

Abstract
The catalytic subunit of DNA dependent protein kinase (DNA-PKcs) and its kinase activity are critical for mediation of non-homologous end-joining (NHEJ) of DNA double-strand breaks (DSB) in mammalian cells after gamma-ray irradiation. Additionally, DNA-PKcs phosphorylations at the T2609 cluster and the S2056 cluster also affect DSB repair and cellular sensitivity to gamma radiation. Previously we reported that phosphorylations within these two regions affect not only NHEJ but also homologous recombination repair (HRR) dependent DSB repair. In this study, we further examine phenotypic effects on cells bearing various combinations of mutations within either or both regions. Effects studied included cell killing as well as chromosomal aberration induction after 0.5–8 Gy gamma-ray irradiation delivered to synchronized cells during the G0/G1 phase of the cell cycle. Blocking phosphorylation within the T2609 cluster was most critical regarding sensitization and depended on the number of available phosphorylation sites. It was also especially interesting that only one substitution of alanine in each of the two clusters separately abolished the restoration of wild-type sensitivity by DNA-PKcs. Similar patterns were seen for induction of chromosomal aberrations, reflecting their connection to cell killing. To study possible change in coordination between HRR and NHEJ directed repair in these DNA-PKcs mutant cell lines, we compared the induction of sister chromatid exchanges (SCEs) by very low fluencies of alpha particles with mutant cells defective in the HRR pathway that is required for induction of SCEs. Levels of true SCEs induced by very low fluence of alpha-particle irradiation normally seen in wild-type cells were only slightly decreased in the S2056 cluster mutants, but were completely abolished in the T2609 cluster mutants and were indistinguishable from levels seen in HRR deficient cells. Again, a single substitution in the S2056 together with a single substitution in the T2609 cluster abolished SCE formation and thus also effectively interferes with HRR.
INTRODUCTION
It is well established that the capacity to promptly repair radiation-induced DNA double-strand breaks (DSB) is crucial for cell survival and genome maintenance after ionizing radiation exposure. The non-homologous end-joining (NHEJ) pathway is operational throughout the cell cycle and is the predominant DSB repair mechanism in mammals, whereas homologous recombination repair (HRR) contributes less to DSB repair after irradiation and is active only during S or G2 phase of cell cycle (1). The NHEJ pathway relies on the DNA dependent protein kinase (DNA-PK) complex, consisting of the Ku70/80 heterodimer and the catalytic subunit DNA-PKcs, to synapse the broken DNA ends and to facilitate processing and ligation of the DNA break. DNA-PKcs is the key regulator of the NHEJ pathway as its kinase activity is essential for NHEJ mediated DSB repair (2). Although many components of NHEJ have been identified as DNA-PKcs substrates, the exact role the enzymatic activity of DNA-PKcs plays in NHEJ has not been fully characterized (3). In addition, DNA-PKcs itself is reported to be regulated by phosphorylation events and DNA-PKcs phosphorylation is critical for NHEJ mediated DSB repair (2).
DNA-PKcs activation after exposure to ionizing radiation or treatment with some radiomimetic chemicals results in rapid phosphorylation at the T2609 phosphorylation cluster and S2056 residue (4–7). DNA-PKcs phosphorylation at the T2609 cluster was initially identified by Mass Spectrometry after in vitro DNA-PK activation and autophosphorylation (4, 7). Further examination, however, revealed that in vivo DNA-PKcs phosphorylation at the T2609 cluster after irradiation or radiomimetic agents is primarily mediated by the related and DSB responsive ataxia-telangiectasia mutated (ATM) kinase and does not depend DNA-PKcs kinase activity (8). Additionally, T2609 cluster phosphorylation in vivo can be elicited by the ataxia-telangiectasia and Rad3 related (ATR) kinase in response to UV radiation and replication stress (9). In contrast, DNA-PKcs phosphorylation at S2056 was identified in vivo from endogenous DNA-PKcs of irradiated HeLa cells (5). S2056 phosphorylation clearly requires DNA-PKcs kinase, as radiation-induced S2056 phosphorylation is drastically decreased in cells expressing kinase-dead (KD) mutant DNA-PKcs, indicating that S2056 is an authentic autophosphorylation site in vivo (5). Adjacent to S2056, 4 additional potential phosphorylation sites have been identified (the S2056 cluster) (6), although it is not clear if they are indeed being phosphorylated in vivo. The differential regulations of the T2609 cluster and S2056 further indicate the potential difference in their functionalities. It seems reasonable to suggest they will participate in DSB repair through distinctive mechanisms with similar or possibly different phenotypic effects.
Studies of DNA-PKcs mutant cell lines defective at the T2609 or S2056 cluster indicate that these phosphorylations are required for full DSB repair capacity and normal cellular resistance to radiation (4–7). Alanine substitution at S2056 or T2609 alone leads to a modest increase in radiosensitivity and defects in DSB repair, whereas alanine substitution at the entire T2609 cluster of phosphorylation sites leads to severely increased radiosensitivity (4, 5, 7). DNA-PKcs phosphorylation at S2056 and at the T2609 cluster apparently does not affect its kinase activity as mutant DNA-PKcs remains as active as wild-type DNA-PKcs in in vitro kinase assays (5, 10). Although the mechanism and phenotypic consequence of DNA-PKcs phosphorylations remain to be clarified, evidence suggests that phosphorylation will lead to conformational changes or serve as docking sites for direct protein–protein interactions (11–13).
We reported previously that V3 cells bearing vectors expressing DNA-PKcs with mutations in the S2056 cluster and the T2609 cluster have clear differences in radiosensitivities when synchronized cells were irradiated with moderate doses in the G0/G1 phase (14, 15). It is important to utilize synchronized cells when comparing populations of different cell lines, because collateral but different changes in cell cycle distributions can easily occur that obscure differences that exist, or lead to misinterpretation of differences that do not exist, but are instead due to the well-known large differences in cell cycle dependent responses. In the previous study, we found the expression of DNA-PKcs with mutations affecting phosphorylations in the T2609 cluster (cell line L-3) resulted in extreme radiosensitivity for cell killing; however, similar mutations in the S2056 cluster (cell line L-12) resulted in intermediate radiosensitivity (15). To clarify their distinctive functionalities in DSB repair and whether there is a coordination between these two regions, we further investigated various mutant cells defective in the S2056 cluster (L-5, L-12), the T2609 cluster (L-3, L-6, L-14, L-15), and both clusters (L-2, L-4) for their radiosensitivity phenotypes for both cell killing and chromosomal aberration induction after gamma-ray irradiation. We have further investigated the influence of these mutations in the S2056 and the T2609 cluster phosphorylation sites described above with respect to HRR function by comparing sister chromatid exchange (SCE) induction after very low fluence of alpha-particle irradiation (16,17).
MATERIALS AND METHODS
Cell Lines and Tissue Culture
Cell lines used included the Chinese Hamster Ovary (CHO) and AA8 lines that are wild-type with respect to their radiosensitivity phenotypes, along with the four HRR deficient mutant strains irs-3, CL-V4B, irs-1 and irs-1SF and the NHEJ deficient mutant (V3) cells, all of which are listed in Table 1. Cell strains derived from DNA-PKcs null V3 cells (18) were also used with complemented human DNA-PKcs cDNA containing amino acid substitutions at various positions at the S2056 and T2609 clusters are described in Fig. 1. Properties of these cell lines have been described previously (14, 15). The cells were maintained at 37°C in a humidified 95% air/5% CO2 atmosphere in Eagle’s minimal essential medium (MEM) supplemented with 10% heat-inactivated (56°C for 30 min) fetal bovine serum (FBS), penicillin (50 U/ml) and streptomycin (50 μg/ml). When the cultures approached 30% confluence in T-25 tissue culture flasks or Mylar dishes, the normal growth medium was replaced twice at 24 h intervals with isoleucine-deficient (IL−) MEM containing 5% of three-times dialyzed FBS to synchronize cells in the G0/G1 phase (19). IL− treated G0/G1-phase synchronized cell populations were analyzed by FACScan flow cytometer and CellQuest software (BD Biosciences). Cells were labeled with BrdU (30 μM) for 15 min. and then fixed in 70% ethanol/PBS. BrdU was detected using Alexa Fluor 488-conjugated anti-BrdU antibody (Invitrogen) as previously reported (15).
TABLE 1.
CHO Mutant Cell Lines with Their Defective Genes and Origins
| Cell line | Defective gene | Origin |
|---|---|---|
| Wild type | ||
| CHO | None | |
| AA8 | None | |
| HR mutants | ||
| irs-3 | Rad51C | V-79 |
| CL-V4B | Rad51C | V-79 |
| irs-1 | xrcc2 | V-79 |
| irs-1SF | xrcc3 | AA8 |
| NHEJ mutant | ||
| V3 | DNA-PKcs | AA8 |
FIG. 1.
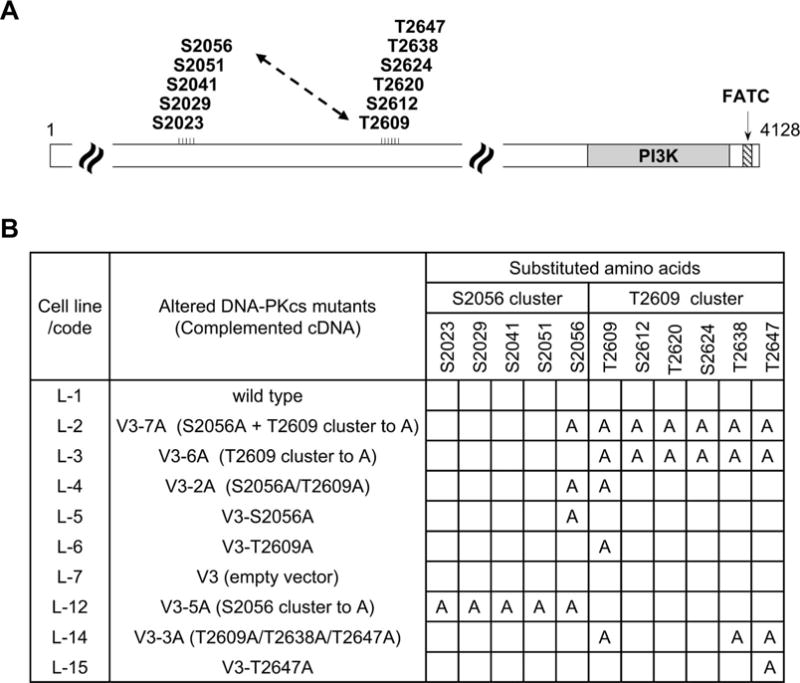
A schematic illustration of mutated DNA-PKcs with alanine substitutions on the S2056 and T2609 cluster. Panel A: Schematic diagram of DNA-PKcs structure includes the S2056 phosphorylation cluster, and the T2609 phosphorylation cluster. Panel B: Cell lines derived from DNA-PKcs null CHO V3 cells with complemented human DNA-PKcs cDNA containing amino acid substitutions at various positions in the DNA-PKcs constructs.
Irradiation and Colony Formation
Cells synchronized in G0/G1 by pre-incubation in IL− medium were gamma-ray irradiated with a JL Shepherd and Associates irradiator that emitted 137Cs γ rays such that at the position of the cells the dose rate was 2.5 Gy/min. Survival curves were obtained by measuring the colony-forming ability of cells irradiated over a range of doses as indicated for the particular experiment. Cells were plated immediately after irradiation onto 100 mm plastic Petri dishes and incubated for 8–10 days for colony formation. The colonies were fixed with 100% ethanol and stained with 0.1% crystal violet solution. A colony with more than 50 cells was scored as a survivor (20). For alpha-particle irradiation, cells were cultured on Mylar dishes and were placed over a Mylar window in the exposure well of a specially constructed irradiator that provides a uniform source of well-characterized 307 MeV alpha particles (21). The source consisted of 296 MBq of 238PuO2 electrodeposited onto one side of a 100 mm diameter stainless steel disk. The cells were irradiated from below in a helium environment, and the alpha-particles traversed a reciprocating collimator before reaching the Mylar window. The target-to-source distance is 42 mm helium gas, 6 mm in air and 1.5 μm in Mylar. Dose was controlled by timer and a precision photographic shutter, which allow exposure (21). For the alpha-particle irradiations, the highest doses to the monolayers were 2.8 mGy which corresponds to approximately 0.016 alpha-particle traversals per cell nucleus so even for the highest doses only one cell in about 60 cells experiences an alpha-particle traversal. Virtually no cells are killed by this dose of alpha particles. Since for wild-type cells this dose produces an average of some 3 SCEs per cell (0.15 SCE per chromosome × 21 chromosomes per cell) nearly all the SCEs are produced in cells that did not experience a direct alpha particle (i.e., in bystander cells) and are not false SCEs that can occur from the direct production of an aberration like a chromosome type translocation by direct ionization (22).
Chromosome Analysis
Synchronized G0/G1 phase cells were subcultured to four T-25 tissue culture flasks and cultured in fresh medium containing 10 μM BrdU for the first and second cell cycles after irradiation depending on whether chromosomal aberrations or SCEs were to be examined. For assessment of chromosomal aberration induction, at 4 h intervals beginning 13 h after subculture, colcemid was added to one of 4 tissue culture flasks to arrest cells in the first mitosis after irradiation; total sampling time thus covered 16 h. Most of cells moved into the first round of mitosis approximately 13–15 h postirradiation. The cells were fixed in methanol acetic acid (3:1) and chromosomes were spread by the air dry method (23). The fluorescence plus Giemsa differential chromatid staining technique was used to identify cells in their first or second post irradiation mitosis (24). In this way we were able to ensure only first post-irradiation mitoses were being included for aberration scoring after gamma irradiations. SCEs after alpha-particle irradiations were analyzed only after completion of both the first and second transits of cells through S phase, and second division mitotic cells were scored at the time of the peak mitotic indices for the cultures. It is important to note that, as mentioned above, for the BrdU labeling schedule used and the very low fluence of alpha particles virtually no “false SCEs” are generated.
RESULTS
The dose-survival responses for colony forming ability were measured for IL− treated G0/G1 synchronized Chinese hamster ovary wild-type (CHO and AA8) and the extremely radiosensitive V3 cells, which were employed to serve as baseline radiosensitivities for the native wild-type cells or the DNA-PKcs null mutant cells not containing vectors. The cell lines used to compare relative radiosensitivities among the mutant constructs were the V3 line containing either an empty vector, or a vector containing wild-type human DNA-PKcs cDNA. With these controls, our experimental V3 cell lines complemented with DNA-PKcs cDNA carrying various site-directed mutations in the S2056 and T2609 clusters were tested. Dose-survival responses are shown in Fig. 2A–D.
FIG. 2.
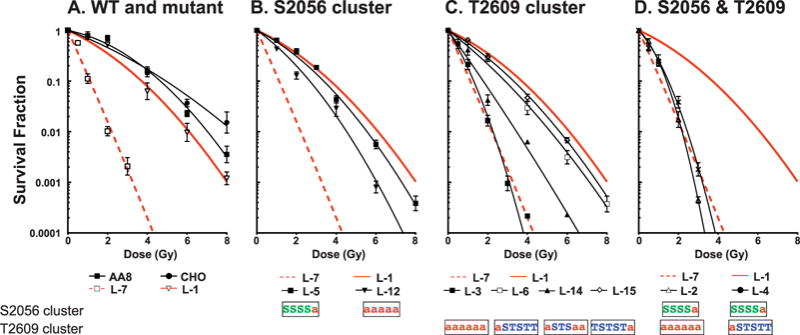
Survival curves of site-directed mutant cell lines with alanine substitutions in the S2056 and T2609 clusters. G0/G1 synchronized cells were irradiated by 137Cs gamma rays and reseeded for analysis of colony formation. The results are means ± SEMs from more than three independent experiments with each cell line. Panel A: Wild-type (CHO, AA8, L-1) and DNA-PKcs deficient (L-7); panel B: V3 complemented with T2056 cluster mutants (L-5, L-12); panel C: V3 complemented with T2609 cluster mutants (L-3, L-6, L-14, L-15); panel D: V3 complemented with double mutants at both S2056 and T2609 clusters (L-2, L-4). The amino acid substitution to alanine is indicated with a red “a” in the S2056 or T2609 clusters.
To facilitate the comparison of relative radiosensitivities among different DNA-PKcs mutant cell lines, the D10 values (radiation dose resulting in 10% survival) were estimated from the survival curves and the values were then normalized relative to that the wild-type CHO cells. For example, the relative radiosensitivities of DNA-PKcs null V3 cells and the derivative L1 cells harboring a wild-type DNA-PKcs cDNA were 0.23 and 0.75, respectively. The relative radiosensitivity phenotypes calculated in this way were plotted (see Fig. 3, left-hand side), where it can be seen that blocking phosphorylation solely within the T2609 cluster had a much greater influence on radiosensitivity than blocking of sites solely within the S2056 cluster. Leaving the S2056 cluster intact but increasing the number of sites blocked in the T2609 cluster caused increasing radiosensitivity. For the S2609 cluster by itself, with one site blocked (L-15 and L-6) only a small radiosensitization occurred; with 3 sites blocked (L-14) a greater degree of radiosensitization occurred; and with all six sites blocked virtually the full radiosensitization equivalent to DNA-PKcs null V3 cells was seen. For the S2056 cluster by itself, a blocking of only one site (L-5) had essentially no effect, and only by blocking all five sites (L-12) could a sensitization midway between wild-type and DNA-PKcs null cells be achieved. Of special interest are the sensitivities of L-4, L-5 and L-6 cells. In L-4 cells a single substitution in each of the S2056 and T2609 clusters rendered the cells as hypersensitive as DNA-PKcs null V3 cells, but a single substitution in either (but not both) the clusters L-5 and L-6 cells had essentially no effect on radiosensitivity.
FIG. 3.
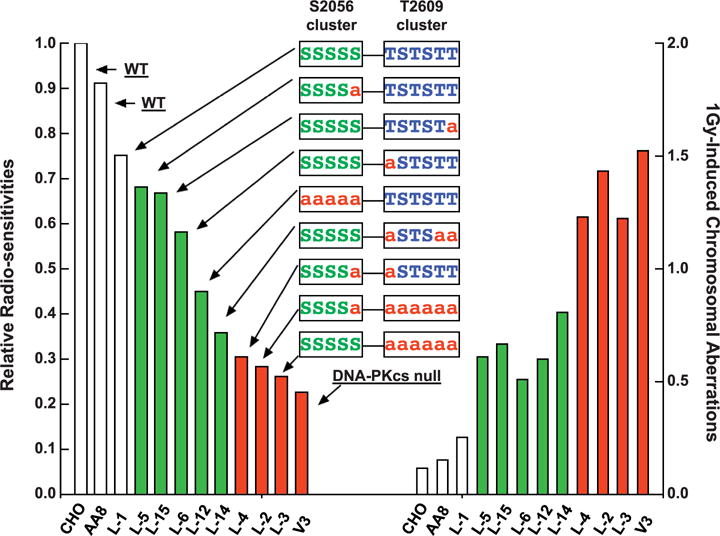
Relationship between radiosensitivities for survival and radiation induced chromosomal aberrations in DNA-PKcs mutant cell lines. Relative sensitivity to 137Cs γ radiation was calculated using the D10 value of each cell line divided by the D10 value of wild-type CHO cells (left-side panel). Total chromosomal aberrations, after exposure to 1 Gy 137Cs γ radiation, were scored and normalized to the basal level of chromosomal aberrations in unirradiated cells (right-side panel).
In addition to clonogenic survival measurement, chromosomal aberrations after increasing doses of gamma radiation were analyzed in DNA-PKcs mutant cell lines (Table 2, Fig. 4 and Supplemental Fig. S1; http://dx.doi.org/10.1667/RR14679.1.S1). In general, variation in the radiosensitivity for the production of chromosomal aberrations in DNA-PKcs mutant cell lines after gamma irradiation correlated with their sensitivities with respect to cell killing (loss of colony forming ability) (Fig. 3). Similar correlation between cell killing and induction of chromosomal aberrations had been reported over many years by numerous investigators (25–32). It is also noteworthy that the mutant constructs with increased radiosensitivity also showed a dramatic increase in the proportion of aberrations that were of the chromatid-type along with the general increase sensitivity for all aberration types after G0/G1 irradiation. Ordinarily, the vast majority of aberrations after G0/G1 irradiation are of the chromosome type. This large increase in chromatid-type aberrations after G0/G1 irradiation of DSB repair mutants has been reported on numerous occasions for hyper-radiosensitive cells (33–38).
TABLE 2.
Chromosome Aberrations after Gamma Irradiation of Various DNA-PKcs Mutant Cells in G0/G1 Phase
| Cell line | Dose (Gy) | Cells scored | Total chromosome aberrations (mean ± SEM) | Chromatid type
|
Chromosome type
|
||
|---|---|---|---|---|---|---|---|
| Breaks | Exchanges | Breaks | Ring and dicentrics | ||||
| CHO | 0 | 477 | 10 (0.021 ± 0.059) | 10 (0.021) | 0 (0) | 0 (0) | 0 (0) |
| 0.25 | 53 | 1 (0.019) | 1 (0.019) | 0 (0) | 0 (0) | 0 (0) | |
| 0.5 | 318 | 21 (0.067 ± 0.025) | 14 (0.044) | 2 (0.006) | 5 (0.016) | 0 (0) | |
| 1 | 477 | 66 (0.138 ± 0.020) | 49 (0.103) | 3 (0.006) | 9 (0.019) | 5 (0.010) | |
| V3 | 0 | 212 | 13 (0.061 ± 0.021) | 6 (0.028) | 2 (0.009) | 3 (0.014) | 2 (0.009) |
| 0.25 | 212 | 61 (0.288 ± 0.041) | 17 (0.080) | 4 (0.019) | 15 (0.071) | 25 (0.118) | |
| 0.5 | 132 | 113 (0.856 ± 0.057) | 26 (0.197) | 13 (0.098) | 32 (0.242) | 42 (0.318) | |
| 1 | 132 | 201 (1.523 ± 0.132) | 38 (0.288) | 25 (0.189) | 56 (0.424) | 82 (0.621) | |
| L-1 | 0 | 159 | 12 (0.075 ± 0.011) | 5 (0.031) | 1 (0.006) | 4 (0.025) | 2 (0.013) |
| 0.25 | 159 | 24 (0.151 ± 0.044) | 8 (0.050) | 5 (0.031) | 3 (0.019) | 8 (0.050) | |
| 0.5 | 133 | 34 (0.256 ± 0.080) | 5 (0.038) | 1 (0.008) | 13 (0.098) | 15 (0.113) | |
| 1 | 133 | 43 (0.323 ± 0.109) | 10 (0.075) | 3 (0.023) | 15 (0.113) | 15 (0.113) | |
| L-2 | 0 | 159 | 13 (0.082 ± 0.012) | 3 (0.019) | 0 (0) | 8 (0.050) | 2 (0.013) |
| 0.25 | 159 | 31 (0.195 ± 0.013) | 7 (0.044) | 0 (0) | 11 (0.069) | 13 (0.082) | |
| 0.5 | 159 | 66 (0.415 ± 0.019) | 10 (0.189) | 3 (0.019) | 22 (0.138) | 31 (0.195) | |
| 1 | 159 | 241 (1.516 ± 0.016) | 39 (0.245) | 40 (0.252) | 67 (0.421) | 95 (0.597) | |
| L-3 | 0 | 212 | 25 (0.118 ± 0.029) | 5 (0.024) | 1 (0.005) | 9 (0.042) | 10 (0.047) |
| 0.25 | 159 | 75 (0.472 ± 0.001) | 18 (0.113) | 6 (0.038) | 22 (0.138) | 29 (0.182) | |
| 0.5 | 212 | 158 (0.745 ± 0.125) | 45 (0.212) | 20 (0.094) | 44 (0.208) | 49 (0.231) | |
| 1 | 212 | 271 (1.278 ± 0.235) | 91 (0.429) | 69 (0.325) | 58 (0.274) | 53 (0.250) | |
| L-4 | 0 | 159 | 28 (0.176 ± 0.025) | 7 (0.044) | 5 (0.031) | 10 (0.063) | 8 (0.050) |
| 0.25 | 159 | 86 (0.541 ± 0.124) | 16 (0.101) | 15 (0.094) | 23 (0.145) | 32 (0.201) | |
| 0.5 | 159 | 129 (0.811 ± 0.469) | 26 (0.164) | 27 (0.170) | 41 (0.258) | 46 (0.289) | |
| 1 | 159 | 239 (1.503 ± 0.328) | 37 (0.233) | 64 (0.403) | 63 (0.396) | 75 (0.472) | |
| L-5 | 0 | 159 | 14 (0.088 ± 0.013) | 0 (0) | 2 (0.013) | 9 (0.170) | 3 (0.019) |
| 0.25 | 106 | 23 (0.217) | 3 (0.028) | 2 (0.019) | 11 (0.104) | 7 (0.066) | |
| 0.5 | 159 | 66 (0.415 ± 0.087) | 14 (0.088) | 5 (0.031) | 23 (0.145) | 34 (0.214) | |
| 1 | 159 | 111 (0.698 ± 0.136) | 23 (0.145) | 10 (0.063) | 24 (0.151) | 54 (0.340) | |
| L-6 | 0 | 212 | 20 (0.094 ± 0.033) | 10 (0.047) | 3 (0.014) | 7 (0.033) | 0 (0) |
| 0.25 | 212 | 63 (0.297 ± 0.079) | 19 (0.090) | 9 (0.042) | 36 (0.170) | 19 (0.090) | |
| 0.5 | 212 | 73 (0.344 ± 0.069) | 18 (0.085) | 13 (0.061) | 22 (0.104) | 26 (0.123) | |
| 1 | 212 | 128 (0.604 ± 0.033) | 31 (0.146) | 5 (0.024) | 29 (0.137) | 63 (0.297) | |
| L-12 | 0 | 159 | 24 (0.151 ± 0.039) | 12 (0.075) | 2 (0.013) | 8 (0.050) | 2 (0.013) |
| 0.25 | 159 | 24 (0.151 ± 0.011) | 12 (0.075) | 0 (0) | 6 (0.038) | 6 (0.038) | |
| 0.5 | 159 | 74 (0.465 ± 0.062) | 25 (0.157) | 15 (0.094) | 14 (0.088) | 20 (0.126) | |
| 1 | 159 | 119 (0.748 ± 0.139) | 29 (0.182) | 20 (0.126) | 14 (0.088) | 56 (0.352) | |
| L-14 | 0 | 159 | 20 (0.126 ± 0.006) | 7 (0.044) | 5 (0.031) | 5 (0.031) | 3 (0.019) |
| 0.25 | 159 | 38 (0.239 ± 0.003) | 5 (0.031) | 0 (0) | 13 (0.082) | 19 (0.119) | |
| 0.5 | 159 | 95 (0.597 ± 0.132) | 20 (0.126) | 14 (0.088) | 23 (0.145) | 32 (0.201) | |
| 1 | 159 | 148 (0.931 ± 0.299) | 30 (0.189) | 30 (0.189) | 34 (0.214) | 54 (0.340) | |
| L-15 | 0 | 159 | 18 (0.132 ± 0.022) | 5 (0.031) | 2 (0.013) | 5 (0.031) | 6 (0.038) |
| 0.25 | 159 | 45 (0.283 ± 0.039) | 8 (0.050) | 4 (0.025) | 7 (0.044) | 26 (0.163) | |
| 0.5 | 159 | 72 (0.453 ± 0.142) | 7 (0.044) | 8 (0.050) | 22 (0.138) | 35 (0.220) | |
| 1 | 159 | 117 (0.736 ± 0.041) | 15 (0.094) | 20 (0.126) | 32 (0.201) | 50 (0.314) | |
FIG. 4.
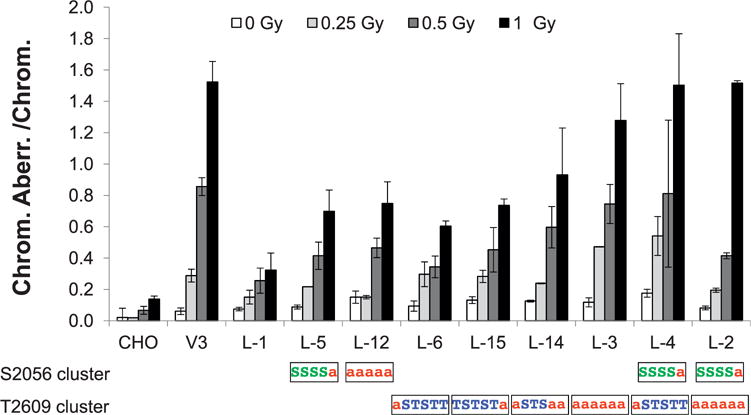
137Cs gamma-ray-induced chromosomal aberrations in DNA-PKcs mutant cells. Wild-type CHO, DNA-PKcs deficient V3 and DNA-PKcs mutant cells were irradiated with doses of 0, 0.25, 0.5 or 1 Gy of γ rays during G0/G1 phase. Total chromosome aberrations (means ± SEMs) were analyzed and scored from more than three independent experiments. The amino acid substitution to alanine is indicated with a red “a” in the S2056 or T2609 clusters.
To study the effect of DNA-PKcs phosphorylation on HRR directed repair, we examined the frequencies of induced sister chromatid exchanges (SCE) in bystander cells irradiated with low fluencies of alpha particles. The extremely low fluence of alpha-particle radiation we used in this study involved an alpha particle traversing approximately 1% of cells, leaving the vast majority of cells (>99%) without direct ionization inside the cells. Hence, the effects were resulted principally from lesions other than directly occurring DSBs from ionizations along or very near the alpha-particle track. These secondary lesions in so-called bystander cells require HRR DSB repair for production of SCEs (39). The induction of SCEs in wild-type CHO cells increased linearly with alpha particle doses below ~0.7 mGy and then reached a plateau for doses greater than 2.8 mGy (Fig. 5). Essentially no induced SCEs were observed in HRR repair deficient cell lines after these extremely low-alpha-particle irradiations (Fig. 5A) (16). A significantly higher induction of SCEs was observed in V3 cells (DNA-PKcs null mutant) for doses up to about 0.35 mGy, then SCE frequencies decreased sharply for doses of 2.8 mGy or higher (Fig. 5B) as previously reported (14, 17).
FIG. 5.
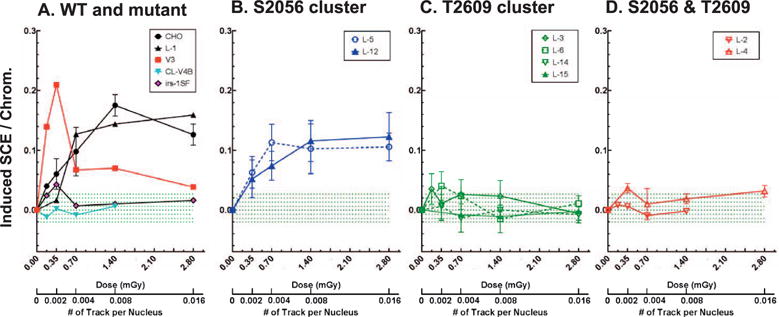
Induction of sister chromatid exchanges after exposure to extremely low fluence of alpha particle. Panel A: Wild-type (CHO), V3 cells complemented with wild-type DNA-PKcs (L-1) and cells deficient in HRR (CL-V4B, irs-1SF) were irradiated with extremely low fluence of alpha particles. SCEs were scored and normalized to the basal level of SCEs. Means ± SEMs from more than three independent experiments were given. Panels B–D: V3 cells complemented with mutant DNA-PKcs defective in the S2056 cluster (L-5, L-12), the T2609 cluster (L-3, L-6, L-14, L-15), or both clusters (L-2, L-4) were alpha-particle irradiated and analyzed for SCEs. The green dotted lines indicate the distribution range of induced SCE in V3 cells complemented with mutant DNA-PKcs defective in the T2609 clusters.
The formation of alpha-particle-induced SCEs displayed distinctive patterns among S2056 and T2609 cluster mutant cells. Both S2056 cluster mutated L-5 and L-12 cells displayed increasing SCEs up to ~0.7 mGy, but plateaued with ~25% lower SCE frequencies than wild-type CHO cells (Fig. 5B). No induction SCEs was observed in T2609 cluster mutant cells (L-3, L-6, L-14, L-15) where the response was similar to that of HRR deficient mutant cells as previously reported (15, 16). Further, there was no dependency for SCEs to the number or the position of alanine substitutions in the T2609 cluster and double mutants in the S2056 and T2609 clusters (L-2, L-4).
DISCUSSION
We have previously shown that DNA-PKcs kinase activity as well as phosphorylations within the S2056 and the T2609 clusters are crucial determinants for radiosensitivity and formation of chromosomal aberrations in response to a moderate dose of gamma radiation (14, 15). In the current study, we further investigated DNA-PKcs deficient V3 cell lines expressing DNA-PKcs mutants defective in either the S2056 cluster (L-5, L-12) or the T2609 cluster (L-3, L-6, L-14, L-15) as well as double mutants at both clusters (L-2, L-4) for comparison of radiosensitivities by cell killing and chromosomal aberrations production after 137Cs gamma-ray irradiation. We have also analyzed SCE induction after exposure to very low fluence of alpha-particle radiation (16, 17) among these DNA-PKcs mutant cells described above and determine their implication in HRR driven repair.
Our results suggest that there is an additive or cumulative effect of DNA-PKcs phosphorylation within the T2609 cluster region. Increasing alanine replacement within the T2609 cluster conferred increasing radiosensitivities (Fig. 2C) as well as increasing frequencies of 1 Gy induced chromosomal aberrations (Fig. 3). Increasing alanine replacement within the S2056 cluster also resulted in slightly increased radiosensitivity (Fig. 2B) as well as slightly increasing frequencies of 1 Gy induced chromosomal aberrations (Fig. 3), but there is less cumulative effect of DNA-PKcs phosphorylation within the S2056 cluster than the T2609 cluster. These results suggest that individual phosphorylations within the T2609 cluster are all functional and contribute to DSB repair and radiosensitivity. On the other hand, it is possible that not all the phosphorylation sites within the S2056 cluster are functionally active in DSB repair. Indeed, within the S2056 cluster region, except S2056 phosphorylation has been confirmed by mass spectrometry and phospho-specific antibody, the in vitro and in vivo status of other potential sites have not been verified (5, 6, 40).
It is also apparent that there is a synergism or coordinative effect between S2056 and T2609 cluster in radiosensitivity and DSB repair, which is consistent with the fact that they are regulated by DNA-PKcs autophosphorylation and ATM kinase, respectively, in response to radiation or radiomimetic chemicals (5, 8). While single site mutation at S2056A (L-5) or T2609A (L-6) alone only produced mild radiosensitivities, double mutant cells (L-4) carried alanine replacement at both S2056A and T2609A resulted in severe radiosensitivity and significant increase in chromosomal aberrations. These results suggest that DNA-PKcs phosphorylation at S2056 and T2609 contributes to DSB repair through distinctive mechanism. Recent evidences suggest that autophosphorylation of DNA-PKcs in vitro will lead to a protein conformational change (11) and could potentially modulate DNA-PKcs association with its interacting proteins (12, 13, 41, 42).
In the V3 mutant cells, where there is no DNA-PKcs present, the levels of induced SCE at the lowest doses are actually much greater than in wild-type cells with normal levels of DNA-PKcs present. This is consistent with our understanding for the competition and balance between NHEJ and HRR and a shift of DSB repair toward HRR pathway in NHEJ defective cells (43). On the contrary, no SCEs were produced in every case involving a mutation affecting the phosphorylation of serine or threonine residues in the T2609 cluster. This result suggests HRR is suppressed as it is required for SCE formation. The T2609 cluster phosphorylation is required to facilitate HRR since single alanine substitution at T2609 (L-6) or T2647 (L-15) drastically decreased the frequency of alpha-particle-induced SCEs, which resembled that observed in HRR deficient cells as previously reported (14, 16). Mutations in the S2056 cluster, however, were irrelevant to SCE formation. Alanine substitution at S2056 (L-5) or entire S2056 cluster (L-12) only slightly decreased SCE frequencies as compared to wild-type CHO cells. It was reported that elimination of the S2056 cluster phosphorylation was able to reverse HRR inhibition in T2609 cluster mutant cells via HRR reporter assay (6). However, we observed very low SCE frequencies in cells with double mutants at both the S2056 and the T2609 cluster (L-2, L-4). It is possible that alanine substitution at S2056 alone is not sufficient to relieve HRR inhibition in S2609 cluster mutant cells. The different conclusion drawn could be due to the distinctive character of each experimental approach. The SCE frequency allows direct measurement of the endogenous HRR byproduct whereas HRR reporter assay measures the frequency of recombination from one single integrated HRR reporter.
The biological effects of extremely low fluence of alpha-particle irradiation may be mediated through the interaction of free radicals, including oxygen and nitrogen species (ROS/RNS) with proteins, lipids, nucleic acids: these free radicals may be generated from primary ionizing events or through secondary amplification systems (44). Generation of intracellular ROS/RNS has been demonstrated to occur for short periods of time after very low fluence of alpha particles in the bystander cells without direct nuclear or cellular hits by the alpha particles (45). ROS/RNS including H2O2, nitric oxide and peroxynitrite are membrane-permeable and are sufficient to diffuse over significant distance within cells to react with proteins, lipids and DNA (44–46). DNA damages occurring among bystander cells are consistent of base damage repair or cross-link DNA repair, as measured by increases in hypoxanthine-guanine phosphoribosyltransferase point mutations, chromatid-type aberrations and SCEs (14, 16, 17, 26, 47–51). The lack of SCE induction after extremely low-fluence alpha-particle irradiation was previously reported in Chinese hamster deficient in Rad51 paralogs as well as the essential HRR protein BRCA2 (14, 47). The lack of SCE induction in L-3, L-6, L-14 and L-15 cells, and together with combined S2056 cluster cells (L-2, L-4) suggest that each individual phosphorylation residue at the T2609 cluster might contribute specifically and distinctively for completion of HRR repair process.
Supplementary Material
Acknowledgments
This work was supported by grants DE-FG02-05ER64089 (JBL) and DE-FG02-07ER64350 (JSB) from the U.S. Department of Energy Low Dose Radiation Research Program, CA166677 (BPC) from the National Cancer Institute/National Institutes of Health.
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR14679.1) contains supplementary information that is available to all authorized users.
References
- 1.Thompson LH. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res. 2012;751:158–246. doi: 10.1016/j.mrrev.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, et al. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol. 1999;19:3877–84. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 2014;17:21–9. doi: 10.1016/j.dnarep.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2001;16:2333–8. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709–15. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- 6.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, et al. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol. 2005;25:10842–52. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, Reddy YV, Wang W, Woods T, Douglas P, et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol. 2003;23:5836–48. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–7. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- 9.Yajima H, Lee KJ, Chen BP. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol. 2006;26:7520–8. doi: 10.1128/MCB.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Yajima H, Huynh H, Zheng J, Callen E, et al. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol. 2008;193:295–305. doi: 10.1083/jcb.201009074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–23. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wechsler T, Chen BP, Harper R, Morotomi-Yano K, Huang BC, et al. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci U S A. 2004;101:1247–52. doi: 10.1073/pnas.0307765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KJ, Shang ZF, Lin YF, Sun J, Morotomi-Yano K, et al. The Catalytic Subunit of DNA-Dependent Protein Kinase Coordinates with Polo-Like Kinase 1 to Facilitate Mitotic Entry. Neoplasia. 2015;17:329–38. doi: 10.1016/j.neo.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin YF, Nagasawa H, Little JB, Kato TA, Shih HY, et al. Differential radiosensitivity phenotypes of DNA-PKcs mutations affecting NHEJ and HRR systems following irradiation with gamma-rays or very low fluences of alpha particles. PLoS One. 2014;9:e93579. doi: 10.1371/journal.pone.0093579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagasawa H, Little JB, Lin YF, So S, Kurimasa A, et al. Differential role of DNA-PKcs phosphorylations and kinase activity in radiosensitivity and chromosomal instability. Radiat Res. 2011;175:83–9. doi: 10.1667/RR2092.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagasawa H, Wilson PF, Chen DJ, Thompson LH, Bedford JS, et al. Low doses of alpha particles do not induce sister chromatid exchanges in bystander Chinese hamster cells defective in homologous recombination. DNA Repair (Amst) 2008;7:515–22. doi: 10.1016/j.dnarep.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 17.Nagasawa H, Peng Y, Wilson PF, Lio YC, Chen DJ, et al. Role of homologous recombination in the alpha-particle-induced bystander effect for sister chromatid exchanges and chromosomal aberrations. Radiat Res. 2005;164:141–7. doi: 10.1667/rr3420. [DOI] [PubMed] [Google Scholar]
- 18.Whitmore GF, Varghese AJ, Gulyas S. Cell cycle responses of two X-ray sensitive mutants defective in DNA repair. Int J Radiat Biol. 1989;56:657–65. doi: 10.1080/09553008914551881. [DOI] [PubMed] [Google Scholar]
- 19.Tobey RA, Ley KD. Isoleucine-mediated regulation of genome repliction in various mammalian cell lines. Cancer Res. 1971;31:46–51. [PubMed] [Google Scholar]
- 20.Stackhouse MA, Bedford JS. An ionizing radiation-sensitive mutant of CHO cells:irs-20. II. Dose-rate effects and cellular recovery processes. Radiat Res. 1993;136:250–4. [PubMed] [Google Scholar]
- 21.Metting NF, Koehler AM, Nagasawa H, Nelson JM, Little JB. Design of a benchtop alpha particle irradiator. Health Phys. 1995;68:710–5. doi: 10.1097/00004032-199505000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Muhlmann-Diaz MC, Bedford JS. Comparison of gamma-ray-induced chromosome ring and inversion frequencies. Radiat Res. 1995;143:175–80. [PubMed] [Google Scholar]
- 23.Hsu TC, Klatt O. Mammalian chromosomes in vitro. IX. On genetic polymorphism in cell populations. J Natl Cancer Inst. 1958;21:437–73. [PubMed] [Google Scholar]
- 24.Perry P, Wolff S. New Giemsa method for the differential staining of sister chromatids. Nature. 1974;251:156–8. doi: 10.1038/251156a0. [DOI] [PubMed] [Google Scholar]
- 25.Cornforth MN, Bedford JS. A quantitative comparison of potentially lethal damage repair and the rejoining of interphase chromosome breaks in low passage normal human fibroblasts. Radiat Res. 1987;111:385–405. [PubMed] [Google Scholar]
- 26.Nagasawa H, Little JB, Inkret WC, Carpenter S, Raju MR, et al. Response of X-ray-sensitive CHO mutant cells (xrs-6c) to radiation. II. Relationship between cell survival and the induction of chromosomal damage with low doses of alpha particles. Radiat Res. 1991;126:280–8. [PubMed] [Google Scholar]
- 27.Nagasawa H, Fornace D, Little JB. Induction of sister-chromatid exchanges by DNA-damaging agents and 12-O-tetradecanoylphorbol-13-acetate (TPA) in synchronous Chinese hamster ovary (CHO) cells. Mutat Res. 1983;107:315–27. doi: 10.1016/0027-5107(83)90173-2. [DOI] [PubMed] [Google Scholar]
- 28.Dewey WC, Humphrey RM. Relative radiosensitivity of different phases in the life cycle of L-P59 mouse fibroblasts and ascites tumor cells. Radiat Res. 1962;16:503–30. [PubMed] [Google Scholar]
- 29.Dewey WC, Miller HH, Leeper DB. Chromosomal aberrations and mortality of x-irradiated mammalian cells: emphasis on repair. Proc Natl Acad Sci U S A. 1971;68:667–71. doi: 10.1073/pnas.68.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrano AV. Chromosome aberrations and radiation-induced cell death. I. Transmission and survival parameters of aberrations. Mutat Res. 1973;17:341–53. doi: 10.1016/0027-5107(73)90006-7. [DOI] [PubMed] [Google Scholar]
- 31.Bedford JS, Mitchell JB, Griggs HG, Bender MA. Radiation-induced cellular reproductive death and chromosome aberrations. Radiat Res. 1978;76:573–86. [PubMed] [Google Scholar]
- 32.Nagasawa H, Little JB. Induction of chromosome aberrations and sister chromatid exchanges by X rays in density-inhibited cultures of mouse 10T1/2 cells. Radiat Res. 1981;87:538–51. [PubMed] [Google Scholar]
- 33.Bedford JS, Dewey WC. Radiation Research Society. 1952–2002. Historical and current highlights in radiation biology: has anything important been learned by irradiating cells? Radiat Res. 2002;158:251–91. doi: 10.1667/0033-7587(2002)158[0251:hachir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 34.Bender MA, Griggs HG, Bedford JS. Mechanisms of chromosomal aberration production. 3. Chemicals and ionizing radiation. Mutat Res. 1974;23:197–212. doi: 10.1016/0027-5107(74)90140-7. [DOI] [PubMed] [Google Scholar]
- 35.Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, et al. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–9. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- 36.Little JB, Nagasawa H. Effect of confluent holding on potentially lethal damage repair, cell cycle progression, and chromosomal aberrations in human normal and ataxia-telangiectasia fibroblasts. Radiat Res. 1985;101:81–93. [PubMed] [Google Scholar]
- 37.Nagasawa H, Latt SA, Lalande ME, Little JB. Effects of X-irradiation on cell-cycle progression, induction of chromosomal aberrations and cell killing in ataxia telangiectasia (AT) fibroblasts. Mutat Res. 1985;148:71–82. doi: 10.1016/0027-5107(85)90209-x. [DOI] [PubMed] [Google Scholar]
- 38.Nagasawa H, Little JB. Comparison of kinetics of X-ray-induced cell killing in normal, ataxia telangiectasia and hereditary retinoblastoma fibroblasts. Mutat Res. 1983;109:297–308. doi: 10.1016/0027-5107(83)90054-4. [DOI] [PubMed] [Google Scholar]
- 39.Nagasawa H, Little JB. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 1992;52:6394–6. [PubMed] [Google Scholar]
- 40.Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst) 2010;9:1307–14. doi: 10.1016/j.dnarep.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagasawa H, Cremesti A, Kolesnick R, Fuks Z, Little JB. Involvement of membrane signaling in the bystander effect in irradiated cells. Cancer Res. 2002;62:2531–4. [PubMed] [Google Scholar]
- 42.Davis AJ, Chi L, So S, Lee KJ, Mori E, et al. BRCA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle. Nucleic Acids Res. 2014;42:11487–501. doi: 10.1093/nar/gku824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–42. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmidt-Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Signal transduction and cellular radiation responses. Radiat Res. 2000;153:245–57. doi: 10.1667/0033-7587(2000)153[0245:stacrr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 45.Narayanan PK, Goodwin EH, Lehnert BE. Alpha particles initiate biological production of superoxide anions and hydrogen peroxide in human cells. Cancer Res. 1997;57:3963–71. [PubMed] [Google Scholar]
- 46.Lander HM, Ogiste JS, Teng KK, Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270:21195–8. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 47.Nagasawa H, Little JB. Bystander effect for chromosomal aberrations induced in wild-type and repair deficient CHO cells by low fluences of alpha particles. Mutat Res. 2002;508:121–9. doi: 10.1016/s0027-5107(02)00193-8. [DOI] [PubMed] [Google Scholar]
- 48.Huo L, Nagasawa H, Little JB. HPRT mutants induced in bystander cells by very low fluences of alpha particles result primarily from point mutations. Radiat Res. 2001;156:521–5. doi: 10.1667/0033-7587(2001)156[0521:hmiibc]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 49.Nagasawa H, Huo L, Little JB. Increased bystander mutagenic effect in DNA double-strand break repair-deficient mammalian cells. Int J Radiat Biol. 2003;79:35–41. [PubMed] [Google Scholar]
- 50.Nagasawa H, Cremesti A, Kolesnick R, Fuks Z, Little JB. Involvement of membrane signaling in the bystander effect in irradiated cells. Cancer Res. 2002;62:2531–34. [PubMed] [Google Scholar]
- 51.Burdak-Rothkamm S, Short SC, Folkard M, Rothkamm K, Prise KM. ATR-dependent radiation-induced gamma H2AX foci in bystander primary human astrocytes and glioma cells. Oncogene. 2007;26:993–1002. doi: 10.1038/sj.onc.1209863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.