

Abstract
Matrix metalloproteinase-9 (MMP-9) is robustly elevated in the first week post-myocardial infarction (MI). Targeted deletion of the MMP-9 gene attenuates cardiac remodeling post-MI by reducing macrophage infiltration and collagen accumulation through increased apoptosis and reduced inflammation. In this study, we used a translational experimental design to determine whether selective MMP-9 inhibition early post-MI would be an effective therapeutic strategy in mice. We enrolled male C57BL/6J mice (3–6 months old, n = 116) for this study. Mice were subjected to coronary artery ligation. Saline or MMP-9 inhibitor (MMP-9i; 0.03 μg/day) treatment was initiated at 3 h post-MI and the mice were sacrificed at day (D) 1 or 7 post-MI. MMP-9i reduced MMP-9 activity by 31 ± 1% at D1 post-MI (p < 0.05 vs saline) and did not affect survival or infarct area. Surprisingly, MMP-9i treatment increased infarct wall thinning and worsened cardiac function at D7 post-MI. While MMP-9i enhanced neutrophil infiltration at D1 and macrophage infiltration at D7 post-MI, CD36 levels were lower in MMP-9i compared to saline, signifying reduced phagocytic potential per macrophage. Escalation and prolongation of the inflammatory response at D7 post-MI in the MMP-9i group was evident by increased expression of 18 pro-inflammatory cytokines (all p < 0.05). MMP-9i reduced cleaved caspase 3 levels at D7 post-MI, consistent with reduced apoptosis and defective inflammation resolution. Because MMP-9i effects on inflammatory cells were significantly different from previously observed MMP-9 null mechanisms, we evaluated pre-MI (baseline) systemic differences between C57BL/6J and MMP-9 null plasma. By mass spectrometry, 34 plasma proteins were significantly different between groups, revealing a previously unappreciated altered baseline environment pre-MI when MMP-9 was deleted. In conclusion, early MMP-9 inhibition delayed inflammation resolution and exacerbated cardiac dysfunction, highlighting the importance of using translational approaches in mice.
Keywords: MMP-9, Proteomics, Neutrophil, Apoptosis, Myocardial infarction, Macrophage, Inhibitor
1. Introduction
Myocardial Infarction (MI) caused by coronary artery occlusion results in remodeling of the left ventricle (LV) that includes altered LV size, shape, structure, and function [1]. While the remodeling process post-MI is not completely understood, an exacerbated inflammatory response and excess extracellular matrix (ECM) degradation are associated with increased risk of progression to heart failure [2]. Matrix metalloproteinases (MMPs) are zinc dependent enzymes that regulate the wound healing cascade initiated after cardiac injury by cleaving ECM substrates to remove damaged and necrotic tissue and by cleaving non-ECM substrates to regulate protein function [3,4]. To date, 11 MMPs and 4 tissue inhibitors of metalloproteinases (TIMPs) have been measured post-MI [3,5]. Of these, MMP-9 has been the most extensively evaluated [4,6,7].
MMP-9 (92 kDa type IV collagenase or gelatinase B) is activated early in both permanent occlusion and ischemia/reperfusion MI models, and concentrations remain elevated over the first week post-MI in mice, rabbits, dogs, pigs, and humans [4,8]. Targeted deletion of the MMP-9 gene attenuates LV remodeling and improves survival post-MI [6,9,10]. MMP-9 deficient mice have reduced cardiac rupture rates, reduced macrophage infiltration rates, and increased neovascularization [6,9,10]. We have recently identified a novel mechanism by which MMP-9 regulates cardiac remodeling through proteolytic processing of CD36, a member of the class B scavenger receptor family [11]. In the absence of MMP-9, intact CD36 stimulates neutrophil apoptosis and enhances macrophage phagocytic potential promoting an early resolution of inflammation [11].
While MMP-9 gene deletion has proven beneficial post-MI in mice, macrophage-specific transgenic overexpression of MMP-9 also improves LV function by attenuating the post-MI inflammatory response, suggesting that timing and concentration of MMP-9 may dictate divergent mechanisms of response [7]. MMP-9 is secreted by a wide number of post-MI relevant cell types, including neutrophils, macrophages, myocytes, and fibroblasts [4]. It is possible that neutrophil-derived and macrophage-derived MMP-9, for example, have distinct roles.
First generation MMP inhibitors lacked specificity and selectivity, leading to less effective in vivo use and inconclusive results [12]. The Fields laboratory has developed a potent and highly selective MMP-9 inhibitor (MMP-9i) [13]. This triple-helical peptide inhibitor has a sequence of (Gly-Pro-Hyp)4-Gly-mep-Flp-Gly-Pro-Pro-GlyΨ(PO2H-CH2)Val-Val-Gly-Glu-Gln-Gly-Glu-Gln-Gly-Pro-Pro-Gly-mep-Flp-(Gly-Pro-Hyp)4-NH2. At 37 °C, this inhibitor has greater inhibitor capacity for recombinant active MMP-9 (Ki of 0.98 ± 0.09 nM) compared to recombinant active MMP-2 (Ki of 2.24 ± 0.11 nM) [13]. An in vivo dose of 0.03 μg/kg was selected for MMP-9 inhibition, with dosing for 7 days. The objective of this study was to use a translational research approach to explore the therapeutic potential of inhibiting MMP-9 during early post-MI LV remodeling.
2. Materials and methods
A detailed description of the materials and methods is available in the Supplemental methods.
2.1. Mice
All animal procedures were performed based on the “Guide for the Care and Use of Laboratory Animals” (Eighth Edition) and were approved by the Institutional Animal Care and Use Committees at the University of Texas Health Science Center at San Antonio and the University of Mississippi Medical Center. In this study, 3- to 6-month old C57BL/6J male mice were purchased from The Jackson Laboratory and housed within the university animal facilities. LV function was evaluated with the Vevo 2100™ system (VisualSonics, Toronto, Ontario, Canada). Infusion of the MMP-9i (0.03 μg/day) was initiated at 3 h post-MI by implantation of osmotic mini-pumps, and inhibition continued through 7 days. Day (D) 0 no MI mice were used as negative controls and saline treated MI mice were used as positive MI controls [3,13].
2.2. Tissue harvest and infarct area evaluation
The heart was collected as described before [3,13,14]. Infarct area was calculated as the percentage of infarct area to total LV area using Adobe Photoshop [15].
2.3. MMP-9 activity assay
MMP-2 and MMP-9 activities were measured in D1 plasma samples from saline or MMP-9i treated mice using a selective MMP-2/MMP-9 fluorogenic triple-helical substrate (EMD Millipore, CBA 003) [16].
2.4. Protein extraction and immunoblotting
The LV was homogenized and total protein extracted into two fractions: soluble and insoluble. Protein levels were quantified by Bradford assay (BioRad, Hercules, CA). Total protein from the soluble and insoluble fractions was immunoblotted onto nitrocellulose membranes (Bio-Rad). Protein intensities were quantified by densitometry using the IQ-TL image analysis software (GE Healthcare, Waukesha, WI).
2.5. Gelatin zymography
Samples (25 μg soluble fraction total protein) were loaded onto nondenaturing 10% polyacrylamide gels containing 0.1% gelatin, electrophoresed, renatured, and developed as described previously [17]. Gel images were acquired and inverted, and densitometry was measured using the IQ-TL image analysis software (GE Healthcare, Waukesha, WI) to quantify activity.
2.6. Real time RT2-PCR
Gene array for inflammatory cytokines and receptors (Qiagen, PAMM-011A, Qiagen, Valencia, CA, USA) and for ECM and adhesion molecules (PAMM-013A) were performed to quantify gene expression.
2.7. Immunohistochemistry
The middle section of the LV was paraffin-embedded and sectioned at 5 μm for quantification of neutrophils or macrophages by immunohistochemistry and collagen by picrosirius red staining, as described previously [11,13,14,18,19]. Images were captured at 40× magnification with Image-Pro software (Media Cybernetics, Bethesda, MD, USA) and quantification was calculated as the percentage of positively stained area to total section area.
2.7.1. Plasma proteomics
The samples were digested with trypsin, and N-linked glycopeptides were isolated using the solid phase extraction of glycopeptides (SPEG) method as previously reported [20,21]. The purpose of using the SPEG approach was to eliminate albumin and other highly prevalent nonglycosylated proteins commonly found in plasma. Peptides were analyzed by LC-MS/MS using a Q Exactive mass spectrometer (Thermo Fisher, Waltham, MA, USA). MS/MS spectra were searched with SEQUEST using Proteome Discoverer. Identified proteins were included in the analysis if the peptide contained an NXT/S motif and the protein had a membrane or extracellular localization consensus sequence [11, 22]. The volcano plot was constructed with Metaboanalyst 3.0 software (www.metaboanalyst.ca/) using peak intensity values [23–26].
2.8. Statistical analyses
Data are presented as mean ± SEM. Survival rates were analyzed by Kaplan-Meier survival analysis and compared by log-rank test. Rupture rates were analyzed by Fisher's exact test. Two group comparisons were analyzed by Student's t-test. Multiple group comparisons were analyzed using one-way ANOVA, two-way ANOVA, or nonparametric Kruskal-Wallis test with appropriate post-test.
3. Results
3.1. MMP-9i reduced MMP-9 activity early post-MI without affecting MMP-2 or MMP-9 expression
Since MMP-9 levels increase early post-MI, we initiated MMP-9i treatment at 3 h post-MI, a time that would be clinically relevant. At D1 post-MI, plasma from the MMP-9i group showed 31 ± 1% reduction in MMP-9 recruitable activity (Fig. 1A). In the saline group, MMP-9 gene levels were elevated at D1 post-MI and returned to baseline by D7, and this pattern was not affected by MMP-9i (Fig. 1B). No significant differences were observed in MMP-9 gene expression among MMP-9i and saline groups at D1 and D7 post-MI. This result indicates that MMP-9i did not affect gene levels, which was expected since the inhibitor is a transition state analog inhibitor that binds to active MMP to block catalysis [27].
Fig. 1.
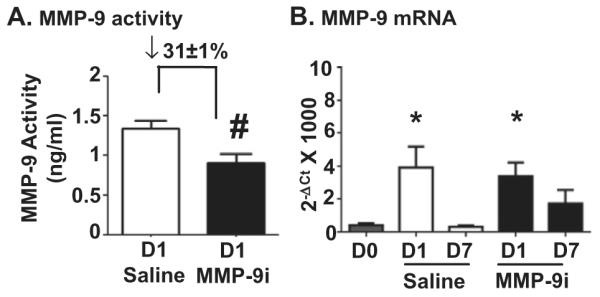
MMP-9 inhibitor (MMP-9i) treatment reduced MMP-9 activity early post-MI without affecting MMP-9 gene expression. (A) MMP-9 activity was reduced by 31 ± 1% in the inhibitor treated group, compared to saline MI. n = 10/group. (B) RT2-PCR analysis showed increased MMP-9 expression levels in both saline and MMP-9i LV infarct (LVI) compared to D0. n = 6/group. *p < 0.05 vs D0, #p < 0.05 vs saline.
As MMP-9i can also inhibit MMP-2 at higher doses, we measured MMP-2 gene expression and protein production [27]. MMP-2 gene and protein (Fig. 2A) levels significantly increased in both saline and MMP-9i groups compared to D0 in the infarcted LV (LVI) at D7 post-MI (all p < 0.05), demonstrating that MMP-9i did not affect MMP-2 concentrations. To determine if the dose of MMP-9i used was also inhibiting MMP-2 activity, we measured the formation of cleavage products of two MMP-2 and MMP-9 substrates (collagen I and fibronectin) by in vivo substrate hydrolysis assessment. Both MMP-2 and MMP-9 proteolytically process collagen I to generate a 30 kDa fragment, which is then further cleaved by MMP-9 but not by MMP-2 [8]. By immunoblotting, the fragment pattern observed (increased intensity of the 30 kDa fragment in MMP-9i group compared to saline group) was indicative of inhibition of MMP-9 activity only (Fig. 2B). Both MMP-2 and MMP-9 cleave fibronectin. MMP-2 generates fragments with molecular weights of 180 kDa, 160 kDa, 90 kDa, and 65 kDa [28]. MMP-9 generates fragments with molecular weights of 166 kDa, 120 kDa, and 70 kDa [29]. We observed increased levels of full length fibronectin in both LVI groups and increased levels of the 180 kDa fragment in the MMP-9i group compared to saline group, indicating MMP-2 activity was preserved in the MMP-9i group while MMP-9 was inhibited (Fig. 2C). Because fibronectin is readily soluble and found in high concentrations in the circulation, observing full length fibronectin in the soluble fraction and increased amounts of fibronectin fragments in the insoluble fraction may indicate that the fragments are locally produced and sequestered in the ECM. In addition, gelatin zymography demonstrated that while both LVI groups showed increased pro and active MMP-9 activity compared to day 0, there were no significant differences in MMP-9 activity between the MMP-9i and saline infarct groups, confirming that administration of the MMP-9 inhibitor did not produce a compensatory increase in MMP-9 protein and that reduced MMP-9 activity was due to binding of the inhibitor to the protease (Fig. 3). Combined, these results demonstrated that the dose of MMP-9i given was sufficient to inhibit MMP-9 and not MMP-2.
Fig. 2.

MMP-9i inhibited MMP-9 but not MMP-2. (A) MMP-2 gene expression at D7 post-MI was evaluated by RT2-PCR. MMP-2 gene levels were not significantly different between MMP-9i and saline treated LV infarct (LVI) groups. MMP-2 protein increased in the insoluble fraction of both saline and MMP-9i LVI compared to D0. (B) The production of the 30 kDa collagen I cleavage fragment, which is generated by both MMP-2 and MMP-9 but only degraded by MMP-9, was higher in the soluble fraction of the MMP-9i LVI compared to saline LVI. (C) Full-length fibronectin was higher in the soluble fraction (left) of both LVI groups, compared to the D0 control. The 180 kDa fibronectin cleavage fragment (arrow) was higher in the insoluble fraction (right) of the MMP-9i LVI compared with the saline treated LVI. Recombinant fibronectin was used as the positive control (labeled as +). n = 6/group. *p < 0.05 vs D0, #p < 0.05 vs saline.
Fig. 3.
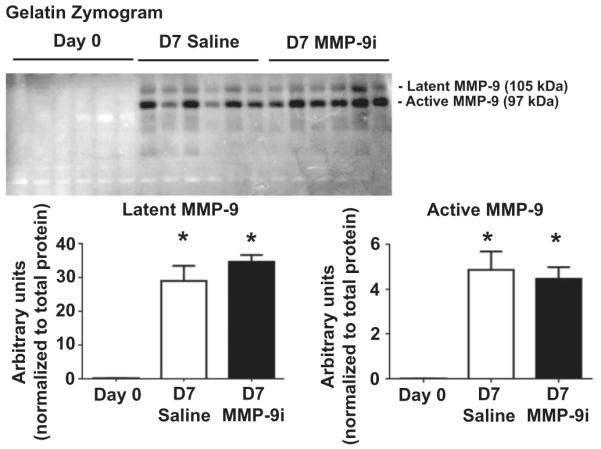
By gelatin zymography, MMP-9 recruitable activity increased post-MI in both saline and MMP-9i treated groups. (A) Representative gelatin zymogram (inverted image) showing pro and active MMP-9 in the D7 post-MI LV infarct. No significant difference in MMP-9 activity was observed between MMP-9i and saline MI groups. n = 6/group. *p < 0.05 vs D0.
3.2. MMP-9 inhibition does not affect survival rate, cardiac rupture rate, or infarct area
A total of 12 out of 36 saline treated mice survived 7 days after MI (33%), and 12 out of 44 MMP-9i treated mice survived 7 days after MI (27%; p = 0.56; Fig. 4A). This result is in contrast to MMP-9 deletion, which improves survival rate post-MI [9]. Cardiac rupture occurred between 3 and 7 days post-MI, consistent with previous reports, and was similar among the groups (Fig. 4B; p = 0.87) [30,31]. Infarct areas were similar between saline and MMP-9i in both D1 and D7 MI groups (Fig. 4C, p = 0.50), an expected result since the inhibitor was started at 3 h post-MI, a time beyond significant myocardial salvage. This demonstrates that both groups of mice were given similar ischemic injuries. Of note, the infarct areas for the MI groups was approximately 55%, which may explain the observed survival rates [14,32,33].
Fig. 4.
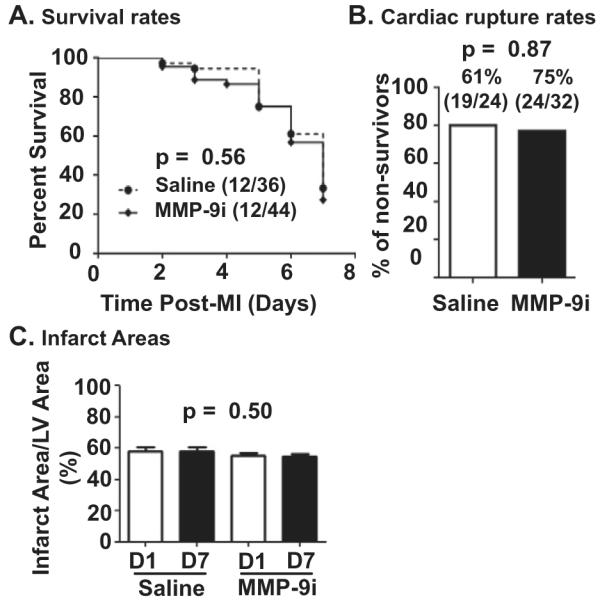
MMP-9 inhibition does not affect survival, cardiac rupture, or infarct area. (A) Survival was similar between saline and MMP-9i treated mice. (B) Cardiac rupture rates were similar between saline and MMP-9i treated mice. In the saline group, 19 out of 24 non-surviving mice had ruptured hearts; whereas in the MMP-9i group, 24 out of 32 non-surviving mice had ruptured hearts. (C) Infarct areas were similar between saline and MMP- 9i treated mice at D1 and D7 post-MI. n = 12/group.
3.3. MMP-9 inhibition increased infarct wall thinning and exacerbated LV dysfunction at D7 post-MI
Post-MI infarct wall thinning was increased and ejection fraction was reduced in both saline and MMP-9i groups, and these effects were greater with MMP-9i (p < 0.05, Table 1). This suggests that MMP-9 functions early post-MI include a blend of detrimental and cardioprotective roles. Worsened LV function with MMP-9i is in contrast to MMP-9 null mice studies, where global gene deletion attenuated LV enlargement [34].
Table 1.
MMP-9 inhibition worsens function of the post-myocardial infarction (MI) left ventricle (LV).
| Day 0 | Day 7 MI saline |
Day 7 MI MMP-9i |
|
|---|---|---|---|
| Body weight; g | 30 ± 1 | 28 ± 1 | 28 ± 1 |
| LV mass/body weight; mg/g | 3.2 ± 0.1 | 4.4 ± 0.3* | 4.0 ± 0.1* |
| Wet lung weight/body weight; mg/g | 5.0 ± 0.5 | 10.0 ± 1.0* | 8.0 ± 1.0* |
| Infarct wall thickness (systolic; mm) | 1.06 ± 0.03 | 0.55 ± 0.03* | 0.45 ± 0.03*# |
| LV remodeling index | 0.44 ± 0.04 | 1.41 ± 0.07* | 1.33 ± 0.12* |
| End diastolic volume; μl | 45 ± 1 | 168 ± 7* | 149 ± 14* |
| End systolic volume; μl | 14 ± 1 | 148 ± 7* | 141 ± 13* |
| Ejection fraction; % | 69 ± 1 | 12 ± 1* | 5 ± 1*# |
Values are presented as mean ± SEM, n = 12/group.
p < 0.05 vs D0, and
p < 0.05 vs saline treatedmice. Remodeling Index = end diastolic volume / LV mass. LV - left ventricle; MI - myocardial infarction.
3.4. MMP-9 inhibition regulated ECM remodeling at D7 post-MI
Since significant differences in LV function were observed, we evaluated ECM gene and protein levels. A total of 84 ECM genes were analyzed in the infarcts at D1 and D7 post-MI and compared to D0 LV. There were 65 genes at D1 and 60 genes at D7 statistically different between treatment groups (all p < 0.05, Table 2, Supplemental Table S2, Supplemental Fig. 1).
Table 2.
ECM and adhesion gene expression changes between infarcted LV (saline or MMP-9i groups) and no MI (D0 LV controls) as well as between MMP-9i treated and vehicle control LV infarct (MMP-9i vs saline). The increase, decrease, and no change were based on quantitative measurements that were statistically different based on RT2-PCR analysis for n = 6 LV/group (see Supplemental Table 2 for quantitative data on all 84 genes). Qualitative directions of change for each group are shown here. Out of 84 genes measured, the 65 genes at D1 and 60 genes at D7 shown below were significantly different among groups (all p < 0.05).
| Saline vs Day 0 |
MMP–9i vs Day 0 |
MMP–9i vs Saline |
Interpretation | |
|---|---|---|---|---|
| Day 1 post–MI | ||||
|
Adamts2, Col2a1, Ctgf, Itgam, Itgb3, Mmp3,
Mmp8, Mmp9, Mmp10, Sele, Sell, Selp, Spp1, Thbs1, Tnc, Vcan |
↑ | ↑ | ↑ with MI | |
|
Ctnna2, Cdh1, Cdh3, Hapln1, Hc, Icam1, Itga3,
Itga5, Itgae, Itgb1, Lama1, Mmp1a, Mmp7, Mmp1. Mmp14, Ncam2, Spock1, Syt1, Timp1 |
↑ | MI effect blunted by MMP–9i |
||
| Itgav | ↑ | ↑ | MI effect exacerbated by MMP–9i |
|
| Adamts1, Cd44 | ↑ | MMP–9i effect | ||
|
| ||||
|
Adamts5, Ctnna1, Ctnnb1, Cdh4, Col4a3, Fbln1,
Itgax, Lama2, Lama3, Lamb2, Lamb3, Lamc1, Mmp2, Mmp11, Mmp15, Sgce, Sparc, Thbs2, Thbs3, Timp2, Vtn |
↓ | ↓ | ↓ with MI | |
| Cdh2, Col3a1, Vcam1 | ↓ | MI effect blunted by MMP–9i |
||
| Col6a1 | ↓ | ↓ | ↓ | MI effect exacerbated by MMP–9i |
|
| ||||
| Col5a1, Itga4 | ↓ | MMP–9i effect | ||
|
| ||||
| Day 7 post–MI | ||||
|
Col1a1, Col2a1, Hapln1, Itgae, Itgav, Ncam1,
Postn, Timp1, Timp2, Timp3, Thbs1, Thbs2, Thbs3 |
↑ | ↑ | ↑ with MI | |
| Ctnna2 | ↑ | MI effect blunted by MMP–9i |
||
|
Adamts2, Cd44, Cdh1, Cdh3, Col4a2, Ctgf, Ecm1, Emilin1, Fn1, Hc, Itga2, Itga4, Itgam, Itgb2, Itgb3, Lama1, Mmp8, Mmp12, Mmp14, Sell, Selp, Sparc, Spp1, Tgfbi, Tnc, Vcam1, Vcan |
↑ | MMP–9i effect | ||
|
Adamts1, Col3a1, Col4a1, Col5a1, Icam1, Itga5,
Itgb1 |
↑ | ↑ | MMP–9i effect | |
|
| ||||
| Cdh4, Itgax, Lamb3, Mmp15, Vtn | ↓ | ↓ | ↓ with MI | |
| Ctnnb1, Itga3, Lamb2, Pecam1, Sele | ↓ | MI effect blunted by MMP–9i |
||
| Ctnna1, Cdh2 | ↓ | ↓ | ↓ | MI effect exacerbated by MMP–9i |
Arrows depict direction of change. D - day. MI - Myocardial infarction. The bold black line separates the genes with increased expression fromthe genes that showed decreased expression at D1 and D7 post-MI. Gene names are provided in the Supplemental methods.
Collagens, including Col1a1, Col2a1, Col3a1, Col4a1, Col4a2, and Col5a1, were increased in both saline LVI and MMP-9i LVI at D7 post-MI compared to D0 controls. Of the 6 collagen genes that were significantly different in both groups, Col 3a1, 4a1, 4a2, and 5a1 were significantly upregulated in MMP-9i LVI compared to saline LVI at D7 post-MI, indicating that MMP-9 inhibition may alter scar formation properties (Table 2). Since gene levels were significantly elevated in the MMP-9i group, we evaluated collagen deposition. By picrosirius red staining, collagen deposition was 21% decreased in the MMP-9i LVI at D7 post-MI, compared to saline MI (p < 0.05, Fig. 5A). The reduced collagen deposition in the MMP-9i group in the presence of increased gene expression signifies a feedback loop between collagen protein and gene expression, impaired collagen crosslinking, or increased collagen degradation by other MMPs. This data is consistent with findings in the MMP-9 null mice, where reduced collagen deposition was observed with MMP-9 deletion at D7 post-MI compared to wild type infarcts [34].
Fig. 5.
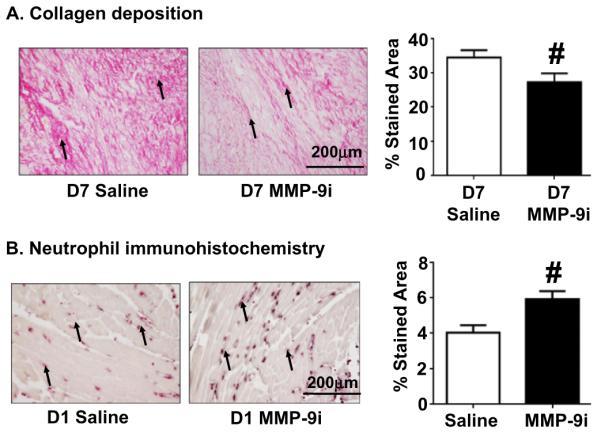
MMP-9 inhibition reduced collagen deposition at D7 post-MI and increased neutrophil infiltration at D1 post-MI. (A) Collagen deposition in the infarct area was decreased in the MMP-9i group at D7 post-MI compared to saline mice. (B) Neutrophil infiltration into LVI was increased in MMP-9i group at D1 post-MI compared to saline mice. Arrows depict positive staining. Scale bar is 200 μm. n = 12/group. #p < 0.05 vs saline.
MMP deletion results in the elevation of other MMPs as a compensatory mechanism [34]. Of the 12 MMPs and 3 TIMPs measured to evaluate possible compensatory mechanisms with MMP-9 inhibition, Mmp1a, Mmp7, Mmp12, Mmp14, and Timp1 gene expression was blunted in the MMP-9i group at D1 post-MI. At D7 post-MI, Mmp8, Mmp12, and Mmp14 levels increased in the MMP-9i group, indicative of increased ECM degradation and consistent with reduced collagen deposition and increased wall thinning observed at D7 post-MI [3,35,36].
3.5. MMP-9 inhibition increased inflammatory cytokine expression at D7 post-MI
Since progression and resolution of the inflammatory response influences overall LV remodeling and function, we measured 84 inflammatory genes to monitor the post-MI inflammatory response at D1 and D7. At D1 post-MI, 16 genes increased and 11 genes decreased in response to MI (compared to D0), indicating responses that were independent of MMP-9i (all p < 0.05, Table 3, Supplemental Table S3, Supplemental Fig. 2).
Table 3.
Inflammatory gene expression changes between infarcted (saline orMMP-9i groups) and no MI (D0 controls), as well as between MMP-9 inhibitor treated and vehicle control groups (MMP-9i vs saline). The increase, decrease, and no change were based on quantitative measurements that were statistically different based on RT2-PCR analysis for n = 6 LV infarct/group (see Supplemental Table 3 for quantitative data on all 84 genes). Qualitative directions of change for each group are shown here. Out of 84 genes measured, only these 40 genes at D1 and 38 genes at D7 were significantly different among groups (all p < 0.05).
| Saline vs Day 0 |
MMP–9i vs Day 0 |
MMP–9i vs Saline |
Interpretation | |
|---|---|---|---|---|
| Day 1 post–MI | ||||
| (a): Cxcl5, Il11 (p): Ccl2, Ccl3, Ccl7, Ccl9, Ccr1, Cxcl1, Il1b, Il1r1, Il1r2, Il2rg, Il6st, Il8rb, Itgam, Spp1 |
↑ | ↑ | ↑ with MI | |
| (a): Il10 (p): Ccl4, Ccr8, Il1f6, Lta |
↑ | MI effect blunted by MMP–9i |
||
| (a): Il3 (p): Xcr1 |
↑ | MMP–9i effect | ||
|
| ||||
| (a): Bcl6, Il10rb (p): Abcf1, C3, Ccl11, Cxcl9, Ccr10, Il15, Mif, Scye1, Tollip |
↓ | ↓ | ↓ with MI | |
| (p): Ccl25 | ↓ | MI effect blunted by MMP–9i |
||
| (a): Tgfb1 (p): Cxcl10 |
↑ | ↓ | MI effect blunted by MMP–9i |
|
| (p): Il16 | ↓ | ↓ | ↓ | MI effect exacerbated by MMP–9i |
| (p): Cxcl11, Tnfrsf1a | ↓ | MMP–9i effect | ||
|
| ||||
| Day 7 post–MI | ||||
| (a): Il10ra, Ccl17 (p): Il1r1, Cx3cl1, Cxcr3 |
↑ | ↑ | ↑ with MI | |
| (p): Casp1, Lta | ↑ | MI effect blunted by MMP–9i |
||
| (p): Il18, Il6ra | ↑ | ↑ | ↑ | MI effect exacerbated by MMP–9i |
| (a): Il11 (p): Ccl4, Ltb, Ccl8, Ccr5, Itgam, Itgb2, Spp1, Pf4 |
↑ | MMP–9i effect | ||
| (a): Ccl6 (p): Ccl2 |
↑ | MMP–9i effect | ||
| (a): Cxcl5 (p): Cxcr5, Ccl12,Ccl7, Ccl9, Ccr1, Ccr2, Ccr3, Tnfrsf1b |
↑ | ↑ | MMP–9i effect | |
|
| ||||
| (a): Il10rb, Tollip (p): Ccl11, Ccl24, Il15, Ccr9, Scye1 |
↓ | ↓ | ↓ with MI | |
| (p): Il16 | ↓ | ↑ | MI effect blunted by MMP–9i |
|
| (p): Abcf1 | ↓ | ↓ | ↓ | MI effect exacerbated by MMP–9i |
Arrows depict direction of change. (a) - anti-inflammatory; D - day; MI-myocardial infarction; (p) - pro-inflammatory. The bold black line separates the genes with increased expression from the genes that showed decreased expression at D1 and D7 post-MI. Gene names are provided in the Supplemental methods.
MMP-9i blunted the expression of Ccl4, Ccl25, Ccr8, Cxcl10, Il1f6, Lta, Il-10 and Tgfβ1, and exacerbated the levels of Il16, compared to the saline D1 group. A total of 4 genes (Cxcl11, Il3, Tnfrsf1a, and Xcr1) showed no change in the saline MI group and significantly changed in the presence of MMP-9i (all p < 0.05). Of note, Cxcl10, Cxcl11, Il3, and Tgfβ1 are known to be expressed by neutrophils. The inflammatory gene profiles observed at D1 post-MI suggested that neutrophil numbers may be elevated in the infarcts of the MMP-9i group [37].
At D7 post-MI, 4 genes increased (Cx3cl1, Cxcr3, Il1r1, and Il10ra) and 7 genes decreased (Ccl11, Ccl24, Ccr9, Il10rb, Il15, Scye1, Tollip) in response to MI in both saline and MMP-9i MI groups (all p < 0.05). Expression of 3 genes (Casp1, Il16, Lta) was blunted and expression of 3 genes (Abcf1, Il6ra, Il18) was intensified by MMP-9i compared to the saline group. A total of 21 genes showed no change with MI in the saline group and significantly increased in the presence of MMP-9i, of which 18 were pro-inflammatory (including Itgam, Itgb2, and Spp1, all p < 0.05). The inflammatory gene profiles observed at D7 post-MI were consistent with a prolonged inflammatory response in the infarct region and indicated delayed resolution of inflammation with MMP-9 inhibition. Overall, our results suggest that inhibition of MMP-9 early post-MI exacerbated inflammation at both D1 and D7 post-MI, which is in contrast to MMP-9 null mice studies where MMP-9 gene deletion decreased the inflammatory response. Of note, Ccl9, Itgam, Itgb2, Pf4, and Spp1 are known to be expressed by macrophages [38–40]. The increase in macrophage produced cytokines indicated a change in the macrophage population or phenotype at D7 in the MMP-9i group.
3.6. MMP-9 inhibition increased neutrophil infiltration at D1 post-MI
Since MMP-9i induced overexpression of inflammatory genes post-MI and MMP-8, −12 and −14 are associated with leukocyte function, we evaluated neutrophil numbers at D1 and D7 post-MI. By immunohistochemistry, there was a 33% increase in neutrophil numbers in the MMP-9i group at D1 post-MI compared to saline (Fig. 5B). Increased neutrophil infiltration is associated with worsened cardiac function [41].
3.7. MMP-9 inhibition increased macrophage infiltration at D7 post-MI
We further investigated MMP-9i effects on macrophage recruitment and function. We quantified macrophage numbers in the LVI, and MMP-9i treatment resulted in 50% higher macrophage numbers at D7 post-MI, compared to the saline MI control (Fig. 6A). Levels of the leukocyte extravasation facilitator CD18 (Itgb2, β2 integrin) were elevated at d7 post-MI in the MMP-9i LVI compared to saline MI, at both the gene (161%) and protein (66%) levels (Table 2 and Fig. 6B), which explained the enhanced leukocyte infiltration. These results give evidence of prolonged inflammation and are in contrast to global MMP-9 deletion that resulted in decreased macrophage infiltration at D5 post-MI in mice [6].
Fig. 6.
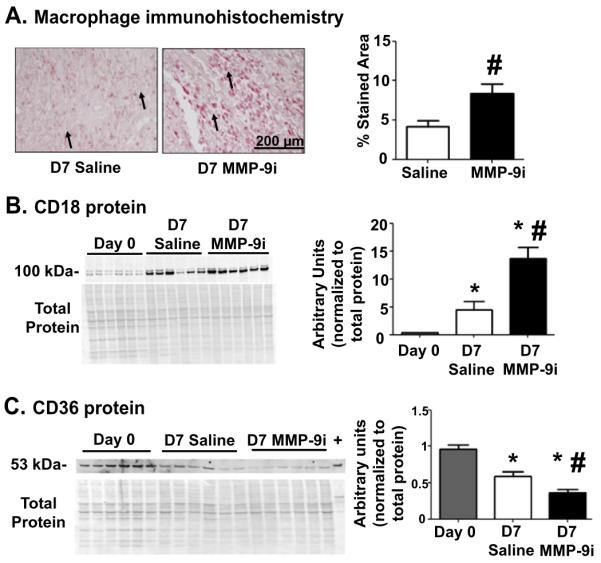
MMP-9 inhibition increased macrophage extravasation, prolonged inflammation, and reduced phagocytic potential at D7 post-MI. (A) Macrophage numbers in the D7 LVI increased with MMP-9 inhibition, suggestive of prolonged inflammation. Arrows depict positive staining. Scale bar is 200 μm. n = 12/group. (B) CD18 was upregulated in the insoluble LVI fraction of the MMP-9i group compared to saline at D7 post-MI, indicating increased macrophage extravasation. n = 6/group. (C) CD36 was downregulated in the soluble fraction of the MMP-9i LVI compared to saline at D7 post-MI, giving evidence of reduced phagocytosis. Peritoneal macrophages were used as a positive control (labeled as +). n = 6/group. *p < 0.05 vs D0 and #p < 0.05 vs saline.
CD36 is a phagocytic marker in macrophages and an MMP-9 substrate [11]. CD36 was significantly reduced in the saline group at D7 post-MI and was reduced 37% further in the MMP-9i group, indicating reduced phagocytic potential of individual macrophages (Fig. 6C). This is in contrast to MMP-9 null mice where CD36 levels were higher post-MI compared to wild type [11].
3.8. MMP-9 inhibition reduced apoptosis at D7 post-MI
Elevated leukocyte numbers and prolonged inflammatory response in the MMP-9i group at D7 post-MI raise the possibility of reduced clearance of apoptotic cells. We measured levels of the apoptosis marker caspase 3 in the LVI and found that it was elevated by 58% in the MMP-9i group compared to saline MI (Fig. 7A). The accumulation of full length caspase 3 was likely due to reduced caspase 3 cleavage, as confirmed by immunohistochemistry (Fig. 7B). TUNEL assay further confirmed that MMP-9i reduced apoptosis by 63% (Fig. 7C). The combined results are consistent with a reduced apoptosis rate at D7. Increased leukocyte accumulation in the infarct would delay inflammation resolution to worsen cardiac function in the MMP-9i group. These results were in contrast to MMP-9 gene deletion studies where apoptosis rates increased at D7 post-MI leading to a blunted inflammatory response [11].
Fig. 7.

MMP-9 inhibition reduced apoptosis at D7 post-MI. (A) MMP-9i increased protein expression of full length caspase 3, an apoptosis marker, in the soluble fraction of the LVI. n = 6/group. (B) Cleaved caspase 3 expression was reduced in the MMP-9i LVI at D7 post-MI compared to saline LVI. Arrows depict positive staining. Scale bar is 200 μm. n = 12/group. (C) MMP-9i reduced TUNEL-positive staining at D7 post-MI. Arrows depict positive staining. Scale bar is 20 μm. n = 12/group. Overall, these data suggest compromised clearance of inflammatory cells. *p < 0.05 vs D0 and #p < 0.05 vs saline.
3.9. MMP-9 deletion alters the baseline environment
Since we observed dramatic differences between our MMP-9 inhibitor results and previous studies using MMP-9 null mice, including differences in leukocyte infiltration that suggested systemic differences, we compared the baseline (D0 no MI) plasma proteomes of MMP-9 null and WT mice. A total of 359 unique N-linked glycopeptides that had both an NXT/S motif and membrane or extracellular protein localization were quantified by mass spectrometry (Supplemental Table S1). A total of 34 glycoproteins showed significant changes in MMP-9 null plasma compared to WT plasma (all p < 0.05). Of these, 21 glycoproteins were at least two-fold changed (up or down). Notably, the top 3 significant ECM proteins depicted by volcano plot were Ecm1, periostin, and fibronectin, all 3 of which are known MMP-9 substrates (Fig. 8) [29,42,43]. For fibronectin, all five peptides identified showed null to WT ratios of <1.0, indicating reduced expression in the absence of MMP-9. The mean ratio of the five peptides was 0.53 ± 0.10 (one sample t-test p value of 0.01). We have previously used PNGase F treatment to confirm in vivo post-MI glycosylation for several of the proteins identified, including fibronectin, γ-sarcoglycan, and periostin [11]. MMP-9 total gene deletion, therefore, altered the baseline environment even before MI occurs, which could explain the downstream mechanistic differences observed between early MMP-9 inhibition and global MMP-9 deletion studies. While previous studies have suggested that the absence of MMP-9 has little effect in the absence of MI, this experiment revealed that MMP-9 null mice have under-appreciated proteome changes that may alter the MI response.
Fig. 8.
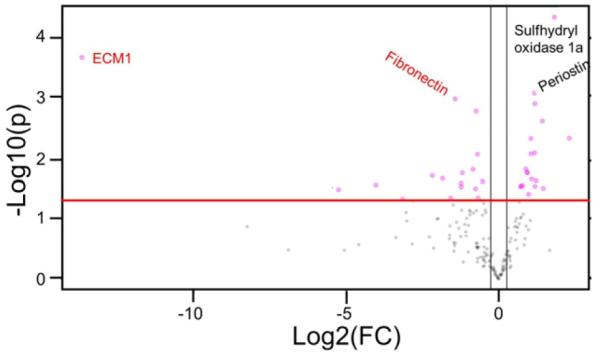
MMP-9 global deletion changes the baseline ECM profile in mice. Extracellular matrix protein 1 (ECM1), fibronectin, and periostin were key ECM protein differences identified between wild type and MMP-9 null plasma at baseline. Important feature analysis visualized with a volcano plot showed the ECM1 and fibronectin were lower and periostin was higher in MMP-9 null plasma from day 0 (no MI) mice. The y threshold was set at 1.3 fold change (red line intersecting the y-axis). The x threshold was set at −0.5 and +0.5 (black line intersecting the x-axis). The pink circles represent features outside the threshold points. Both fold change and p value are log transformed. Proteins in red (Ecm1 and fibronectin) were significantly decreased in MMP-9 null compared to wild type mice (p < 0.05). Proteins in black (periostin and sulfhydryl oxidase 1a) were significantly increased in MMP-9 null compared to wild type mice (p < 0.05). n = 10/group.
4. Discussion
The goal of this study was to evaluate the therapeutic applicability of using a selective MMP-9 inhibitor following MI. Our findings show that while early MMP-9 inhibition had no effect on infarct size or survival, MMP-9i enhanced infarct wall thinning and worsened cardiac dysfunction at D7 post-MI. MMP-9i treatment increased expression of Mmp8, Mmp12, and Mmp14, and decreased collagen deposition at D7 post-MI, indicating decreased efficiency of scar formation and enhanced matrix degradation. MMP-9i treatment increased neutrophil numbers at D1 post-MI and macrophage infiltration at D7 post-MI, revealing strong MMP-9 effects on leukocyte functions. Combined, our results reveal that using a selective translational approach to inhibit MMP-9 results in effects that diverge greatly from those seen with global MMP-9 deletion.
A comparison of results from MMP-9 null and MMP-9i MI studies is shown in Table 4. Similar to MMP-9i phenotype, MMP-9 null mice display reduced collagen deposition post-MI, albeit through different mechanisms [44]. In contrast to the findings of the current study, MMP-9 null mice have reduced rupture, improved survival, and improved LV remodeling post-MI [11,34]. The MMP-9i MI phenotype also includes increased and prolonged inflammatory response and reduced apoptosis rates [11,34]. We observed reduced levels of the macrophage marker CD36 with MMP-9 inhibition. Reduced CD36 indicates reduced phagocytosis and potentially explains the elevated number of macrophages to compensate for an individual cell reduction in phagocytic potential [45]. In addition, CD36 regulates apoptosis and, consistent with this, MMP-9i reduced leukocyte apoptosis at D7 post-MI [46]. The net results of MMP-9 inhibition, therefore, were prolonged inflammation.
Table 4.
Comparative analysis of events pre- and post-MI between MMP-9 null and wild type MMP-9i treated group [6–9,11,18,29,34,43].
| MMP-9 null | MMP-9 inhibitor | |
|---|---|---|
| Infarct area | ↔ | ↔ |
| Survival rate | ↑ | ↔ |
| LV remodeling | Improved | Worsened |
| Rupture rate | ↓ | ↔ |
| Collagen deposition | ↓ | ↓ |
| Inflammatory gene expression | ↓ | ↑ |
| Neutrophils at D1 post-MI | ↔ | ↑ |
| Neutrophils at D7 post-MI | ↓ | ↔ |
| Macrophage infiltration | ↓ | ↑ |
| CD36 levels | ↑ | ↓ |
| Cell apoptosis | ↑ | ↓ |
| Post-MI MMP levels | ↑ MMP-2, -13, TIMP-1 | ↑ MMP-8, -12, -14 |
| Pre-MI MMP levels | ↑ MMP-3, -13 | ↔ |
There are several reasons that may explain the differences in MMP-9 deletion versus inhibition phenotypes. First, MMP-9 deletion removes the entire MMP-9 component, while MMP-9i reduced MMP-9 activity by 30%. Thus, partial blockade may result in a different phenotype. Evidence for this being a relevant mechanism is seen with fibronectin cleavage. MMP-9 deletion dramatically prevents fibronectin cleavage, resulting in downregulation of synthesis to result in reduced full length protein concentrations. MMP-9i, in contrast, partially prevented fibronectin cleavage, resulting in a negative feedback loop that instigated new fibronectin synthesis, resulting in increased fibronectin gene expression (Table 2). Second, global MMP-9 deletion induced before gestation yields a background environment in the LV that has not been previously appreciated. This is corroborated by the plasma proteomic analysis, which showed Ecm1, periostin, and fibronectin (all key MMP-9 substrates) are differentially expressed in an already altered baseline environment. In addition to changing ECM substrate profiles, MMP-9 deletion also changes the MMP and TIMP landscape. MMP-9 null mice showed increased expression of MMP-2, MMP-13, and TIMP-1 in the LV infarct region at day 15 and pre-MI animals show increased levels of MMP-3 and MMP-13 at baseline [34]. While these differences do not result in a physiological difference in the D0 LV, it does likely change the response to MI sufficiently to alter the process of LV remodeling. Third, pharmacological interventions may affect alternative targets resulting in pleiotropic effects. Fourth, differences in drug penetration may yield local drug gradients, which may lead to regional differences in the extent of MMP-9 inhibition, particularly in the first day post-MI when infarct perfusion is low. Overall, whether gene deletion needs to be global and from the time of development to have such effects, or whether transient or conditional gene deletion strategies may also impair the baseline pre-MI LV should be evaluated in future experiments. In addition, extensive testing of a broad range in vivo dose response of the MMP-9 inhibitor may yield further insights.
In conclusion, we have shown that early pharmacological MMP-9 inhibition yields a much different post-MI response than seen with global MMP-9 deletion. The implications of the present study are that pharmacological MMP-9 inhibitors that block partial activity after the onset of MI do not recapitulate gene deletion models. While both models provide mechanistic insight, the findings of the pharmacological inhibition strategy may provide more translational relevance.
Supplementary Material
Acknowledgements and funding sources
We acknowledge technical support from Yuan Tian for the proteomics experiments and funding support from the American Heart Association for 14POST18770012 and 14SDG18860050. We acknowledge support from NIH HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center; from NIH HL075360, HL129823, HL051971, GM104357, GM114833, and CA098799; and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505.
Footnotes
Supplementary data to this article can be found online at doi:10.1016/j.yjmcc.2016.10.005.
Disclosures
None.
References
- [1].Zamilpa R, Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J. Mol. Cell. Cardiol. 2010;48(3):558–563. doi: 10.1016/j.yjmcc.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thompson MM, Squire IB. Matrix metalloproteinase-9 expression after myocardial infarction: physiological or pathological? Cardiovasc. Res. 2002;54(3):495–498. doi: 10.1016/s0008-6363(02)00397-8. [DOI] [PubMed] [Google Scholar]
- [3].Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Bras LE, Lindsey ML. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int. J. Cardiol. 2015;185:198–208. doi: 10.1016/j.ijcard.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology. 2013;28(6):391–403. doi: 10.1152/physiol.00029.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iyer RP, de Castro Bras LE, Jin YF, Lindsey ML. Translating Koch's postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: cardiac metalloproteinase actions (CarMA) postulates. Circ. Res. 2014;114(5):860–871. doi: 10.1161/CIRCRESAHA.114.301673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Jr., Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006;290(1):H232–H239. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- [7].Zamilpa R, Ibarra J, Bras L.E. de Castro, Ramirez TA, Nguyen N, Halade GV, Zhang J, Dai Q, Dayah T, Chiao YA, Lowell W, Ahuja SS, D'Armiento J, Jin YF, Lindsey ML. Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction. J. Mol. Cell. Cardiol. 2012;53(5):599–608. doi: 10.1016/j.yjmcc.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin YF, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Bras LE. A novel collagen matricryptin reduces left ventricular dilation post-myocardial infarction by promoting scar formation and angiogenesis. J. Am. Coll. Cardiol. 2015;66(12):1364–1374. doi: 10.1016/j.jacc.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ramirez TA, Iyer RP, Ghasemi O, Lopez EF, Levin DB, Zhang J, Zamilpa R, Chou YM, Jin YF, Lindsey ML. Aliskiren and valsartan mediate left ventricular remodeling post-myocardial infarction in mice through MMP-9 effects. J. Mol. Cell. Cardiol. 2014;72:326–335. doi: 10.1016/j.yjmcc.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999;5(10):1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- [11].DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML. CD36 is a matrix metalloproteinase-9 substrate that stimulates neutrophil apoptosis and removal during cardiac remodeling. Circ. Cardiovasc. Genet. 2016;9(1):14–25. doi: 10.1161/CIRCGENETICS.115.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Churg A, Wang R, Wang X, Onnervik PO, Thim K, Wright JL. Effect of an MMP-9/ MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax. 2007;62(8):706–713. doi: 10.1136/thx.2006.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Iyer RP, de Castro Bras LE, Cannon PL, Ma Y, DeLeon-Pennell KY, Jung M, Flynn ER, Henry JB, Bratton D, White J, Fulton L, Grady A, Lindsey ML. Defining the sham environment for post myocardial infarction studies in mice. Am. J. Physiol. Heart Circ. Physiol. 2016 doi: 10.1152/ajpheart.00067.2016. (ajpheart 00067 2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ma Y, Chiao YA, Zhang J, Manicone AM, Jin YF, Lindsey ML. Matrix metalloproteinase-28 deletion amplifies inflammatory and extracellular matrix responses to cardiac aging. Microsc. Microanal. 2012;18(1):81–90. doi: 10.1017/S1431927611012220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zamilpa R, Kanakia R, Cigarroa J.t., Dai Q, Escobar GP, Martinez H, Jimenez F, Ahuja SS, Lindsey ML. CC chemokine receptor 5 deletion impairs macrophage activation and induces adverse remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2011;300(4):H1418–1426. doi: 10.1152/ajpheart.01002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lauer-Fields JL, Sritharan T, Stack MS, Nagase H, Fields GB. Selective hydrolysis of triple-helical substrates by matrix metalloproteinase-2 and -9. J. Biol. Chem. 2003;278(20):18140–18145. doi: 10.1074/jbc.M211330200. [DOI] [PubMed] [Google Scholar]
- [17].Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, Entman M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation. 2001;103(17):2181–2187. doi: 10.1161/01.cir.103.17.2181. [DOI] [PubMed] [Google Scholar]
- [18].Yabluchanskiy A, Ma Y, DeLeon-Pennell KY, Altara R, Halade GV, Voorhees AP, Nguyen NT, Jin YF, Winniford MD, Hall ME, Han HC, Lindsey ML. Myocardial infarction superimposed on aging: MMP-9 deletion promotes M2 macrophage polarization. J. Gerontol. A Biol. Sci. Med. Sci. 2016;71(4):475–483. doi: 10.1093/gerona/glv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016;110(1):51–61. doi: 10.1093/cvr/cvw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H. Solid-phase extraction of N-linked glycopeptides. Nat. Protoc. 2007;2(2):334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003;21(6):660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- [22].Tian Y, Koganti T, Yao Z, Cannon P, Shah P, Pietrovito L, Modesti A, Aiyetan P, DeLeon-Pennell K, Ma Y, Halade GV, Hicks C, Zhang H, Lindsey ML. Cardiac extra-cellular proteome profiling and membrane topology analysis using glycoproteomics. Proteomics Clin. Appl. 2014;8(7–8):595–602. doi: 10.1002/prca.201400009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40(Web Server issue):W127–W133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37(Web Server issue):W652–W660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lauer-Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J. Biol. Chem. 2008;283(29):20087–20095. doi: 10.1074/jbc.M801438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Okada Y, Morodomi T, Enghild JJ, Suzuki K, Yasui A, Nakanishi I, Salvesen G, Nagase H. Matrix metalloproteinase 2 from human rheumatoid synovial fibroblasts. Purification and activation of the precursor and enzymic properties. Eur. J. Biochem. 1990;194(3):721–730. doi: 10.1111/j.1432-1033.1990.tb19462.x. [DOI] [PubMed] [Google Scholar]
- [29].Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, Weintraub ST, Lindsey ML. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 2010;10(11):2214–2223. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Flurkey HD, CJ K. American College Laboratory Animal Medicine. second ed Elsevier; Burlington MA: 2007. The Mouse in Biomedical Research. [Google Scholar]
- [31].Y.L. S, Salto-Tellez M, El-Oakley RM, Tang TP, AL ZA, Lim SK. Myocardial infarction in the C57BL/6J mouse: a quantifiable and highly reproducible experimental model. Cardiovasc. Pathol. 2004;2004:91–97. doi: 10.1016/S1054-8807(03)00129-7. [DOI] [PubMed] [Google Scholar]
- [32].DeLeon-Pennell KY, de Castro Bras LE, Iyer RP, Bratton DR, Jin YF, Ripplinger CM, Lindsey ML. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J. Mol. Cell. Cardiol. 2014;76:218–226. doi: 10.1016/j.yjmcc.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Halade GV, Ma Y, Ramirez TA, Zhang J, Dai Q, Hensler JG, Lopez EF, Ghasemi O, Jin YF, Lindsey ML. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2013;305(12):H1830–H1842. doi: 10.1152/ajpheart.00224.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 2000;106(1):55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler. Thromb. Vasc. Biol. 2008;28(12):2108–2114. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- [36].Lu KG, Stultz CM. Insight into the degradation of type-I collagen fibrils by MMP-8. J. Mol. Biol. 2013;425(10):1815–1825. doi: 10.1016/j.jmb.2013.02.002. [DOI] [PubMed] [Google Scholar]
- [37].Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front. Immunol. 2014;5:508. doi: 10.3389/fimmu.2014.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gray JL, Shankar R. Down regulation of CD11b and CD18 expression in atherosclerotic lesion-derived macrophages. Am. Surg. 1995;61(8):674–679. [PubMed] [Google Scholar]
- [39].White FJ, Burghardt RC, Hu J, Joyce MM, Spencer TE, Johnson GA. Secreted phosphoprotein 1 (osteopontin) is expressed by stromal macrophages in cyclic and pregnant endometrium of mice, but is induced by estrogen in luminal epithelium during conceptus attachment for implantation. Reproduction. 2006;132(6):919–929. doi: 10.1530/REP-06-0068. [DOI] [PubMed] [Google Scholar]
- [40].Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during athero-genesis. Arterioscler. Thromb. Vasc. Biol. 2011;31(7):1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, Kubota T, Takeshita A. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc. Res. 2004;64(3):526–535. doi: 10.1016/j.cardiores.2004.07.017. [DOI] [PubMed] [Google Scholar]
- [42].Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J. Mol. Cell. Cardiol. 2016;91:134–140. doi: 10.1016/j.yjmcc.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Iyer RP, Jung M, Lindsey ML. MMP-9 signaling in the left ventricle following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016;311(1):H190–H198. doi: 10.1152/ajpheart.00243.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lopez B, Gonzalez A, Querejeta R, Larman M, Diez J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J. Am. Coll. Cardiol. 2006;48(1):89–96. doi: 10.1016/j.jacc.2006.01.077. [DOI] [PubMed] [Google Scholar]
- [45].Erdman LK, Cosio G, Helmers AJ, Gowda DC, Grinstein S, Kain KC. CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J. Immunol. 2009;183(10):6452–6459. doi: 10.4049/jimmunol.0901374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J. Leukoc. Biol. 2009;86(5):1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.