Kim BS. Mechanisms of idiotype suppression: role of anti-idiotype antibody. Surv Immunol Res. 1982;1:126–132. doi: 10.1007/BF02918336. [DOI] [PubMed] [Google Scholar]
- 6.Rodkey LS. Autoregulation of immune responses via idiotype network interactions. Microbiol Rev. 1980;44:631–659. doi: 10.1128/mr.44.4.631-659.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzioufas AG, Routsias JG. Idiotype, anti-idiotype network of autoantibodies. Pathogenetic considerations and clinical application. Autoimmun Rev. 2010;9:631–633. doi: 10.1016/j.autrev.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Behn U. Idiotypic networks: toward a renaissance? Immunol Rev. 2007;216:142–152. doi: 10.1111/j.1600-065X.2006.00496.x. [DOI] [PubMed] [Google Scholar]
- 9.Coutinho A. Will the idiotypic network help to solve natural tolerance? Trends Immunol. 2003;24:53–54. doi: 10.1016/s1471-4906(02)00035-2. [DOI] [PubMed] [Google Scholar]
- 10.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark LA, Ganesan S, Papp S, van Vlijmen HW. Trends in antibody sequence changes during the somatic hypermutation process. J Immunol. 2006;177:333–340. doi: 10.4049/jimmunol.177.1.333. [DOI] [PubMed] [Google Scholar]
- 12.Bogen B, Ruffini P. Review: to what extent are T cells tolerant to immunoglobulin variable regions? Scand J Immunol. 2009;70:526–530. doi: 10.1111/j.1365-3083.2009.02340.x. [DOI] [PubMed] [Google Scholar]
- 13.Bogen B, Malissen B, Haas W. Idiotope-specific T cell clones that recognize syngeneic immunoglobulin fragments in the context of class II molecules. Eur J Immunol. 1986;16:1373–1378. doi: 10.1002/eji.1830161110. [DOI] [PubMed] [Google Scholar]
- 14.Eyerman MC, Zhang X, Wysocki LJ. T cell recognition and tolerance of antibody diversity. J Immunol. 1996;157:1037–1046. [PubMed] [Google Scholar]
- 15.Janeway CA, Jr, Sakato N, Eisen HN. Recognition of immunoglobulin idiotypes by thymus-derived lymphocytes. Proc Natl Acad Sci USA. 1975;72:2357–2360. doi: 10.1073/pnas.72.6.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jerne NK, Roland J, Cazenave PA. Recurrent idiotopes and internal images. Embo J. 1982;1:243–247. doi: 10.1002/j.1460-2075.1982.tb01154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrater AF, Goidl EA, Thorbecke GJ, Siskind GW. Production of auto-anti-idiotypic antibody during the normal immune response to TNP-ficoll. III. Absence in nu/nu mice: evidence for T-cell dependence of the anti-idiotypic-antibody response. J Exp Med. 1979;150:808–817. doi: 10.1084/jem.150.4.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papamattheou MG, Routsias JG, Karagouni EE, Sakarellos C, Sakarellos-Daitsiotis M, Moutsopoulos HM, Tzioufas AG, Dotsika EN. T cell help is required to induce idiotypic-anti-idiotypic autoantibody network after immunization with complementary epitope 289–308aa of La/SSB autoantigen in non-autoimmune mice. Clin Exp Immunol. 2004;135:416–426. doi: 10.1111/j.1365-2249.2004.02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss S, Bogen B. B-lymphoma cells process and present their endogenous immunoglobulin to major histocompatibility complex-restricted T cells. Proc Natl Acad Sci USA. 1989;86:282–286. doi: 10.1073/pnas.86.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King CA, Wills MR, Hamblin TJ, Stevenson FK. Idiotypic IgM on a B-cell surface requires processing for recognition by anti-idiotypic T cells. Cell Immunol. 1993;147:411–424. doi: 10.1006/cimm.1993.1080. [DOI] [PubMed] [Google Scholar]
- 21.Tite JP, Kaye J, Saizawa KM, Ming J, Katz ME, Smith LA, Janeway CA., Jr Direct interactions between B and T lymphocytes bearing complementary receptors. J Exp Med. 1986;163:189–202. doi: 10.1084/jem.163.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reitan SK, Hannestad K. The primary IgM antibody repertoire: a source of potent idiotype immunogens. Eur J Immunol. 2001;31:2143–2153. doi: 10.1002/1521-4141(200107)31:7<2143::aid-immu2143>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Jacobsen JT, Lunde E, Sundvold-Gjerstad V, Munthe LA, Bogen B. The cellular mechanism by which complementary Id(+) and anti-Id antibodies communicate: T cells integrated into idiotypic regulation. Immunol Cell Biol. 2010;55:515–522. doi: 10.1038/icb.2009.118. [DOI] [PubMed] [Google Scholar]
- 24.Snyder CM, Aviszus K, Heiser RA, Tonkin DR, Guth AM, Wysocki LJ. Activation and tolerance in CD4(+) T cells reactive to an immunoglobulin variable region. J Exp Med. 2004;200:1–11. doi: 10.1084/jem.20031234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munthe LA, Corthay A, Os A, Zangani M, Bogen B. Systemic autoimmune disease caused by autoreactive B cells that receive chronic help from Ig V region-specific T cells. J Immunol. 2005;175:2391–2400. doi: 10.4049/jimmunol.175.4.2391. [DOI] [PubMed] [Google Scholar]
- 26.Weiss S, Bogen B. MHC class II-restricted presentation of intracellular antigen. Cell. 1991;64:767–776. doi: 10.1016/0092-8674(91)90506-t. [DOI] [PubMed] [Google Scholar]
- 27.Munthe LA, Kyte JA, Bogen B. Resting small B cells present endogenous immunoglobulin variable-region determinants to idiotope-specific CD4(+) T cells in vivo. Eur J Immunol. 1999;29:4043–4052. doi: 10.1002/(SICI)1521-4141(199912)29:12<4043::AID-IMMU4043>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Nayak R, Mitra-Kaushik S, Shaila MS. Perpetuation of immunological memory: a relay hypothesis. Immunology. 2001;102:387–395. doi: 10.1046/j.1365-2567.2001.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vani J, Nayak R, Shaila MS. Maintenance of antigen-specific immunological memory through variable regions of heavy and light chains of anti-idiotypic antibody. Immunology. 2007;120:486–496. doi: 10.1111/j.1365-2567.2006.02519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geha RS. Presence of circulating anti-idiotype-bearing cells after booster immunization with tetanus toxoid (TT) and inhibition of anti-TTantibody synthesis by auto-anti-idiotypic antibody. J Immunol. 1983;130:1634–1639. [PubMed] [Google Scholar]
- 31.Sapir T, Shoenfeld Y. Facing the enigma of immunomodulatory effects of intravenous immunoglobulin. Clin Rev Allergy Immunol. 2005;29:185–199. doi: 10.1385/CRIAI:29:3:185. [DOI] [PubMed] [Google Scholar]
- 32.Wallmann J, Pali-Scholl I, Jensen-Jarolim E. Anti-Ids in Allergy: Timeliness of a Classic Concept. World Allergy Organiz J. 2010;3:195–201. doi: 10.1097/WOX.0b013e3181e61ebf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kojima K, Matsuyama T, Tanaka H. Suppression of in vitro human antithyroglobulin antibody secretion by private and cross-reactive anti-idiotypic antibodies. Clin Immunol Immunopathol. 1986;39:337–344. doi: 10.1016/0090-1229(86)90097-8. [DOI] [PubMed] [Google Scholar]
- 34.Horsfall AC, Mumford PA, Venables PJ, Maini RN. Anti-idiotypic induced suppression of Sjogren’s syndrome associated anti-La autoantibody secretion in vitro. Clin Exp Immunol. 1988;71:62–66. [PMC free article] [PubMed] [Google Scholar]
- 35.Koide J, Takeuchi T, Hosono O, Takano M, Abe T. Suppression of in vitro production of anti-U1-ribonucleoprotein antibody by monoclonal anti-idiotypic antibody to anti-U1-ribonucleoprotein antibody. Scand J Immunol. 1988;28:687–696. doi: 10.1111/j.1365-3083.1988.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 36.Bolland S, Pearse RN, Kurosaki T, Ravetch JV. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity. 1998;8:509–516. doi: 10.1016/s1074-7613(00)80555-5. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Oliveira-Dos-Santos AJ, Mariathasan S, Bouchard D, Jones J, Sarao R, Kozieradzki I, Ohashi PS, Penninger JM, Dumont DJ. The inositol polyphosphate 5-phosphatase ship is a crucial negative regulator of B cell antigen receptor signaling. J Exp Med. 1998;188:1333–1342. doi: 10.1084/jem.188.7.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brauweiler AM, Cambier JC. Autonomous SHIP-dependent FcgammaR signaling in pre-B cells leads to inhibition of cell migration and induction of cell death. Immunol Lett. 2004;92:75–81. doi: 10.1016/j.imlet.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 39.Ashman RF, Peckham D, Stunz LL. Fc receptor off-signal in the B cell involves apoptosis. J Immunol. 1996;157:5–11. [PubMed] [Google Scholar]
- 40.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2:580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 41.Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J Exp Med. 1999;189:179–185. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura A, Yuasa T, Ujike A, Ono M, Nukiwa T, Ravetch JV, Takai T. Fcgamma receptor IIB-deficient mice develop Goodpasture’s syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. J Exp Med. 2000;191:899–906. doi: 10.1084/jem.191.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su K, Yang H, Li X, Gibson AW, Cafardi JM, Zhou T, Edberg JC, Kimberly RP. Expression profile of FcgammaR-IIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. 2007;178:3272–3280. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tackenberg B, Jelcic I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, Lunemann JD. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci USA. 2009;106:4788–4792. doi: 10.1073/pnas.0807319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazatchkine MD, Kaveri SV. Immunomodulation of auto-immune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747–755. doi: 10.1056/NEJMra993360. [DOI] [PubMed] [Google Scholar]
- 46.Rossi F, Dietrich G, Kazatchkine MD. Anti-idiotypes against autoantibodies in normal immunoglobulins: evidence for network regulation of human autoimmune responses. Immunol Rev. 1989;110:135–149. doi: 10.1111/j.1600-065x.1989.tb00031.x. [DOI] [PubMed] [Google Scholar]
- 47.Negi VS, Elluru S, Siberil S, Graff-Dubois S, Mouthon L, Kazatchkine MD, Lacroix-Desmazes S, Bayry J, Kaveri SV. Intravenous immunoglobulin: an update on the clinical use and mechanisms of action. J Clin Immunol. 2007;27:233–245. doi: 10.1007/s10875-007-9088-9. [DOI] [PubMed] [Google Scholar]
- 48.Rossi F, Jayne DR, Lockwood CM, Kazatchkine MD. Anti-idiotypes against anti-neutrophil cytoplasmic antigen autoantibodies in normal human polyspecific IgG for therapeutic use and in the remission sera of patients with systemic vasculitis. Clin Exp Immunol. 1991;83:298–303. doi: 10.1111/j.1365-2249.1991.tb05631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans MJ, Suenaga R, Abdou NI. Detection and purification of antiidiotypic antibody against anti-DNA in intravenous immune globulin. J Clin Immunol. 1991;11:291–295. doi: 10.1007/BF00918187. [DOI] [PubMed] [Google Scholar]
- 50.Dietrich G, Kazatchkine MD. Normal immunoglobulin G (IgG) for therapeutic use (intravenous Ig) contain anti-idiotypic specificities against an immunodominant, disease-associated, cross-reactive idiotype of human anti-thyroglobulin autoantibodies. J Clin Invest. 1990;85:620–625. doi: 10.1172/JCI114483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dietrich G, Pereira P, Algiman M, Sultan Y, Kazatchkine MD. A monoclonal anti-idiotypic antibody against the antigen-combining site of anti-factor VIII autoantibodies defines and idiotope that is recognized by normal human polyspecific immunoglobulins for therapeutic use (IVIg) J Autoimmun. 1990;3:547–557. doi: 10.1016/s0896-8411(05)80020-4. [DOI] [PubMed] [Google Scholar]
- 52.Berchtold P, Dale GL, Tani P, McMillan R. Inhibition of autoantibody binding to platelet glycoprotein IIb/IIIa by anti-idiotypic antibodies in intravenous gammaglobulin. Blood. 1989;74:2414–2417. [PubMed] [Google Scholar]
- 53.Fuchs S, Feferman T, Meidler R, Margalit R, Sicsic C, Brenner T, Laub O, Souroujon MC. Immunosuppression of EAMG by IVIG is mediated by a disease-specific anti-immunoglobulin fraction. Ann NY Acad Sci. 2008;1132:244–248. doi: 10.1196/annals.1405.032. [DOI] [PubMed] [Google Scholar]
- 54.Shetty S, Bhave M, Ghosh K. Acquired hemophilia a: diagnosis, aetiology, clinical spectrum and treatment options. Autoimmun Rev. 2011;10:311–316. doi: 10.1016/j.autrev.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 55.Sultan Y, Kazatchkine MD, Maisonneuve P, Nydegger UE. Anti-idiotypic suppression of autoantibodies to factor VIII (antihaemophilic factor) by high-dose intravenous gamma-globulin. Lancet. 1984;2:765–768. doi: 10.1016/s0140-6736(84)90701-3. [DOI] [PubMed] [Google Scholar]
- 56.Gilles JG, Grailly SC, De Maeyer M, Jacquemin MG, VanderElst LP, Saint-Remy JM. In vivo neutralization of a C2 domain-specific human anti-Factor VIII inhibitor by an anti-idiotypic antibody. Blood. 2004;103:2617–2623. doi: 10.1182/blood-2003-07-2207. [DOI] [PubMed] [Google Scholar]
- 57.Waterman SA, Gordon TP, Rischmueller M. Inhibitory effects of muscarinic receptor autoantibodies on parasympathetic neurotransmission in Sjogren’s syndrome. Arthritis Rheum. 2000;43:1647–1654. doi: 10.1002/1529-0131(200007)43:7<1647::AID-ANR31>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 58.Goldblatt F, Gordon TP, Waterman SA. Antibody-mediated gastrointestinal dysmotility in scleroderma. Gastroenterology. 2002;123:1144–1150. doi: 10.1053/gast.2002.36057. [DOI] [PubMed] [Google Scholar]
- 59.Cavill D, Waterman SA, Gordon TP. Antiidiotypic antibodies neutralize autoantibodies that inhibit cholinergic neurotransmission. Arthritis Rheum. 2003;48:3597–3602. doi: 10.1002/art.11343. [DOI] [PubMed] [Google Scholar]
- 60.Dalakas M. Intravenous immune globulin therapy for neurologic diseases. Ann Intern Med. 1997;126:721–730. doi: 10.7326/0003-4819-126-9-199705010-00008. [DOI] [PubMed] [Google Scholar]
- 61.Liblau R, Gajdos P, Bustarret FA, el Habib R, Bach JF, Morel E. Intravenous gamma-globulin in myasthenia gravis: interaction with anti-acetylcholine receptor autoantibodies. J Clin Immunol. 1991;11:128–131. doi: 10.1007/BF00918680. [DOI] [PubMed] [Google Scholar]
- 62.Bain PG, Motomura M, Newsom-Davis J, Misbah SA, Chapel HM, Lee ML, Vincent A, Lang B. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47:678–683. doi: 10.1212/wnl.47.3.678. [DOI] [PubMed] [Google Scholar]
- 63.Imbach P, Barandun S, d’Apuzzo V, Baumgartner C, Hirt A, Morell A, Rossi E, Schoni M, Vest M, Wagner HP. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981;1:1228–1231. doi: 10.1016/s0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- 64.Lacroix-Desmazes S, Mouthon L, Spalter SH, Kaveri S, Kazatchkine MD. Immunoglobulins and the regulation of autoimmunity through the immune network. Clin Exp Rheumatol. 1996;14(Suppl 15):S9–S15. [PubMed] [Google Scholar]
- 65.Abdou NI, Suenaga R, Hatfield M, Evans M, Hassanein KM. Antiidiotypic antibodies against anti-DNA antibodies in sera of families of lupus patients. J Clin Immunol. 1989;9:16–21. doi: 10.1007/BF00917123. [DOI] [PubMed] [Google Scholar]
- 66.Abdou NI, Wall H, Lindsley HB, Halsey JF, Suzuki T. Network theory in autoimmunity.In vitro suppression of serum anti-DNA antibody binding to DNA by anti-idiotypic antibody in systemic lupus erythematosus. J Clin Invest. 1981;67:1297–1304. doi: 10.1172/JCI110158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taniguchi O, Chia DS, Barnett EV. Auto-anti-anti-DNA antibodies from SLE patients and normals. J Rheumatol. 1984;11:291–297. [PubMed] [Google Scholar]
- 68.Zouali M, Eyquem A. Expression of anti-idiotypic clones against auto-anti-DNA antibodies in normal individuals. Cell Immunol. 1983;76:137–147. doi: 10.1016/0008-8749(83)90356-8. [DOI] [PubMed] [Google Scholar]
- 69.Williams WM, Isenberg DA. Naturally occurring anti-idiotypic antibodies reactive with anti-DNA antibodies in systemic lupus erythematosus. Lupus. 1998;7:164–175. doi: 10.1191/096120398678919958. [DOI] [PubMed] [Google Scholar]
- 70.Silvestris F, Bankhurst AD, Searles RP, Williams RC., Jr Studies of anti-F(ab’)2 antibodies and possible immunologic control mechanisms in systemic lupus erythematosus. Arthritis Rheum. 1984;27:1387–1396. doi: 10.1002/art.1780271209. [DOI] [PubMed] [Google Scholar]
- 71.Williams RC, Jr, Malone CC, Huffman GR, Silvestris F, Croker BP, Ayoub EM, Massengill S. Active systemic lupus erythematosus is associated with depletion of the natural generic anti-idiotype (anti-F(ab’)2) system. J Rheumatol. 1995;22:1075–1085. [PubMed] [Google Scholar]
- 72.Routsias JG, Touloupi E, Dotsika E, Moulia A, Tsikaris V, Sakarellos C, Sakarellos-Daitsiotis M, Moutsopoulos HM, Tzioufas AG. Unmasking the anti-La/SSB response in sera from patients with Sjogren’s syndrome by specific blocking of anti-idiotypic antibodies to La/SSB antigenic determinants. Mol Med. 2002;8:293–305. [PMC free article] [PubMed] [Google Scholar]
- 73.Weisbart RH, Noritake DT, Wong AL, Chan G, Kacena A, Colburn KK. A conserved anti-DNA antibody idiotype associated with nephritis in murine and human systemic lupus erythematosus. J Immunol. 1990;144:2653–2658. [PubMed] [Google Scholar]
- 74.Spertini F, Leimgruber A, Morel B, Khazaeli MB, Yamamoto K, Dayer JM, Weisbart RH, Lee ML. Idiotypic vaccination with a murine anti-dsDNA antibody: phase I study in patients with nonactive systemic lupus erythematosus with nephritis. J Rheumatol. 1999;26:2602–2608. [PubMed] [Google Scholar]
- 75.Isenberg DA, Shoenfeld Y, Madaio MP, Rauch J, Reichlin M, Stollar BD, Schwartz RS. Anti-DNA antibody idiotypes in systemic lupus erythematosus. Lancet. 1984;2:417–422. doi: 10.1016/s0140-6736(84)92904-0. [DOI] [PubMed] [Google Scholar]
- 76.Galeazzi M, Bellisai F, Sebastiani GD, Morozzi G, Marcolongo R, Houssiau F, Cervera R, Levy Y, George J, Sherer Y, et al. Association of 16/6 and SA1 anti-DNA idiotypes with anticardiolipin antibodies and clinical manifestations in a large cohort of SLE patients. European Concerted Action on the Immunogenetics of SLE. Clin Exp Rheumatol. 1998;16:717–720. [PubMed] [Google Scholar]
- 77.Blank M, Manosroi J, Tomer Y, Manosroi A, Kopolovic J, Charcon-Polak S, Shoenfeld Y. Suppression of experimental systemic lupus erythematosus (SLE) with specific anti-idiotypic antibody-saporin conjugate. Clin Exp Immunol. 1994;98:434–441. doi: 10.1111/j.1365-2249.1994.tb05509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pan ZJ, Anderson CJ, Stafford HA. Anti-idiotypic antibodies prevent the serologic detection of antiribosomal P autoantibodies in healthy adults. J Clin Invest. 1998;102:215–222. doi: 10.1172/JCI1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stafford HA, Anderson CJ, Reichlin M. Unmasking of antiribosomal P autoantibodies in healthy individuals. J Immunol. 1995;155:2754–2761. [PubMed] [Google Scholar]
- 80.Anderson CJ, Neas BR, Pan Z, Taylor-Albert E, Reichlin M, Stafford HA. The presence of masked antiribosomal P autoantibodies in healthy children. Arthritis Rheum. 1998;41:33–40. doi: 10.1002/1529-0131(199801)41:1<33::AID-ART5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 81.Stea EA, Routsias JG, Clancy RM, Buyon JP, Moutsopoulos HM, Tzioufas AG. Anti-La/SSB antiidiotypic antibodies in maternal serum: a marker of low risk for neonatal lupus in an offspring. Arthritis Rheum. 2006;54:2228–2234. doi: 10.1002/art.21954. [DOI] [PubMed] [Google Scholar]
- 82.Sikorska HM. Anti-thyroglobulin anti-idiotypic antibodies in sera of patients with Hashimoto’s thyroiditis and Graves’ disease. J Immunol. 1986;137:3786–3795. [PubMed] [Google Scholar]
- 83.Tandon N, Jayne DR, McGregor AM, Weetman AP. Analysis of anti-idiotypic antibodies against anti-microsomal antibodies in patients with thyroid autoimmunity. J Autoimmun. 1992;5:557–570. doi: 10.1016/0896-8411(92)90153-h. [DOI] [PubMed] [Google Scholar]
- 84.Paschke R, Teuber J, Enger I, Schmeidl R, Schwedes U, Usadel KH. Evidence for a role of anti-idiotypic antibodies in the induction of remission in Graves’ disease. J Autoimmun. 1990;3:441–448. doi: 10.1016/s0896-8411(05)80011-3. [DOI] [PubMed] [Google Scholar]
- 85.Balazs C, Molnar I. In vitro suppression of anti-TSH receptor antibody by autologous anti-idiotypic antibody in patients with Graves’ disease. Acta Microbiol Immunol Hung. 1995;42:163–169. [PubMed] [Google Scholar]
- 86.Tada H, Izumi Y, Watanabe Y, Takano T, Fukata S, Kuma K, Hidaka Y, Amino N. Blocking type anti-tSH receptor antibodies detected by radioreceptor assay in Graves’ disease. Endocr J. 2001;48:703–710. doi: 10.1507/endocrj.48.703. [DOI] [PubMed] [Google Scholar]
- 87.Kohler H, Kaveri S, Kieber-Emmons T, Morrow WJ, Muller S, Raychaudhuri S. Idiotypic networks and nature of molecular mimicry: an overview. Meth Enzymol. 1989;178:3–35. doi: 10.1016/0076-6879(89)78003-4. [DOI] [PubMed] [Google Scholar]
- 88.Oak S, Gilliam LK, Landin-Olsson M, Torn C, Kockum I, Pennington CR, Rowley MJ, Christie MR, Banga JP, Hampe CS. The lack of anti-idiotypic antibodies, not the presence of the corresponding autoantibodies to glutamate decarboxylase, defines type 1 diabetes. Proc Natl Acad Sci USA. 2008;105:5471–5476. doi: 10.1073/pnas.0800578105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Winter WE, Schatz DA. Autoimmune markers in diabetes. Clin Chem. 2011;57:168–175. doi: 10.1373/clinchem.2010.148205. [DOI] [PubMed] [Google Scholar]
- 90.Reijonen H, Daniels TL, Lernmark A, Nepom GT. GAD65-specific autoantibodies enhance the presentation of an immunodominant T-cell epitope from GAD65. Diabetes. 2000;49:1621–1626. doi: 10.2337/diabetes.49.10.1621. [DOI] [PubMed] [Google Scholar]
- 91.Banga JP, Moore JK, Duhindan N, Madec AM, Van Endert PM, Orgiazzi J, Endl J. Modulation of antigen presentation by autoreactive B cell clones specific for GAD65 from a type I diabetic patient. Clin Exp Immunol. 2004;135:74–84. doi: 10.1111/j.1365-2249.2004.02343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jaume JC, Parry SL, Madec AM, Sonderstrup G, Baekkeskov S. Suppressive effect of glutamic acid decarboxylase 65-specific autoimmune B lymphocytes on processing of T cell determinants located within the antibody epitope. J Immunol. 2002;169:665–672. doi: 10.4049/jimmunol.169.2.665. [DOI] [PubMed] [Google Scholar]
- 93.Panto F, Giordano C, Amato MP, Pugliese A, Donatelli M, D’Acquisto G, Galluzzo A. The influence of high dose intravenous immunoglobulins on immunological and metabolic pattern in newly diagnosed type I diabetic patients. J Autoimmun. 1990;3:587–592. doi: 10.1016/s0896-8411(05)80025-3. [DOI] [PubMed] [Google Scholar]
- 94.Colagiuri S, Leong GM, Thayer Z, Antony G, Dwyer JM, Kidson W, Wakefield D. Intravenous immunoglobulin therapy for autoimmune diabetes mellitus. Clin Exp Rheumatol. 1996;14(Suppl 15):S93–S97. [PubMed] [Google Scholar]
- 95.Pocecco M, De Campo C, Cantoni L, Tedesco F, Panizon F. Effect of high doses intravenous IgG in newly diagnosed diabetic children. Helv Paediatr Acta. 1987;42:289–295. [PubMed] [Google Scholar]
- 96.Heinze E, Thon A, Vetter U, Gaedicke G, Zuppinger K. Gamma-globulin therapy in 6 newly diagnosed diabetic children. Acta Paediatr Scand. 1985;74:605–606. doi: 10.1111/j.1651-2227.1985.tb11039.x. [DOI] [PubMed] [Google Scholar]
- 97.Urakami T, Hanaoka Y, Fujita H, Kitagawa T. High-dose gammaglobulin therapy in six children with newly diagnosed insulin-dependent diabetes mellitus. Acta Paediatr Jpn. 1987;29:355–360. doi: 10.1111/j.1442-200x.1987.tb00330.x. [DOI] [PubMed] [Google Scholar]
- 98.Heinze E. Immunoglobulins in children with autoimmune diabetes mellitus. Clin Exp Rheumatol. 1996;14(Suppl 15):S99–102. [PubMed] [Google Scholar]
- 99.Casiglia D, Giardina E, Triolo G. IgG auto-anti-idiotype antibodies against antibody to insulin in insulin-dependent (type 1) diabetes mellitus. Detection by capture enzyme linked immunosorbent assay (ELISA) and relationship with anti-insulin antibody levels. Diabetes Res. 1991;16:181–184. [PubMed] [Google Scholar]
- 100.Elias D, Bone AJ, Baird JD, Cooke A, Cohen IR. Insulin-mimicking anti-idiotypic antibodies in development of spontaneous autoimmune diabetes in BB/E rats. Diabetes. 1990;39:1467–1471. doi: 10.2337/diab.39.12.1467. [DOI] [PubMed] [Google Scholar]
- 101.Brown TJ, Christie MR. Detection of a serum factor that blocks antibody binding to glutamate decarboxylase. Diabe-tologia. 1993;(Suppl 1):A92. (Abstr.) [Google Scholar]
- 102.Hall TR, Bogdani M, Leboeuf RC, Kirk EA, Maziarz M, Banga JP, Oak S, Pennington CA, Hampe CS. Modulation of diabetes in NOD mice by GAD65-specific monoclonal antibodies is epitope specific and accompanied by anti-idiotypic antibodies. Immunology. 2008;123:547–554. doi: 10.1111/j.1365-2567.2007.02724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ortqvist E, Brooks-Worrell B, Lynch K, Radtke J, Bekris LM, Kockum I, Agardh CD, Cilio CM, Lethagen AL, Persson B, et al. Changes in GAD65Ab-specific antiidiotypic antibody levels correlate with changes in C-peptide levels and progression to islet cell autoimmunity. J Clin Endocrinol Metab. 2010;95:E310–E318. doi: 10.1210/jc.2010-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haller MJ, Atkinson MA, Schatz DA. Efforts to prevent and halt autoimmune beta cell destruction. Endocrinol Metab Clin North Am. 2010;39:527–539. doi: 10.1016/j.ecl.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agardh CD, Cilio CM, Lethagen A, Lynch K, Leslie RD, Palmer M, Harris RA, Robertson JA, Lernmark A. Clinical evidence for thesafetyof GAD65 immunomodulation in adult-onset autoimmune diabetes. J Diabetes Compl. 2005;19:238–246. doi: 10.1016/j.jdiacomp.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 106.Ludvigsson J, Casas R, Vaarala O, Forsander G, Ivarsson S, Johansson C, Lindh A, Nilsson N, Åman J, Örtqvist E, et al. The Swedish GAD-vaccination Trial: outcomes of a phase II safety and efficacy trial with DiamydTM for preservation of beta cell function in children with T1D. Diabetologia. 2006;49:1–755. [Google Scholar]
- 107.Ohi H, Yasugi T. Occurrence of C3 nephritic factor and C4 nephritic factor in membranoproliferative glomerulonephritis (MPGN) Clin Exp Immunol. 1994;95:316–321. doi: 10.1111/j.1365-2249.1994.tb06530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Spitzer RE, Stitzel AE, Tsokos GC. Human anti-idiotypic antibody responses to autoantibody against the alternative pathway C3 convertase. Clin Immunol Immunopathol. 1990;57:19–31. doi: 10.1016/0090-1229(90)90019-m. [DOI] [PubMed] [Google Scholar]
- 109.Spitzer RE, Stitzel AE, Tsokos GC. Study of the idiotypic response to autoantibody to the alternative pathway C3/C5 convertase in normal individuals, patients with membrano-proliferative glomerulonephritis, and experimental animals. Clin Immunol Immunopathol. 1992;62:291–294. doi: 10.1016/0090-1229(92)90105-w. [DOI] [PubMed] [Google Scholar]
- 110.Nardi M, Karpatkin S. Antiidiotype antibody against platelet anti-GPIIIa contributes to the regulation of thrombocytopenia in HIV-1-ITP patients. J Exp Med. 2000;191:2093–2100. doi: 10.1084/jem.191.12.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abrams CS, Ruggeri ZM, Taub R, Hoxie JA, Nagaswami C, Weisel JW, Shattil SJ. Anti-idiotypic antibodies against an antibody to the platelet glycoprotein (GP) IIb-IIIa complex mimic GP IIb-IIIa by recognizing fibrinogen. J Biol Chem. 1992;267:2775–2785. [PubMed] [Google Scholar]
- 112.Mehta YS, Ghosh K, Badakere SS, Pathare AV, Mohanty D. Role of antiidiotypic antibodies on the clinical course of idiopathic thrombocytopenic purpura. J Lab Clin Med. 2003;142:113–120. doi: 10.1016/S0022-2143(03)00104-5. [DOI] [PubMed] [Google Scholar]
- 113.Csernok E, Muller A, Gross WL. Immunopathology of ANCA-associated vasculitis. Intern Med. 1999;38:759–765. [PubMed] [Google Scholar]
- 114.Jayne DR, Esnault VL, Lockwood CM. Anti-idiotype antibodies to anti-myeloperoxidase autoantibodies in patients with systemic vasculitis. J Autoimmun. 1993;6:221–226. doi: 10.1006/jaut.1993.1019. [DOI] [PubMed] [Google Scholar]
- 115.Strunz HP, Csernok E, Gross WL. Incidence and disease associations of a proteinase 3-antineutrophil cytoplasmic antibody idiotype (5/7 Id) whose antiidiotype inhibits proteinase 3-antineutrophil cytoplasmic antibody antigen binding activity. Arthritis Rheum. 1997;40:135–142. doi: 10.1002/art.1780400118. [DOI] [PubMed] [Google Scholar]
- 116.Pradhan VD, Ghosh K. Anti-idiotype antibodies in immune regulation of anca associated vasculitis. Indian J Dermatol. 2009;54:258–262. doi: 10.4103/0019-5154.55637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen QY, Rowley MJ, Mackay IR. Anti-idiotypic antibodies to anti-PDC-E2 in primary biliary cirrhosis and normal subjects. Hepatology. 1999;29:624–631. doi: 10.1002/hep.510290344. [DOI] [PubMed] [Google Scholar]
- 118.Dwyer DS, Bradley RJ, Urquhart CK, Kearney JF. Naturally occurring anti-idiotypic antibodies in myasthenia gravis patients. Nature. 1983;301:611–614. doi: 10.1038/301611a0. [DOI] [PubMed] [Google Scholar]
- 119.Lefvert AK, Pirskanen R, Svanborg E. Anti-idiotypic antibodies, acetylcholine receptor antibodies and disturbed neuromuscular function in healthy relatives to patients with myasthenia gravis. J Neuroimmunol. 1985;9:41–53. doi: 10.1016/s0165-5728(85)80005-9. [DOI] [PubMed] [Google Scholar]
- 120.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]