

Abstract
High fidelity of biological systems is frequently achieved by duplication of the essential intracellular machineries or, removal of the entire cell, which becomes unnecessary or even harmful in altered physiological environments. Carefully controlled removal of these cells, without damaging normal cells, requires precise signaling, and is critical to maintaining homeostasis. This review describes how two anionic phospholipids - phosphatidylserine (PS) and cardiolipin (CL) - residing in distinct compartments of the cell, signal removal of ”the unnecessary” using several uniform principles. One of these principles is realized by collapse of inherent transmembrane asymmetry and the externalization of the signal on the outer membrane surface - mitochondria for CL and the plasma membrane for PS – to trigger mitophagy and phagocytosis, respectively. Release from damaged cells of intracellular structures with externalized CL or externalized PS triggers their elimination by phagocytosis. Another of these principles is realized by oxidation of polyunsaturated species of CL and PS. Highly specific oxidation of CL by cytochrome c serves as a signal for mitochondria-dependent apoptosis, while oxidation of externalized PS improves its effectiveness to trigger phagocytosis of effete cells.
Keywords: Phosphatidylserine signaling, cardiolipin signaling, cardiolipin oxidation, Phosphatidylserine oxidation, mitophagy, phagocytosis, apoptosis
“Funny how the unnecessary can seem so important, expanding, contracting, cleaning itself. Whatever it is, it won’t let us in. It folds inside itself like a dying star, in a way it’s superior, as original as every murder.”
From: “The Unnecessary” by Karen Murai, The Paris Review No. 114 (Spring 1990)
Introduction
The subject of “the unnecessary” has been metaphorically and also constructively discussed and used by artists, politicians, and economists with different goals and in different contexts. Perhaps one of the most illuminating examples of this is a well-known principle in Pablo Picasso’s creative life namely that “art is the elimination of the unnecessary”, which he so exquisitely and skillfully applied to his work. We ask, what may be the relevance of this principle to biological processes?
The reliability of biological processes is achieved, to a large extent, through the initial generation of excessive amounts of biological material, including organelles and cells, which upon successful completion of their specific functions become unnecessary. Removal of the unnecessary in a timely manner is essential to prevent the unnecessary from becoming undesired or even harmful. This principle is well illustrated during development, such as in the elimination of the tail in the tadpole during metamorphosis [1] and tissue remodeling of a vertebrate limb bud during development [2], wherein unnecessary cells and tissue are removed through the process of apoptosis. Elimination of unnecessary cells also occurs in inflammation, wherein specialized cells recruited to the inflammation site must be cleared after successful completion of their job. Other examples include, mammary gland involution and ovarian follicle atresia, which are characterized by removal of milk-producing epithelial cells [3] and immature ovarian follicles [4] respectively, through apoptosis. The importance of clearance is underscored by observations that its inhibition results in severe developmental defects, organismal lethality [5–7], autoimmune disease [8, 9], or promotion of tumorigenesis [10]. Thus, robust and reliable mechanisms that direct timely removal of unwanted cells are essential, not just for maintenance of homeostasis, but also for circumventing undesired pathophysiological consequences. Given the remarkable importance of eliminating of the unnecessary in biology, in this brief review, we will consider recent advancements in our understanding of the role that two anionic phospholipids – phosphatidylserine (PS) and cardiolipin (CL) – play in the signaling and clearance of “unnecessary” organelles and cells.
Synthesis and localization of PS and CL
There are at least two different ways in which PS and CL participate in different types of elimination signaling: a) by trans-membrane re-distribution and subsequent appearance on the cell/organelle surfaces thus enabling recognition by specialized receptors and b) by oxidation of polyunsaturated acyl chains of the phospholipid molecules leading to modification of the membranes including their barrier functions. Intriguingly, PS and CL are compartmentalized in cells such that CL is localized almost exclusively in the inner mitochondrial membrane (IMM) whereas PS is essentially absent from the mitochondria [11, 12]. It is tempting to speculate that this separation is an evolutionary attainment to utilize and optimize two spatially distinct yet related membrane signaling functions realized in mitochondria and extra-mitochondrial compartments, respectively.
While CL and PS have distinct locations in the cell, their biogenesis occurs at contiguous sites. PS synthesis occurs in the endoplasmic reticulum (ER), specifically in ER domains proximal to the mitochondria, known as the mitochondria-associated membranes (MAM) [12, 13]. The synthesis of CL is well documented and is described in detail in several recent reviews [11, 14–17].
Following its initial synthesis, CL undergoes acyl chain “remodeling” to generate tissue and organ specific CL molecular species. This secondary stage of synthesis is catalyzed by enzymes including i) a CL-specific phospholipase(s) e.g. Cld1p in yeast cells that are needed to make monolyso-CL (MLCL) [18]; ii) An acyl transferase (tafazzin) that is located in the intermembrane space (IMS) of mitochondria which can reversibly exchange acyl chains between phospholipids and MLCL.[19–23]; iii) An MLCL acyltransferase-1 (MLCLAT-1) that is located in the matrix of the mitochondria, described as a splice variant of trifunctional protein [14, 24–26] and iv) An acyl-CoA:lyso-CL-acyltransferase-1 (ALCAT-1) that is reported to be beyond the outer side of the mitochondrial outer membrane (OMM) in the mitochondria associated membranes (MAMS) of the endoplasmic reticulum (ER) [14, 27, 28]. How and where these enzymes participate in the final synthesis of CL will dictate the location of CL in and around the mitochondria. After remodeling, most of the CL is located in the IMM, specifically concentrated on the inner leaflet of the IMM. CL has multiple binding affinities for the numerous mitochondrial proteins, including mitochondrial respiratory super-complexes [29–33] and respiratory components including cytochrome c [34] and is also believed to aid in the organization and stabilization of the highly curved IMM membrane structure [15, 35]. The diversified association with a multitude of mitochondrial proteins and its reliance on the mitochondrial membrane potential for stability [36, 37] defines CL’s role as a coordinator of numerous mitochondrial functions, including their critical roles as a signal for mitophagy and as an activator of the NLRP3 inflammasome (see below).
The synthesis of PS occurs via two separate synthetic pathways in mammalian cells and is distinct from the yeast PS synthetic pathway [12, 38]. In yeast, PS synthesis occurs via a CDP-diacylglycerol precursor which is also the precursor of CL in both yeast and mammalian CL synthesis. [15, 39–42]. The yeast CL biosynthetic pathway is conserved in mammals, but the biosynthesis of PS in mammalian cells has evolved using two separate and distinct PS biosynthetic pathways (see [38] for a recent review).
Once synthesized, PS travels to the mitochondria, where it is decarboxylated to phosphatidylethanolamine (PE), catalyzed by the IMM enzyme, PS decarboxylase. Knocking out this enzyme in mice resulted in embryonic lethality. Morphological analysis revealed highly abnormal and fragmented mitochondria [43]. It is likely that the accumulation of mitochondrial PS, rather than decreased levels of PE, caused the lethal phenotype. In line with this, knocking out the Psd1 gene in yeast, which codes for the yeast mitochondrial PS decarboxylase, significantly impaired cell viability [43]. Thus, mitochondria might be considered to be “PS averse”, likely due to the interference of PS with the mitophagic and apoptotic signaling by CL.
PS externalization is an engulfment signal
When phagocytosis was first discovered and described by I. Mechnikov in 1883 [44], the molecular dynamics of signaling and the role of PS externalization was unknown. It took more than a century that witnessed a multitude of fundamental discoveries in biology before it was established that one of the hallmarks of the activation of apoptosis and initiation of phagocytosis is the appearance of PS on the outer leaflet of the apoptotic cell membrane [45, 46]. The externalized PS is a recognition signal for specific receptors on the professional phagocytes (including macrophages) which initiates the engulfment of target injured or dying cells [47]. Studies using model systems demonstrate that the presence of externalized PS alone is sufficient to activate phagocytosis. For example, PS-coated single-walled carbon nanotubes are readily phagocytized when compared with non-coated controls [48]. Likewise, integration of PS into the outer surface of viable cells, that are not apoptotic, promoted phagocytosis by macrophages [45, 49].
Externalization of PS is a near-universal feature of apoptosis and is commonly involved in recognition and elimination of apoptotic cell corpses (Figure 1). Of particular importance is the PS-dependent phagocytosis of neutrophils by macrophages, a process that defines the transition from a pro-inflammatory to an anti-inflammatory state [50]. Normally, PS is asymmetrically distributed across the plasma membrane – being located predominantly in the inner leaflet along with phosphatidylethanolamine [51, 52]. The maintenance of this asymmetry is controlled by an ATP-dependent aminophospholipid translocase, also called a flippase [53]. During apoptosis this asymmetry collapses resulting in PS exposure on the external face of the cell membrane. Two possible mechanisms for this re-shuffling of PS are proposed. The activation of a calcium dependent scramblase can rapidly catalyze the inversion of phospholipids that are asymmetrically located on the plasma membrane [54]. The rate of scramblase activity (>10,000 phospholipids per second) far exceeds the ATP dependent flippase activity of (~1–100 ATP per second). Inactivation of the flippase concomitant with scramblase activation, has been proposed as the most efficient way to achieve PS exposure on the outer membrane leaflet [54, 55]. Experimentally, this concept was demonstrated by nitrosative stress under non-apoptotic conditions, wherein selective inhibition of the aminophospholipid translocase (flippase) resulted in PS externalization and recognition of these non-apoptotic cells by macrophages [56] – a phenomenon referred to as “buried alive” [57]. This underscores the uniqueness and sufficiency of PS externalization as an engulfment signal for professional phagocytes.
Figure 1. CL and PS as elimination signals.
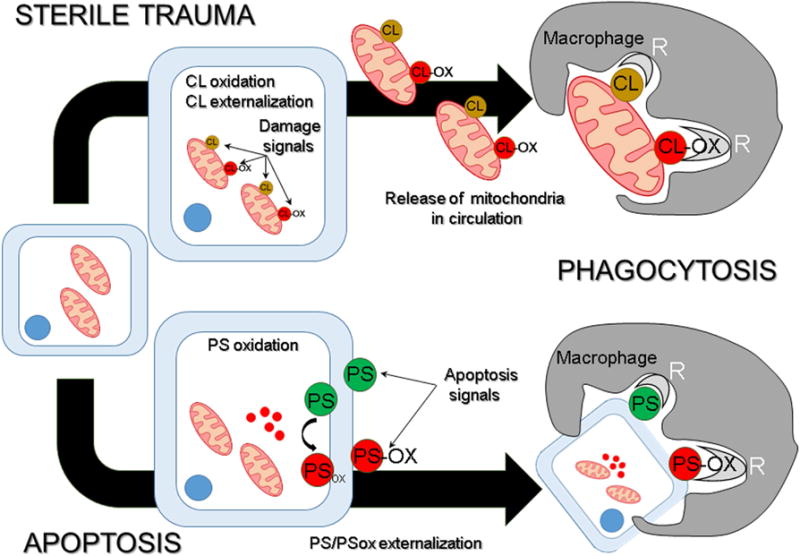
There are at least two different ways in which PS and CL participate in elimination signaling. CL and PS are localized in spatially distinct membranes – the mitochondria and the plasma membrane, respectively - that sets apart their availability as beacons for clearance. During sterile trauma, mitochondria containing CL and peroxidized CL (CLox) are released by damaged cells. When encountered by macrophages, CL and CLox trigger phagocytosis with similar efficacies, whereby these membranes are engulfed, for subsequent degradation in lysosomes. Induction of apoptosis results in externalization of PS on the cell surface. Oxidative stress induced apoptosis is further characterized by lipid peroxidation that results in the presentation of both PS and PSox on the dying cells. While both PS species are targeted by macrophages for engulfment, the efficiency of PSox-dependent clearance is substantially higher than PS-dependent clearance.
Importantly, there are concentration thresholds of PS on the surface, needed for the recognition of apoptotic cells, in order to initiate phagocytosis [58]. The existence of a threshold mechanism is essential for the prevention of excessive phagocytosis of non-apoptotic cells which may transiently present externalize PS on their surface [47, 58, #24, 59, 60].
PS oxidation promotes apoptotic cell clearance
During inflammation, massive production of reactive oxygen species (ROS) creates pro-oxidant environments potentially favoring PS oxidation [61], yet for phagocytosis, externalized PS is not required to be oxidized. However, presenting externalized, oxidized PS strongly enhances the effectiveness of phagocytosis [62]. Cells grown in cultures on standard media do not contain significant amounts of oxidizable PS, yet apoptosis and macrophage phagocytosis can be triggered. Metabolic incorporation of oxidizable polyunsaturated fatty acids (PUFA) such as linoleic acid, (LA) results in the appearance of PS containing PUFA acyl chain(s). During apoptosis, activation of the redox machinery in these cells stimulates the formation of oxidized PS species (PSox) and appearance of PSox on the outer leaflet of the plasma membrane. This is accompanied by a significantly higher clearance of apoptotic cells realized via both increased number of phagocytosis-positive cells and the increased content of engulfed target cells per phagocyte [62].
Structural variations between oxidized and non-oxidized PS raise the possibility of species specific PS-receptor interactions. Indeed, eukaryotic cells have amassed a rich repertoire of distinct receptors on different types of professional phagocytes for recognizing apoptotic cells via PS binding. These include, scavenger receptors A & B, CD36, vitronectin, PS-receptor, CXCL16, MerMFG-E8, Tim-4, BAI-1, lipoprotein receptor–related protein (LRP) and TREM2 [63–68]. The “language” of engulfment is further diversified and enhanced by soluble, PS-binding proteins, including thrombospondin-1, milk fat globule-EGF factor 8 (MFG-E8), growth arrest-specific 6 (GAS-6), and β2-glycoprotein I [47, 69–71]. Such a multiplicity in receptor expression likely reflects ligand specific signaling responses as has been suggested by the differential binding preference of PS for CXCL16, TIM-4 and CD36 and of PSox for BAI-1 and GAS-6 [62]. It is also conceivable that an engulfment “synapse” is formed between the phagocyte and its apoptotic prey requiring more than one ligand-receptor couple [72].
Numerous recent reviews have provided information on macrophage recognition of cells undergoing apoptosis [53, 73]. Evolutionary conservation of receptor multiplicity further underscores that eliminating the unnecessary is vital to homeostasis [74]. While it is commonly accepted that the phagocytosis of apoptotic cells triggers the resolution of inflammation [75, 76], the actual meaning of each individual signal and combinations of different molecular signals in pro-/anti-inflammatory conditions has not been deciphered. With the development of redox (phospho)lipidomics the identification of the role of individual PS and oxidized PS species as “orchestral players” of inflammation can be unraveled [77].
CL asymmetry is a signal for mitophagy
From a signaling perspective, externalization of PS on the cell membrane, is similar to CL externalization on the mitochondrial surface (Figure 1). Factors that determine the need for mitophagy may include maintenance of an adequate membrane potential, oxidation of proteins and lipids and the ability of mitochondria to undergo biogenesis. Mitochondrial externalization of CL serves as an elimination sign initiating mitophagy in analogy to PS externalization which signals phagocytosis, yet the molecular details are very different. Externalized CL can bind to microtubule-associated protein 1 light chain 3 (LC3) which is part of the autophagy machinery [78]. The externalization mechanisms for CL and PS are also very different, but the signaling principal is the same. To appear on the mitochondrial surface, CL has to transgress the IMM, the intermembrane space and finally the OMM. The initiation of CL asymmetry collapse happens when mitophagy is warranted and initiated. Assuming that most of CL in the IMM is not “free”, but rather engaged in different types of lipid-protein and protein-protein interactions [79], one can wonder how CL gets released from its complexes to traverse from the IMM inner side to the outer side of the OIM and becomes a mitophagy beacon. Obviously, the dissipation of mitochondrial membrane potential is the trigger and the driving force of this process. The details of the machinery engaged in the mitophageal translocations of CL are not fully understood. There are several “suspects” for the CL redistribution in the IMM. Given the essentiality of mitochondrial depolarization and potential involvement of redox-driven mechanisms, uncoupling proteins seem to be interesting candidates [80, 81]. The inter-membrane space translocation may be particularly challenging in terms of energetic expenditures. Interestingly, a mitochondrial isoform of nucleoside diphosphate kinase (NDPKD) has been demonstrated as the driving force of this process in mitophagy [82, 83]. The participants of the CL translocating machinery in the OMM are also not definitively identified although mitochondrial scramblase proteins (e.g., scramblase 3) are possibly involved [84, 85].
Intriguingly, while redox mechanisms are believed to act as almost universal triggers of mitochondrial damage and mitophagy, there is no compelling requisite for CL oxidation for mitophagy, which is in sharp contrast to the obligatory dependency of CL oxidation for apoptosis (see below). This may reflect substantial differences in the execution pathways and physiological roles of mitophagy and apoptosis as two distinctive cell survival and cell death mechanisms, respectively. Overall, the mechanisms that control CL asymmetry and translocations are still enigmatic, although there is clear evidence to indicate that mitochondrial damage is associated with presentation of CL on the OMM surface to signal and trigger intracellular mitochondrial clearance by mitophagy [84].
CL oxidation elicits intrinsic apoptosis
While mitophagy is a pro-survival rescue pathway, the continued accumulation of mitochondrial impairments and the insufficiency and/or failure of repair mechanisms may necessitate the elimination of the entire cell through the activation of the apoptotic program. This requires not only the presence of externalized CL, but also oxidation of its polyunsaturated acyl chain(s). Historically, the involvement of CLs in (BAX-mediated) apoptosis had been proposed and supported by the work of Kuwana et al. [86]. A later study from the Orrenius laboratory, clearly demonstrated that there is no absolute requirement for CL for the execution of the apoptotic program in yeast cells (S. cerevisae) where BAX-dependent apoptosis was occurring in CL synthase deficient mutants (Crd ∆) [87]. This apparent discrepancy may be reconciled if one considers that yeast cells grown under standard laboratory conditions simply do not contain oxidizable, polyunsaturated CL species [88–90]. Indeed, subsequent detailed studies revealed that CL peroxidation, catalyzed by cytochrome c, is required for the execution of intrinsic apoptosis in mammalian cells that have oxidizable CL species [91]. Detailed analysis of mechanisms and pathways involved in CL oxidation in apoptosis has been recently discussed in several reviews [37, 81, 92–95]. Briefly, externalization of CL in damaged mitochondria strongly enhances its availability for the intermembrane space hemoprotein, cytochrome c. This encounter results in the formation of cytochrome c/CL complex in which the normally hexa-coordinated hemoprotein adopts the penta-coordinate organization allowing for the emergence of the peroxidase catalytic competence [91]. As a consequence, the closest PUFA-phospholipid target, CL, gets peroxidized. Studies from several laboratories have confirmed and developed this paradigm. There is thus, a transformative structural shift for cytochrome c from being an electron carrier shuttling electrons between respiratory complexes III and IV while located on the outer surface of the IMM, to a CL-specific peroxidase function in apoptosis [34, 91, 96–99].
The peroxidase activity and CL oxidation precede and are required for mitochondria-initiated apoptosis whereby the sources of oxidizing equivalents are derived from dis-coordinated electron transport [33,36,82]. Structural details of cytochrome c/CL complexes and their significance for the peroxidase activation are still under detailed scrutiny whereby the dependence of the peroxidase activity on the level of protein unfolding are being explored using sophisticated state-of-the-art technologies including, solution NMR and solid state NMR [100, 101]. These developments in understanding CL oxidation and its role in apoptosis has also offered new opportunities for therapeutic interventions, and new types of regulators acting as suppressors of CL oxidation have been generated. For example, a series of mitochondria-targeted hemigramicidin nitroxides (eg., XJB131, XJB125) – acting as scavengers of electrons - have been designed, synthesized and formulated and found to have high therapeutic effectiveness in acute traumatic brain injury [102] and total body irradiation, among others. Another groups of regulators – imidazole substituted fatty acids displayed anti-apoptotic activity by preventing/suppressing cytochrome c/CL peroxidase activity via Fe-chelation [103].
One of the difficult and controversial issues originating from in vivo studies was the large diversification of CL oxidation products – dozens or even hundreds of different types of structurally distinctive oxidation products [104]. For some time, it remained unclear which of them are, indeed, directly related to the execution of apoptosis. This conundrum has been recently resolved by using a protocol of redox opto-lipidomics [77]. It has been demonstrated that only few out of a large number of detectable CL oxidation products represent real apoptotic signals. In fact, in cells exposed to several pro-apoptotic treatments such as actinomycin D, staurosporine, ionizing radiation or 10-nonylacridine orange, only C18:2 mono-oxygenated species of CLs were identified as predictive biomarkers of apoptosis [77].
Phagocytosis of extracellular mitochondria
When an effective biological solution evolves, it is frequently adapted and widely used. Autophagy of mitochondria and phagocytic removal of cells both depend on similar signaling systems although the processes are quite different. Interestingly, recent studies demonstrated that macrophages have evolved to retain mechanisms to target CLs on extracellular mitochondria for phagocytosis [105]. While the details and machinery of eliminating extracellular mitochondria remain a subject of interest, oxidation of CL has no role in extracellular mitochondrial phagocytosis. Similar to what has been shown for PS exposition on the surface of viable, non-apoptotic cells (see above), the integration of mitochondrial CL onto protein-free liposomes or onto healthy mitochondria that do not have surface CL also triggers engulfment through macrophage receptor CD36 [105]. The mechanisms of clearance in these model systems differs from the usual intracellular LC3-mediated mitophagy signaling of CL, wherein the CL externalized mitochondrial membrane does not leave the cell [84]. Thus, while both PS and CL can in principle signal clearance, there are spatial constraints that restrain signaling cross-talk between these lipids under normal physiology conditions, with CL being restricted to the mitochondria and PS being present in most other cellular membranes, most notably the plasma membrane, allowing CL to exclusively signal LC3 mediated mitophagy.
CL dependent intracellular clearance of damaged mitochondria by mitophagy, is a survival mechanism, and is therefore expected to occur without untoward activation of inflammatory cytokines. There are, however, several reports that under pathophysiological conditions, cells release CL-presenting mitochondria and mitochondrial membranes. Examples include, activation of neutrophils, stress, necrosis, and acute trauma [106–108]. Similarly, infections challenge the immune system by externalizing CL on the surface of bacterial membranes ([108] These CL-containing membranes are unique in that they lack the pro-phagocytic and anti-inflammatory signal PS [38, 51]. Moreover, they are also rich in immune-cell activating mtDNA or bacterial DNA, in addition to formyl peptides [109], which activate immune surveillance via an array of extracellular pattern recognition receptors, including Toll-like receptors (TLRs) [110], and intracellular inflammasomes platforms for pro-inflammatory cytokine production [111].
Hence, rapid clearance of these CL-expressing entities is essential to avoid an uncontrolled innate response, which can progress into local and systemic inflammatory maladies, culminating in chronic autoimmune disease. Macrophages address this eventuality by employing the same family of engulfment receptors discussed above for PS dependent phagocytosis. Among these, scavenger receptor CD36 stands out in its ability to target membranes, organelles and cells expressing eukaryotic or prokaryotic CL for clearance as evidenced using macrophages from CD36 null animals [105]. Interestingly, activation of many of the macrophage phagocyte receptors including CD36 is upregulated through activation of TLRs, and, moreover, there is also evidence to suggest cross-talk between TLRs and engulfment receptors [112, 113]. Thus, the host immune system has evolved to allow a balanced approach to appropriately deal with the unnecessary “self”, the undesired “self”, and the undesired “non-self” or foreign entities. In this scenario, both phagocytosis and immune surveillance systems work in parallel to remove the unnecessary and undesired, so as to maintain cellular and tissue homeostasis, while at the same time preventing any deviations of the inflammatory responses.
In addition to signaling for removal of mitochondria and bacteria by phagocytosis, extracellular CLs have also been suggested to play a role in regulating inflammatory responses elicited by bacteria. In a recent study, it was determined that the ability of the gram-negative bacterial antigen, lipopolysaccharide (LPS) to activate inflammatory cytokine production via macrophage TLR4 receptors was significantly dampened in the presence of mitochondrial or bacterial CLs [105]. In contrast, substituting PS for CL showed no effects on cytokine production. This effect of CL was determined to operate by an extracellular mechanism as a consequence of competition between CL and LPS for the MD2 subunit of the TRL4/MD2 complex, in a manner similar to that reported for immature lipid A [114]. Thus, there are topographical differences between CL dependent cytokine suppression that acts by extracellular signaling via TLRs, and PS dependent cytokine suppression that occurs through intracellular mechanisms [115–117].
NF-κB, a transcription factor which acts downstream of cell surface receptors including macrophage TLRs drives inflammation through various pathways, including the priming of the NLRP3 inflammasome [118]. How the inflammasome is kept in check following its activation has remained unclear. Tschopp and co-workers showed that mitophagy/autophagy blockade resulted in the accumulation of damaged mitochondria, which in turn activated the NLRP3 inflammasome [118]. Recent studies have revealed a regulatory mechanism whereby NF-κB can restrain NLRP3 activation through mitophagy with specific recognition of the damaged organelles by p62 [119]. NF-κB exerted its anti-inflammatory activity by inducing delayed accumulation of the autophagy receptor p62 and macrophage-specific p62 ablation caused pronounced accumulation of damaged mitochondria and excessive IL-1β-dependent inflammation. Thus, mitophagy/autophagy might also prevent NLRP3 inflammasome activation from going awry.
It will be of interest to further examine the intracellular signaling events, including transcriptional responses, engendered by CL during removal of unnecessary organelles within the cell (mitophagy), or ingestion of extruded organelles (by other cells).
In contrast to the immunosuppressive function of extracellular CLs, there is evidence to indicate that intracellular CLs plays a direct role in activation of NLRP3 inflammasomes [120], which are tasked with the activation of inflammatory cytokines IL-1 and IL-18 as a prelude to pyroptosis in response to an intracellular pathogen. Thus, the signaling effects of CL are unique to its spatial location in the cell. Moreover, the consequences are unique and contrasting as well. For example, in the cytoplasm, CL plays a positive role in the maintenance of cell health by removal of damaged mitochondria during mitophagy. Cytoplasmic CL plays a suicidal role in cells infected with intracellular pathogens, through the activation of pyroptosis. The extracellular functions of CL are activated only under pathophysiological conditions resulting from infection (bacterial CLs), sterile cell damage (mitochondrial CLs) or infection induced host cell damage (bacterial CLs + mitochondrial CLs). Since macrophages cannot differentiate between bacterial and mitochondrial CLs [105], the signaling consequences are identical in each of these cases. Extracellular CL thus plays a crucial role in pathophysiology, by promoting removal of mitochondrial membranes that lack PS as an elimination signal (as opposed to other intracellular membranes which have PS). Extracellular CL also plays another important role in restoration of tissue homeostasis following gram-negative bacterial infections by subduing further activation of TLR4 (Figure 2).
Figure 2. Role of cardiolipins in homeostasis.
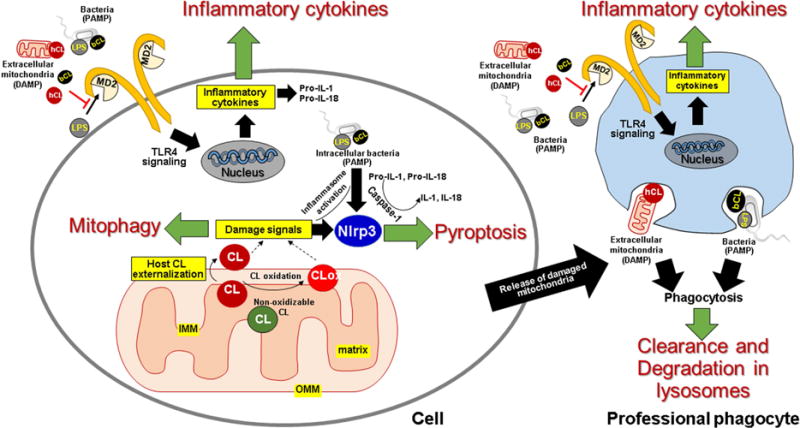
Intra- or extracellular signaling by CL is triggered by its externalization to the mitochondrial surface. Intracellular CL: In healthy cells, CL is exclusively localized in the inner mitochondrial membrane (IMM) specifically on the inner leaflet of the IMM that faces the matrix. Damage to the mitochondria results in loss of this asymmetry and movement of the lipid to the outer leaflet of the IMM, to the inner leaflet of the outer mitochondrial membrane (OMM), and finally to the outer leaflet of the OMM. Additionally, oxidative stress results in peroxidation of oxidizable CL (having polyunsaturaed acyl chains) by cytochrome c, to produce CLox, which may also be presented at the mitochondrial surface as a damage signal. Once at the mitochondrial surface, CL in the presence of cytoplasmic LC3, signals clearance of the damaged organelle by mitophagy, thereby restoring intracellular homeostasis. Under pathological conditions, such as presence of intracellular pathogens or presence of intracellular pathogen associated molecular patterns (PAMPs) like lipopolysaccharide (LPS), mitochondrial CL and/or CLox promotes activation of the NLRP3 dependent inflammasome pathway that directs cell death by pyroptosis. Extracellular CL: Mitochondria released following cell damage provide human CL (hCL) as a source of extracellular damage associated molecular patterns (DAMPs), which challenge host cells, including professional phagocytes. Similarly, infections challenge these cells with bacterial CL (bCL) and LPS. Both hCL and bCL signal engulfment of mitochondrial and bacterial membranes by professional phagocytes for subsequent degradation in lysosomes. hCL and bCL also interfere with binding of LPS to MD2, thereby attenuating Toll-like receptor (TLR)-4 dependent inflammatory cytokine production. The attenuation of cytokine response and phagocytic clearance both contribute to restoration of homeostasis.
Concluding remarks
Reliability in biology frequently requires an excess of constituents including metabolites, enzymes, organelles, and cells. During times of change and stress, such as growth, injury, or infection, this excess can become unnecessary and detrimental. Elimination of the unnecessary applies to many facets of biology. Here we described how PS and CL, two distinct phospholipids, have evolved to signal transformative yet similar cellular processes that eliminate “the unnecessary”. Discerning this information has involved generations of scientists who are curious about life’s control mechanisms. Looking forward, in this research field, the progress being made in lipidomics and bioinformatics is likely to continue and expand while integrating knowledge from other research fields. It is thus a good time to understand “the unnecessary”. As discussed here, lipid signaling also involves topographical molecular constraints to prevent promiscuous or inadvertent activation of phagocytosis and/or mitophagy in healthy cells. Indeed, available data suggest that one and the same signal, CL, may signal for widely disparate cellular and organismal outcomes depending upon its intra- or extracellular localization.
In pathological scenarios, e.g., burn injury or physical trauma or infection, the constraints placed by compartmentalization may break down and both PS and CL may inevitably co-signal through macrophage receptors. This is expected to occur during both sterile and non-sterile (pathogen-induced) tissue damage, which is characterized by the release of intracellular membranes and organelles, including both intact mitochondria and membrane fragments from damaged mitochondria [106–108]. The immunological consequence of simultaneous PS and CL signaling in this case would depend on several factors including, the proportion of each lipid, receptor expression pattern and activation status of the macrophages or other professional phagocytes being engaged [121–123], and the relative affinities for PS and CL for the expressed receptors.
Active research in the field of phospholipid signaling to eliminate the unnecessary has already revealed many essential mechanisms and pathways involved in these important processes in isolated model biochemical systems and cells. A more challenging task is the assessment of the role of these processes in vivo. It is likely that not only high analytical power of LC-MS protocols in the identification of different individual signals, but also more integral approaches that afford on-line recording of the signaling process may be useful. Among them, a promising technique may be registration of different types of chemiluminescence originating from the excited states of oxidatively modified lipids [124]. Assuming that (phospho)lipid hydroperoxides are unavoidable primary molecular products of lipid peroxidation and that their decomposition gives rise to multiple secondary products with carbonyl functions in the excited state, it is likely that chemiluminescence protocols – particularly with physical enhancers of chemiluminescence responses – may be used for continuous monitoring of the peroxidation process in live cells. Given that highly portable chemiluminometers have become available, this approach may find promising applications in precision medicine.
Supplementary Material
Highlights.
Phospholipid (PL) signaling in elimination of organelles and cells is reviewed.
Anionic PL membrane asymmetry generates distinct signals for elimination.
Oxidation of cardiolipin is a required stage in mitochondrial apoptosis.
Oxidation of externalized phosphatidylserine enhances phagocytosis efficiency.
Acknowledgments
The authors are supported, in part, by NIH: PO1HL114453, CA165065, ES020693, U19AIO68021, NS076511, NS061817; NIOSH: OH008282, Human Frontier Science Program HFSP-RGP00132014, the Barth Syndrome Foundation, Inc. and the Barth Syndrome Foundation of Canada and the Swedish Research Council (BF).
Abbreviations
- CL
cardiolipin
- IMM
inner mitochondria membrane
- IMS
mitochondrial inter membrane space
- LC
liquid chromatography
- MAMS
mitochondria associated membranes
- MLCL
monolyso-cardiolipin
- MS
mass spectrometry
- NPDKD
nucleoside diphosphate kinase
- OMM
outer mitochondrial membrane
- PS
phosphatidylserine
- PSox
oxidized phosphatidylserine
- PUFA
polyunsaturated fatty acids
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shi YB, Fu L, Hsia SC, Tomita A, Buchholz D. Thyroid hormone regulation of apoptotic tissue remodeling during anuran metamorphosis. Cell research. 2001;11:245–252. doi: 10.1038/sj.cr.7290093. [DOI] [PubMed] [Google Scholar]
- 2.Saunders JW., Jr Death in embryonic systems. Science (New York, NY) 1966;154:604–612. doi: 10.1126/science.154.3749.604. [DOI] [PubMed] [Google Scholar]
- 3.Watson CJ. Involution: apoptosis and tissue remodelling that convert the mammary gland from milk factory to a quiescent organ. Breast cancer research: BCR. 2006;8:203. doi: 10.1186/bcr1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J, Carlock C, Zhou C, Nakae S, Hicks J, Adams HP, Lou Y. IL-33 is required for disposal of unnecessary cells during ovarian atresia through regulation of autophagy and macrophage migration. Journal of immunology (Baltimore, Md: 1950) 2015;194:2140–2147. doi: 10.4049/jimmunol.1402503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 6.White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science (New York, NY) 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 8.Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nature genetics. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 9.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (New York, NY) 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 10.Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. Annual review of physiology. 1997;59:349–363. doi: 10.1146/annurev.physiol.59.1.349. [DOI] [PubMed] [Google Scholar]
- 11.Horvath SE, Daum G. Lipids of mitochondria. Progress in lipid research. 2013;52:590–614. doi: 10.1016/j.plipres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic (Copenhagen, Denmark) 2015;16:1–18. doi: 10.1111/tra.12230. [DOI] [PubMed] [Google Scholar]
- 13.Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. The Journal of biological chemistry. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- 14.Lu YW, Claypool SM. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Frontiers in genetics. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren M, Phoon CK, Schlame M. Metabolism and function of mitochondrial cardiolipin. Progress in lipid research. 2014;55:1–16. doi: 10.1016/j.plipres.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Renne MF, Bao X, De Smet CH, de Kroon AI. Lipid Acyl Chain Remodeling in Yeast. Lipid insights. 2015;8:33–40. doi: 10.4137/LPI.S31780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlame M, Greenberg ML. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochimica et biophysica acta. 2016 doi: 10.1016/j.bbalip.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baile MG, Whited K, Claypool SM. Deacylation on the matrix side of the mitochondrial inner membrane regulates cardiolipin remodeling. Molecular biology of the cell. 2013;24:2008–2020. doi: 10.1091/mbc.E13-03-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. The Journal of cell biology. 2006;174:379–390. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Molecular microbiology. 2004;51:149–158. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- 21.Lu YW, Galbraith L, Herndon JD, Lu YL, Pras-Raves M, Vervaart M, Van Kampen A, Luyf A, Koehler CM, McCaffery JM, Gottlieb E, Vaz FM, Claypool SM. Defining functional classes of Barth syndrome mutation in humans. Human molecular genetics. 2016;25:1754–1770. doi: 10.1093/hmg/ddw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L, Vaz FM, Gu Z, Wanders RJ, Greenberg ML. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. The Journal of biological chemistry. 2004;279:44394–44399. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- 23.Schlame M, Rustow B. Lysocardiolipin formation and reacylation in isolated rat liver mitochondria. The Biochemical journal. 1990;272:589–595. doi: 10.1042/bj2720589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carpenter K, Pollitt RJ, Middleton B. Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochemical and biophysical research communications. 1992;183:443–448. doi: 10.1016/0006-291x(92)90501-b. [DOI] [PubMed] [Google Scholar]
- 25.Taylor WA, Hatch GM. Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1) The Journal of biological chemistry. 2009;284:30360–30371. doi: 10.1074/jbc.M109.048322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor WA, Mejia EM, Mitchell RW, Choy PC, Sparagna GC, Hatch GM. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PloS one. 2012;7:e48628. doi: 10.1371/journal.pone.0048628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao J, Liu Y, Lockwood J, Burn P, Shi Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA: lysocardiolipin acyltransferase (ALCAT1) in mouse. The Journal of biological chemistry. 2004;279:31727–31734. doi: 10.1074/jbc.M402930200. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Chen YQ, Li S, Konrad RJ, Cao G. The microsomal cardiolipin remodeling enzyme acyl-CoA lysocardiolipin acyltransferase is an acyltransferase of multiple anionic lysophospholipids. Journal of lipid research. 2009;50:945–956. doi: 10.1194/jlr.M800567-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bazan S, Mileykovskaya E, Mallampalli VK, Heacock P, Sparagna GC, Dowhan W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. The Journal of biological chemistry. 2013;288:401–411. doi: 10.1074/jbc.M112.425876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mejia EM, Cole LK, Hatch GM. Cardiolipin metabolism and the role it plays in heart failure and mitochondrial supercomplex formation. Cardiovascular & hematological disorders drug targets. 2014;14:98–106. doi: 10.2174/1871529x14666140505123753. [DOI] [PubMed] [Google Scholar]
- 31.Mileykovskaya E, Dowhan W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chemistry and physics of lipids. 2014;179:42–48. doi: 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peyta L, Jarnouen K, Pinault M, Guimaraes C, Pais de Barros JP, Chevalier S, Dumas JF, Maillot F, Hatch GM, Loyer P, Servais S. Reduced cardiolipin content decreases respiratory chain capacities and increases ATP synthesis yield in the human HepaRG cells. Biochimica et biophysica acta. 2016;1857:443–453. doi: 10.1016/j.bbabio.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Vartak R, Porras CA, Bai Y. Respiratory supercomplexes: structure, function and assembly. Protein & cell. 2013;4:582–590. doi: 10.1007/s13238-013-3032-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free radical biology & medicine. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis RN, McElhaney RN. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochimica et biophysica acta. 2009;1788:2069–2079. doi: 10.1016/j.bbamem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Amoscato AA, Sparvero LJ, He RR, Watkins S, Bayir H, Kagan VE. Imaging Mass Spectrometry of Diversified Cardiolipin Molecular Species in the Brain. Analytical Chemistry. 2014;86:6587–6595. doi: 10.1021/ac5011876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kagan VE, Tyurina YY, Tyurin VA, Mohammadyani D, Angeli JP, Baranov SV, Klein-Seetharaman J, Friedlander RM, Mallampalli RK, Conrad M, Bayir H. Cardiolipin signaling mechanisms: collapse of asymmetry and oxidation. Antioxidants & redox signaling. 2015;22:1667–1680. doi: 10.1089/ars.2014.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vance JE, Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochimica et biophysica acta. 2013;1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 39.Koprivnjak T, Zhang D, Ernst CM, Peschel A, Nauseef WM, Weiss JP. Characterization of Staphylococcus aureus cardiolipin synthases 1 and 2 and their contribution to accumulation of cardiolipin in stationary phase and within phagocytes. Journal of bacteriology. 2011;193:4134–4142. doi: 10.1128/JB.00288-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin TY, Weibel DB. Organization and function of anionic phospholipids in bacteria. Applied microbiology and biotechnology. 2016;100:4255–4267. doi: 10.1007/s00253-016-7468-x. [DOI] [PubMed] [Google Scholar]
- 41.Sandoval-Calderon M, Geiger O, Guan Z, Barona-Gomez F, Sohlenkamp C. A eukaryote-like cardiolipin synthase is present in Streptomyces coelicolor and in most actinobacteria. The Journal of biological chemistry. 2009;284:17383–17390. doi: 10.1074/jbc.M109.006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CR, Guan Z. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:04–16509. doi: 10.1073/pnas.1212797109. 1kobayashi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vance JE, Steenbergen R. Metabolism and functions of phosphatidylserine. Progress in lipid research. 2005;44:207–234. doi: 10.1016/j.plipres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Tauber AI. Metchnikoff and the phagocytosis theory, Nature reviews. Molecular cell biology. 2003;4:897–901. doi: 10.1038/nrm1244. [DOI] [PubMed] [Google Scholar]
- 45.Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. The Journal of biological chemistry. 2001;276:1071–1077. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- 46.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. Journal of immunology (Baltimore, Md: 1950) 1992;148:2207–2216. [PubMed] [Google Scholar]
- 47.Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunological reviews. 2016;269:44–59. doi: 10.1111/imr.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konduru NV, Tyurina YY, Feng W, Basova LV, Belikova NA, Bayir H, Clark K, Rubin M, Stolz D, Vallhov H, Scheynius A, Witasp E, Fadeel B, Kichambare PD, Star A, Kisin ER, Murray AR, Shvedova AA, Kagan VE. Phosphatidylserine targets single-walled carbon nanotubes to professional phagocytes in vitro and in vivo. PloS one. 2009;4:e4398. doi: 10.1371/journal.pone.0004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kagan VE, Gleiss B, Tyurina YY, Tyurin VA, Elenstrom-Magnusson C, Liu SX, Serinkan FB, Arroyo A, Chandra J, Orrenius S, Fadeel B. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. Journal of immunology (Baltimore, Md: 1950) 2002;169:487–499. doi: 10.4049/jimmunol.169.1.487. [DOI] [PubMed] [Google Scholar]
- 50.Fadeel B, Ahlin A, Henter JI, Orrenius S, Hampton MB. Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood. 1998;92:4808–4818. [PubMed] [Google Scholar]
- 51.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annual review of biophysics. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 52.Sharom FJ. Flipping and flopping–lipids on the move. IUBMB life. 2011;63:736–746. doi: 10.1002/iub.515. [DOI] [PubMed] [Google Scholar]
- 53.Bevers EM, Williamson PL. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiological reviews. 2016;96:605–645. doi: 10.1152/physrev.00020.2015. [DOI] [PubMed] [Google Scholar]
- 54.Pomorski TG, Menon AK. Lipid somersaults: Uncovering the mechanisms of protein-mediated lipid flipping. Progress in lipid research. 2016;64:69–84. doi: 10.1016/j.plipres.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 56.Tyurina YY, Basova LV, Konduru NV, Tyurin VA, Potapovich AI, Cai P, Bayir H, Stoyanovsky D, Pitt BR, Shvedova AA, Fadeel B, Kagan VE. Nitrosative stress inhibits the aminophospholipid translocase resulting in phosphatidylserine externalization and macrophage engulfment: implications for the resolution of inflammation. The Journal of biological chemistry. 2007;282:8498–8509. doi: 10.1074/jbc.M606950200. [DOI] [PubMed] [Google Scholar]
- 57.Fadeel B, Orrenius S, Pervaiz S. Buried alive: a novel approach to cancer treatment. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:1–4. doi: 10.1096/fj.03-0510hyp. [DOI] [PubMed] [Google Scholar]
- 58.Borisenko GG, Matsura T, Liu SX, Tyurin VA, Jianfei J, Serinkan FB, Kagan VE. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells–existence of a threshold. Archives of biochemistry and biophysics. 2003;413:41–52. doi: 10.1016/s0003-9861(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 59.Elliott MR, Ravichandran KS. The Dynamics of Apoptotic Cell Clearance. Developmental cell. 2016;38:147–160. doi: 10.1016/j.devcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal, Nature reviews. Immunology. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 61.Fadeel B, Kagan VE. Apoptosis and macrophage clearance of neutrophils: regulation by reactive oxygen species. Redox report: communications in free radical research. 2003;8:143–150. doi: 10.1179/135100003225001511. [DOI] [PubMed] [Google Scholar]
- 62.Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, Kapralov AA, Macphee CH, Kagan VE. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell death and differentiation. 2014;21:825–835. doi: 10.1038/cdd.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. The Journal of experimental medicine. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 65.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, Ohashi K, Mizuno K, Arita H. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. The Journal of biological chemistry. 1997;272:29411–29414. doi: 10.1074/jbc.272.47.29411. [DOI] [PubMed] [Google Scholar]
- 67.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 68.Shimaoka T, Kume N, Minami M, Hayashida K, Kataoka H, Kita T, Yonehara S. Molecular cloning of a novel scavenger receptor for oxidized low density lipoprotein, SR-PSOX, on macrophages. The Journal of biological chemistry. 2000;275:40663–40666. doi: 10.1074/jbc.C000761200. [DOI] [PubMed] [Google Scholar]
- 69.Lauber K, Keppeler H, Munoz LE, Koppe U, Schroder K, Yamaguchi H, Kronke G, Uderhardt S, Wesselborg S, Belka C, Nagata S, Herrmann M. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell death and differentiation. 2013;20:1230–1240. doi: 10.1038/cdd.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maiti SN, Balasubramanian K, Ramoth JA, Schroit AJ. Beta-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells. Binding to lipoprotein receptor-related protein receptor family members. The Journal of biological chemistry. 2008;283:3761–3766. doi: 10.1074/jbc.M704990200. [DOI] [PubMed] [Google Scholar]
- 71.Moodley Y, Rigby P, Bundell C, Bunt S, Hayashi H, Misso N, McAnulty R, Laurent G, Scaffidi A, Thompson P, Knight D. Macrophage recognition and phagocytosis of apoptotic fibroblasts is critically dependent on fibroblast-derived thrombospondin 1 and CD36. The American journal of pathology. 2003;162:771–779. doi: 10.1016/S0002-9440(10)63874-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Molecular cell. 2004;14:277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- 73.Biermann M, Maueroder C, Brauner JM, Chaurio R, Janko C, Herrmann M, Munoz LE. Surface code–biophysical signals for apoptotic cell clearance. Physical biology. 2013;10:065007. doi: 10.1088/1478-3975/10/6/065007. [DOI] [PubMed] [Google Scholar]
- 74.Fadeel B, Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Critical reviews in biochemistry and molecular biology. 2009;44:264–277. doi: 10.1080/10409230903193307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunological reviews. 2016;273:357–370. doi: 10.1111/imr.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mao G, Qu F, St Croix CM, Tyurina YY, Planas-Iglesias J, Jiang J, Huang Z, Amoscato AA, Tyurin VA, Kapralov AA, Cheikhi A, Maguire J, Klein-Seetharaman J, Bayir H, Kagan VE. Mitochondrial Redox Opto-Lipidomics Reveals Mono-Oxygenated Cardiolipins as Pro-Apoptotic Death Signals. ACS chemical biology. 2016;11:530–540. doi: 10.1021/acschembio.5b00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hsu P, Shi Y. Regulation of autophagy by mitochondrial phospholipids in health and diseases. Biochimica et biophysica acta. 2016 doi: 10.1016/j.bbalip.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Planas-Iglesias J, Dwarakanath H, Mohammadyani D, Yanamala N, Kagan VE, Klein-Seetharaman J. Cardiolipin Interactions with Proteins. Biophysical journal. 2015;109:1282–1294. doi: 10.1016/j.bpj.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwok KH, Ho PW, Chu AC, Ho JW, Liu HF, Yiu DC, Chan KH, Kung MH, Ramsden DB, Ho SL. Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free radical biology & medicine. 2010;49:1023–1035. doi: 10.1016/j.freeradbiomed.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 81.Maguire JJ, Tyurina YY, Mohammadyani D, Kapralov AA, Anthonymuthu TS, Qu F, Amoscato AA, Sparvero LJ, Tyurin VA, Planas-Iglesias J, He RR, Klein-Seetharaman J, Bayir H, Kagan VE. Known unknowns of cardiolipin signaling: The best is yet to come. Biochimica et biophysica acta. 2016 doi: 10.1016/j.bbalip.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet-Rousselle C, Dar HH, Verma M, Tyurin VA, Kapralov AA, Cheikhi A, Mao G, Stolz D, St Croix CM, Watkins S, Shen Z, Li Y, Greenberg ML, Tokarska-Schlattner M, Boissan M, Lacombe ML, Epand RM, Chu CT, Mallampalli RK, Bayir H, Schlattner U. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell death and differentiation. 2016;23:1140–1151. doi: 10.1038/cdd.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schlattner U, Tokarska-Schlattner M, Ramirez S, Tyurina YY, Amoscato AA, Mohammadyani D, Huang Z, Jiang J, Yanamala N, Seffouh A, Boissan M, Epand RF, Epand RM, Klein-Seetharaman J, Lacombe ML, Kagan VE. Dual function of mitochondrial Nm23-H4 protein in phosphotransfer and intermembrane lipid transfer: a cardiolipin-dependent switch. The Journal of biological chemistry. 2013;288:111–121. doi: 10.1074/jbc.M112.408633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature cell biology. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kowalczyk JE, Beresewicz M, Gajkowska B, Zablocka B. Association of protein kinase C delta and phospholipid scramblase 3 in hippocampal mitochondria correlates with neuronal vulnerability to brain ischemia. Neurochemistry international. 2009;55:157–163. doi: 10.1016/j.neuint.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 86.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 87.Iverson SL, Enoksson M, Gogvadze V, Ott M, Orrenius S. Cardiolipin is not required for Bax-mediated cytochrome c release from yeast mitochondria. The Journal of biological chemistry. 2004;279:1100–1107. doi: 10.1074/jbc.M305020200. [DOI] [PubMed] [Google Scholar]
- 88.Joshi AS, Zhou J, Gohil VM, Chen S, Greenberg ML. Cellular functions of cardiolipin in yeast. Biochimica et biophysica acta. 2009;1793:212–218. doi: 10.1016/j.bbamcr.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kito M, Aibara S, Kato M, Hata T. Differences in fatty acid composition among phosphatidylethanolamine, phosphatidylglycerol and cardiolipin of Escherichia coli. Biochimica et biophysica acta. 1972;260:475–478. doi: 10.1016/0005-2760(72)90062-8. [DOI] [PubMed] [Google Scholar]
- 90.Schlame M, Brody S, Hostetler KY. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. European journal of biochemistry. 1993;212:727–735. doi: 10.1111/j.1432-1033.1993.tb17711.x. [DOI] [PubMed] [Google Scholar]
- 91.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature chemical biology. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 92.Kagan VE, Chu CT, Tyurina YY, Cheikhi A, Bayir H. Cardiolipin asymmetry, oxidation and signaling. Chemistry and physics of lipids. 2014;179:64–69. doi: 10.1016/j.chemphyslip.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaminskyy VO, Zhivotovsky B. Free radicals in cross talk between autophagy and apoptosis. Antioxidants & redox signaling. 2014;21:86–102. doi: 10.1089/ars.2013.5746. [DOI] [PubMed] [Google Scholar]
- 94.Legrand-Poels S, Esser N, L’Homme L, Scheen A, Paquot N, Piette J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochemical pharmacology. 2014;92:131–141. doi: 10.1016/j.bcp.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 95.Tyurina YY, Domingues RM, Tyurin VA, Maciel E, Domingues P, Amoscato AA, Bayir H, Kagan VE. Characterization of cardiolipins and their oxidation products by LC-MS analysis. Chemistry and physics of lipids. 2014;179:3–10. doi: 10.1016/j.chemphyslip.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hannibal L, Tomasina F, Capdevila DA, Demicheli V, Tortora V, Alvarez-Paggi D, Jemmerson R, Murgida DH, Radi R. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry. 2016;55:407–428. doi: 10.1021/acs.biochem.5b01385. [DOI] [PubMed] [Google Scholar]
- 97.Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, Tyurina YY, Wipf P, Atkinson J, Greenberger JS, Chapkin RS, Belikova NA. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: a new strategy in anti-apoptotic drug discovery. Molecular nutrition & food research. 2009;53:104–114. doi: 10.1002/mnfr.200700402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muenzner J, Pletneva EV. Structural transformations of cytochrome c upon interaction with cardiolipin. Chemistry and physics of lipids. 2014;179:57–63. doi: 10.1016/j.chemphyslip.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vladimirov YA, Proskurnina EV, Alekseev AV. Molecular mechanisms of apoptosis. structure of cytochrome c-cardiolipin complex. Biochemistry Biokhimiia. 2013;78:1086–1097. doi: 10.1134/S0006297913100027. [DOI] [PubMed] [Google Scholar]
- 100.Kobayashi H, Nagao S, Hirota S. Characterization of the Cytochrome c Membrane-Binding Site Using Cardiolipin-Containing Bicelles with NMR. Angewandte Chemie (International ed in English) 2016 doi: 10.1002/anie.201607419. [DOI] [PubMed] [Google Scholar]
- 101.Mandal A, Hoop CL, DeLucia M, Kodali R, Kagan VE, Ahn J, van der Wel PC. Structural Changes and Proapoptotic Peroxidase Activity of Cardiolipin-Bound Mitochondrial Cytochrome c. Biophysical journal. 2015;109:1873–1884. doi: 10.1016/j.bpj.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RS, Kochanek PM, Wipf P, Kagan VE, Bayir H. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature neuroscience. 2012;15:1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jiang J, Bakan A, Kapralov AA, Silva KI, Huang Z, Amoscato AA, Peterson J, Garapati VK, Saxena S, Bayir H, Atkinson J, Bahar I, Kagan VE. Designing inhibitors of cytochrome c/cardiolipin peroxidase complexes: mitochondria-targeted imidazole-substituted fatty acids. Free radical biology & medicine. 2014;71:221–230. doi: 10.1016/j.freeradbiomed.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, Mohammadyani D, Klein-Seetharaman J, Jackson TC, Kochanek PM, Pitt BR, Greenberger JS, Vladimirov YA, Bayir H, Kagan VE. A mitochondrial pathway for biosynthesis of lipid mediators. Nature chemistry. 2014;6:542–552. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Balasubramanian K, Maeda A, Lee JS, Mohammadyani D, Dar HH, Jiang JF, St Croix CM, Watkins S, Tyurin VA, Tyurina YY, Kloditz K, Polimova A, Kapralova VI, Xiong Z, Ray P, Klein-Seetharaman J, Mallampalli RK, Bayir H, Fadeel B, Kagan VE. Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity. Science signaling. 2015;8:ra95. doi: 10.1126/scisignal.aaa6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dorio A, Cerella C, De Nicola M, D’Alessio M, Gualandi G, Ghibelli L. Non-apoptogenic Ca2+-related extrusion of mitochondria in anoxia/reoxygenation stress. Annals of the New York Academy of Sciences. 2007;1099:512–515. doi: 10.1196/annals.1387.067. [DOI] [PubMed] [Google Scholar]
- 107.Goldmann O, Medina E. The expanding world of extracellular traps: not only neutrophils but much more. Frontiers in immunology. 2012;3:420. doi: 10.3389/fimmu.2012.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakajima A, Kurihara H, Yagita H, Okumura K, Nakano H. Mitochondrial Extrusion through the cytoplasmic vacuoles during cell death. The Journal of biological chemistry. 2008;283:24128–24135. doi: 10.1074/jbc.M802996200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Uematsu S, Akira S. Toll-like receptors and innate immunity. Journal of molecular medicine (Berlin, Germany) 2006;84:712–725. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 111.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling, Nature reviews. Immunology. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 112.Erdman LK, Cosio G, Helmers AJ, Gowda DC, Grinstein S, Kain KC. CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. Journal of immunology (Baltimore, Md: 1950) 2009;183:6452–6459. doi: 10.4049/jimmunol.0901374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annual review of biochemistry. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 114.Scior T, Lozano-Aponte J, Figueroa-Vazquez V, Yunes-Rojas JA, Zahringer U, Alexander C. Three-dimensional mapping of differential amino acids of human, murine, canine and equine TLR4/MD-2 receptor complexes conferring endotoxic activation by lipid A, antagonism by Eritoran and species-dependent activities of Lipid IVA in the mammalian LPS sensor system. Computational and structural biotechnology journal. 2013;7:e201305003. doi: 10.5936/csbj.201305003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 116.Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YP, Eagle AR, Dunn SE, Awakuni JU, Nguyen KD, Steinman L, Michie SA, Chawla A. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nature medicine. 2009;15:1266–1272. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.N A-G, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 119.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bashir S, Sharma Y, Elahi A, Khan F. Macrophage polarization: the link between inflammation and related diseases. Inflammation research: official journal of the European Histamine Research Society. 2016;65:1–11. doi: 10.1007/s00011-015-0874-1. [DOI] [PubMed] [Google Scholar]
- 122.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Frontiers in bioscience: a journal and virtual library. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 124.Vladimirov YA, Proskurnina EV, Izmailov DY. Chemiluminescence as a method for detection and study of free radicals in biological systems. Bulletin of experimental biology and medicine. 2007;144:390–396. doi: 10.1007/s10517-007-0340-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.