

Abstract
PIK3CA , the gene that encodes the catalytic subunit of phosphatidylinositol 3-kinase α (PI3Kα), is frequently mutated in breast and other types of cancer. A specific inhibitor that targets the mutant forms of PI3Kα could maximize treatment efficiency while minimizing side-effects. Herein we describe the identification of novel binding pockets that may provide an opportunity for the design of mutant selective inhibitors. Using a fragment-based approach, we screened a library of 352 fragments (MW <300 Da) for binding to PI3Kα by X-ray crystallography. Five novel binding pockets were identified, each providing potential opportunities for inhibitor design. Of particular interest was a binding pocket near Glu542, which is located in one of the two most frequently mutated domains.
Keywords: PIK3CA, PI3K, PIP2, PIP3, fragment-based drug discovery
Graphical Abstract
1. Introduction
Phosphatidylinositol 3-kinase (PI3K) inhibitors have been gaining momentum as a viable cancer treatment strategy, as documented by the recent FDA approval of the first PI3K inhibitor, idelalisib (1, Fig. 1a). It was approved for use in combination with rituximab in relapsed chronic lymphoid leukemia, follicular non-Hodgkin’s B-cell lymphoma and small lymphocytic lymphoma.1 Idelalisib selectively targets the PI3Kδ isoform, which is overexpressed in these hematological malignancies.2–4 During a Phase III clinical trial, the addition of idelalisib to rituximab produced a rapid and relatively long-lasting response in 81% of patients compared with a 13% response-rate in the group receiving rituximab alone.5
Figure 1. PI3K inhibitors and domain organization.

a) Chemical structures of PI3K inhibitors approved or in clinical trials. 1. idelalisib, 2. buparlisib, 3. alpelisib. b) Domain organization of PI3Kα heterodimer. Colored domains are included in the construct used for crystallization.
While the PI3Kδ isoform is overexpressed in hematological malignancies, the PI3Kα isoform is frequently mutated in solid tumors, particularly those of the breast, ovary, colorectum, and head and neck.6–10 As many as 80% of these mutations occur in three hotspot positions within the catalytic subunit, p110α (encoded by the PIK3CA gene) of the heterodimeric enzyme (Fig. 1b). Of these three hotspots, the two helical domain mutants, E542K and E545K, are located in a loop of the helical domain of p110α that binds in a groove of the nSH2 domain of the regulatory subunit, p85α. This interaction is thought to result in the auto-inhibition of the enzyme. Both these mutations disrupt this interaction and relieve the auto-inhibition of PI3Kα.11,12 In contrast, the H1047R mutation is located in the kinase domain and induces conformational changes that increase PI3Kα membrane association and lipid binding.11–13
The most advanced inhibitors in clinical trials for solid tumors harboring PIK3CA mutations include buparlisib (BKM-120, 2), a pan-PI3K inhibitor and alpelisib (BYL-719, 3), a PI3Kα-selective inhibitor (Fig. 1a).14–16 Buparlisib increased progression-free survival by 4 months in a Phase III trial in aromatase inhibitor resistant breast cancer, and responses correlated with the presence of mutations in PIK3CA.17 However, 25% of patients experienced serious side effects, including hyperglycemia and liver toxicity, which limited treatment. Concerns about the toxicity of pan-PI3K inhibitors led to the investigation of PI3Kα isoform selective agents such as alpelisib. Although not expected to completely abrogate adverse effects, treatment with PI3Kα selective inhibitors may increase the therapeutic window, potentially leading to more effective treatments.18
All PI3K isoforms are involved in vital cellular processes such as metabolism, growth, proliferation, and migration. PI3Kα, in particular, plays a central role in the regulation of glucose metabolism and growth.19–21 Many of the observed adverse effects stem from on-target toxicity, so selective targeting of the oncogenic mutant enzymes may be one approach to increase the therapeutic window. This should lead to more tolerable and efficacious treatments. One of the major limitations is the structural similarity of the ATP binding sites between the wild-type and mutant PI3Kα. Comparison of the ATP-binding sites of the wild-type and the H1047R oncogenic mutant reveals no major differences.12,22,23 There is no structural information available for the helical domain mutants; however, given the inability thus far to develop an ATP-competitive inhibitor that is selective for these mutants, it is unlikely that there are any major differences in the ATP-binding site itself.
In the parallel field of protein kinase inhibitor development, selectivity between highly related kinases has posed a significant challenge. The development of Akt inhibitors provides a paradigmatic example. Early on, significant efforts were invested in the search for ATP-competitive Akt inhibitors. However, many of these inhibitors that advanced to the clinic were ultimately discontinued due to a lack of selectivity and the resultant severe toxicity issues.24 The subsequent discovery of MK-2206, an allosteric inhibitor, revolutionized this field.25,26 Adopting a novel binding mechanism, this drug binds to the interface between the kinase and Plekstrin homology domains and stabilizes the inactive conformation of the enzyme. In contrast to the ATP-competitive inhibitors, MK-2206 has been well-tolerated in the clinic, and showed good efficacy.27 Although some ATP-competitive Akt inhibitors with better selectivity are now under clinical evaluation, in general, the allosteric inhibitors show greater selectivity due to the less conserved binding site.28 The auto-inhibited conformation of PI3Kα could potentially be targeted in a similar fashion.
Allosteric inhibitors have a number of potential benefits over the canonical ATP competitive inhibitors.29–31 Allosteric sites tend to be less homologous than ATP-binding sites among related kinases, potentially offering improved selectivity. Furthermore, as opposed to the highly competitive intellectual property space surrounding ATP-like inhibitors, allosteric binding sites allow for the exploration of novel chemical space. Binding sites with different topographies also have the potential to yield inhibitors with significantly improved pharmacokinetic properties. A final limitation of ATP-competitive inhibitors is the need to compete with millimolar concentrations of ATP within the cell, necessitating extremely high potency. Allosteric binding sites avoid this requirement and permit the identification of lower affinity binders as potential drug candidates.29–31
Significant challenges exist, however, in the search for allosteric inhibitors. Due to the nature of their target binding site, typical kinase screening libraries predominantly consist of ATP analogs, biasing the search towards ATP-competitive inhibitors. However, even with a generic, non-target focused library of compounds, it is difficult to specifically screen for allosteric binders, since the enzymatic assays typically used either only detect binding at the ATP-binding site, or do not provide a simple way to discriminate between allosteric and orthosteric binders.29,31 An ATP-consumption based assay, for example, is inherently biased towards the discovery of ATP-competitive inhibitors.
Fragment screening by X-ray crystallography provides a way to identify novel binding sites.31,32 Fragments are small molecular weight compounds (MW < 300 Da) that are frequently used for the discovery of novel drugs. They generally possess good drug-like properties, high ligand efficiency (a measure of affinity corrected by size), and good coverage of available chemical space. Each of these properties makes fragments well-suited to the search for novel binding sites. Though laborious, crystallography is a uniquely valuable approach to fragment screening. It is sensitive, able to detect binders with affinities as low as 10 mM, provides structural information about the binding site and, unlike NMR, is suitable for large molecular weight proteins, such as PI3Kα.31,33,34
The pursuit of allosteric inhibitors for PI3K is an underexplored area, and could hold significant advantages in the search for mutant selective inhibitors. By screening a fragment library of 352 compounds, we have identified 10 novel binding sites on PI3Kα, at least one of which holds promise for the development of oncogenic mutant selective inhibitors.
2. Materials and Methods
2.1 Molecular Modeling
2.1.1 Protein Preparation
The crystal structure of PI3Kα (PDB ID: 4OVU) was prepared using the Protein Preparation Wizard in Maestro (Schrödinger Release 2016-2: Maestro, version 10.6, Schrödinger, LLC, New York, NY, 2016).35 This involves the assignment of bond orders and formal charges and the addition of hydrogens. The hydrogen bonding network within the protein is optimized (including the reorientation of thiol and hydroxyl groups, sampling Asn, Gln and His side chains, and the prediction of the protonation states of His, Asp and Glu), followed by a brief minimization.
2.1.2 Binding site prediction
SiteMap (Schrödinger Release 2016-2: SiteMap, version 3.9, Schrödinger, LLC, New York, NY, 2016) was used to identify potentially druggable binding sites, using default settings.36,37
2.2 Protein expression
SF9 cells were grown in suspension culture in SF-900 III Serum Free Media (Invitrogen) supplemented with 0.5% penicillin-streptomycin at 27°C. At a density of 4 × 106 cells/mL, cells were infected with WT p110α and p85α (or niSH2) viruses at a multiplicity of infection ratio of 3:2. Media was supplemented with J-32, a PI3K inhibitor, as described in Mandelker et al.12 Cells were harvested 48 hours after infection and the cell pellet collected through centrifugation at 500 × g. Protein purification was performed as previously described.12,22,23
2.3 Crystallization and data collection
Crystallization was performed as previously described and improved with successive rounds of macroseeding.12,23 Crystals of p110α/niSH2 were soaked for 30 minutes in mother liquor supplemented with 2–4 fragments each at 2 mM concentration. Fragments were obtained from Zenobia Therapeutics. X-ray diffraction data were collected at Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source or at the Berkeley Center for Structural Biology Beamline 502 at Advanced Light Source. Data from crystals that diffracted to a resolution of 3.5 Å or better were collected and processed with HKL2000 (Supp. Table 1).38
2.4 Structure determination and analysis
The structures were determined by direct refinement using the coordinates of the previously determined WT p110α/niSH2 (PDB ID 4OVU) as a model.23 After rigid body and positional refinement, the fragments were built in the difference electron density maps using the program Coot for model building.39 Iterative rounds of refinement were performed with REFMAC 5.0.40–42 Model quality was assessed using Coot. Visualization, analysis and figure preparation were carried out with PyMOL (The PyMOL Molecular Graphics System, Version 1.6 Schrödinger, LLC).
2.5 Fluorescence Polarization Assay
The fluorescence polarization assay kit was purchased from Echelon Biosciences and adapted to a 96-well format. The fragments were dissolved at 200 mM in dimethyl sulfoxide (DMSO). Fragment dilutions at appropriate concentrations were prepared in 10% (v/v) DMSO and added to the reaction. PI3Kα enzymatic activity was determined in the presence of 10 μM PIP2 and 25 μM ATP and incubated at room temperature for 45 minutes. Increasing concentrations of fragments were used to generate a dose-response curve which was analyzed using GraphPad Prism v6 for OSX to calculate the IC50.
3. Results and Discussion
To identify novel binding sites, we used X-ray crystallography as our primary screening method. We have previously reported the structure of wild-type PI3Kα alone and in complex with its lipid substrate.22,23 The construct used for crystallization contains the full-length p110α, which has five domains: the adaptor-binding domain (ABD), Ras-binding domain (RBD), C2 domain, helical domain and kinase domain; in addition to the truncated regulatory subunit, p85α, which contains just the N-terminal SH2 domain and the inter-SH2 domain (hereafter termed niSH2) (Fig. 1b). This is the minimal portion of p85α required for auto-inhibition and stable protein expression.22,43 We soaked wild-type PI3Kα crystals with fragment cocktails containing 2–4 fragments from the Zenobia Fragment Library (352 compounds, Zenobia Therapeutics Inc.) at 2 mM individual concentrations. A total of 91 crystals were soaked, and we collected 24 datasets that diffracted better than 3.5 Å. In the structures determined with these datasets, we found ligand electron density in 10 distinct regions throughout the protein (Fig. 2a). Interestingly, most of the identified binding sites were located at the interfaces between multiple domains. To reduce the number of binding sites for further exploration, we examined these ten regions in more detail to decide whether they could represent druggable, novel allosteric binding sites. We determined that five of these regions were not in well-defined pockets within the protein; instead, in these cases, the fragment interacted with surface residues of the protein or made isolated interactions with a flexible loop. These five regions were not considered of interest for future drug design and will not be discussed further.
Figure 2. Identification of allosteric binding sites.

a) Location of allosteric binding sites. Fragment binding sites are shown in blue; the substrate (ATP and PIP2) binding sites are shown in gray for reference. PI3K domains are colored as in Fig. 1b. b) Binding site 1. Three fragments, 4-6, were identified in this binding site at the interface between the RBD, kinase and helical domains. The surface representation of the protein is shown in gray. c) Binding site 2. Four fragments, 7-10, were identified in this binding site between the RBD and helical domains. d) Binding site 3. Two fragments, 7 and 11, were found at this binding site located between the ABD and kinase domain. e) Binding site 4. A single fragment, 12, binds at this site located between the C2 and helical domains. f) Phosphopeptide binding site. A total of 12 fragments, 13-24, were bound to this site which overlaps the physiological phosphopeptide site at the interface between the nSH2 and helical domains. This binding site is in close proximity to E542, which is mutated to a lysine residue in many cancers.
Of the five sites, the first binding site was a deep pocket located at the interface of the RBD, kinase and helical domains (Fig. 2b). Three different fragments bound in this pocket: an indole 4, a benzimidazole 5, and a piperidine 6. The fragments were sandwiched between two residues, F666 of the helical domain and R818 of the kinase domain. These two residues are both capable of π-stacking with the aromatic fragments, similar to the ‘hydrophobic clamp’ that has been observed with phosphodiesterase inhibitors.44 The site is bordered by a number of residues that can participate in hydrogen bonding: N170, S629, R662, H670, Q815. The fragments overlap in their binding sites, but each one presents numerous distinct opportunities for further derivatization to increase binding affinity.
The second site is a shallow binding site on the surface of the helical domain, with some residues from the adjacent RBD contributing hydrogen bonding partners (Fig. 2c). The binding site is lined by polar and charged residues: K621, T624, D625 and K656 from the helical domain, with E263 and Y265 extending from a loop of the RBD. Four fragments (7-10), including monocyclic and bicyclic aromatic compounds, bound to this site.
Two other sites, which were very polar in nature, were also identified. Two fragments (6 and 11) bound in one of these sites located at the interface between the kinase domain and the ABD (Fig. 2d). This deep, narrow pocket has R115 and F119 at the base of the site and is lined with polar residues on either side: R704, E707, D746 from the kinase domain; and D84 and T86 from the ABD. A single fragment, 12, was identified in the other binding site located at the interface between the C2 domain, helical domain, and ABD-RBD linker (Fig. 2e). This binding site is completely devoid of hydrophobic residues and is bordered by D133, E135, T462, S464, and Q682. Either of these two polar binding sites could be used as the basis for the development of compounds with good solubility and pharmacokinetic properties.
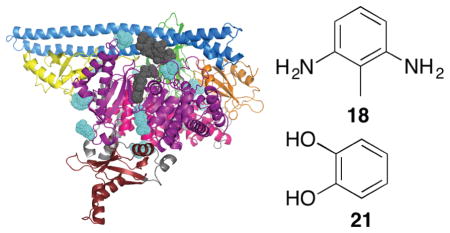
A total of 12 fragments (13-24) were modeled into a fifth binding site lying at the interface between the helical, kinase, C2 and nSH2 domains, also known as the phosphopeptide binding site (Fig. 2f). Following activation of RTKs, the nSH2 domain of PI3K binds to the auto-phosphorylated tyrosine residues leading to the activation of kinase activity.12 This fragment binding site overlaps with the binding site of such phosphorylated peptides. The sides of the binding site are lined by the residues E365, L540, E542, L570, C604 and N605 of p110α and residues E341, N344 and N377 of p85α. The top and bottom of the binding site are defined by residues I543 and F1016 of p110α. The structures of the twelve fragments show some commonalities: six of the fragments are amino-substituted aromatic rings and four of the fragments contain a hydroxyl group (Fig. 2f). The identification of this binding site is particularly interesting because one of the residues lining it, E542, is one of the three most commonly mutated residues in p110α; the most frequent mutation is a substitution to lysine. It is conceivable that an inhibitor could be developed that makes an ionic interaction with the lysine present in this oncogenic mutant; such an inhibitor would bind to the mutant but would not interact with the glutamate present in the wild-type.
To determine if this binding site would be useful for the design of mutant selective inhibitors, we next evaluated the effects of these fragments on PI3K activity. Binding at this site could stabilize the auto-inhibited conformation, thereby inactivating the enzyme, or conversely activate the enzyme in the absence of a phosphorylated peptide. To distinguish between these possibilities, we used a fluorescence polarization assay (Echelon Biosciences), which detects the formation of PIP3.45 The fragments from each of the phosphopeptide binding site cocktails were screened for wild-type PI3Kα inhibition against both p110α/niSH2 (construct used for crystallography) and full-length enzymes at a concentration of 100 μM. At higher concentrations than this, interference with the assay was observed, possibly due to aggregation of the fragments. The results for the niSH2 and full-length constructs were similar. Most fragments tested had less than 25% inhibition at 100 μM, but two compounds, 18 and 21, showed, respectively, 92% and 87% inhibition of full-length PI3Kα at 100 μM (Fig. 2f). These high levels of inhibition are particularly surprising given the small size of the fragments and their intended use only as one component of a larger compound. Furthermore, the IC50 values for 18 and 21 were determined to be 20 ± 4 μM and 33 ± 4 μM, respectively. Although rare, inhibitory activities like these by low molecular weight fragments are not without precedent; La et al. identified fragments that inhibited HIV reverse transcriptase with IC50 values of 20–100 μM.46 The low IC50 values could also reflect potential assay interference. Fragment 21 has previously been identified as a frequent hitter, and as such is unlikely to prove a good lead for the development of inhibitors.47 However, to the best of our knowledge, 18 has not previously been identified as a frequent hitter, and may provide a more useful starting point.
To further investigate these novel binding sites on PI3Kα, we used SiteMap, a program that predicts binding sites on a given protein using a grid-based approach.36,37 The number of predicted binding sites was limited to five. The two top ranked binding sites coincide with the phosphopeptide binding site, also identified through our fragment screen, at the interface between the nSH2 and helical domains. The binding sites extend to encompass the entire interface between p110α and the nSH2 domain of p85α (Fig. 2a). One of the predicted sites extends the binding site along the edge of the helical domain to a pocket between the nSH2 and C2 domains (Fig. 3a). Most of the residues contributed by the nSH2 domain are charged, lysines and aspartates, creating a “charged” base. One side of the site is lined by residues of the helical domain, extending towards E545 of the helical domain, suggesting inhibitors binding at this site could also potentially incorporate some selectivity towards the other helical domain mutant, E545K. The β-loop of the C2 domain from 361–370 then forms the roof of the site. Fragments were identified at one extreme of this site, right up against the helical domain. This SiteMap predicted site defines a larger site that could incorporate drug-like molecules and potentially be used in virtual screening. The other binding site adjacent to the phosphopeptide site extends instead along the boundary of the kinase domain towards the activation loop and into a large pocket formed between the iSH2 domain and p110α. This predicted site is extremely large, and most likely represents several smaller potential binding sites. Of interest for the further development of these fragments into inhibitors is the pocket formed between the nSH2, C2 and kinase domains, with E542 from the helical domain forming the base of the site. The two sides of the site are lined by residues from nSH2 and C2, and the roof of the site is formed by the activation loop of the kinase domain. These two predicted sites adjacent to our experimental fragment sites suggest significant opportunities for fragment growth in either direction to design a potent inhibitor that selectively targets the oncogenic mutant, E542K PI3Kα.
Figure 3. SiteMap predicted binding sites.

The locations and details of the binding sites predicted by SiteMap are shown. PI3K domains are colored according to Fig. 1b. The location of the binding sites identified by SiteMap are highlighted with green spheres. a) Sites 1 and 2 cover most of the interface between p110α and p85α, including the phosphopeptide binding site. b) Site 3 is located at the interface between the ABD and kinase domains. c) Site 4 is located close to the ATP-binding site in the kinase domain. It is a large binding site that has many potential sub-sites for inhibitor binding. d) Site 5 is a smaller binding site located at the interface between the nSH2, C2 and helical domains.
The next highest ranked binding site was located at the interface between the ABD and the kinase domain. The binding site predicted by SiteMap extends to cover the interface between the ABD and kinase domain (Fig. 3b). Molecules binding at this site could potentially disrupt heterodimer formation or stability, however are unlikely to yield mutant selective inhibitors. The next predicted binding site sits behind the ATP-binding site between the kinase, RBD and helical domains (Fig. 3c). This binding site encompasses our experimentally determined binding site 1 (Fig. 2b). The last predicted binding site is between the nSH2 and helical domains, near the linker with the C2 domain (Fig. 3d).
In sum, we have identified a novel binding site containing one of the most frequently mutated amino acid residues in p110α, E542K, found in multiple cancer types. SiteMap predicted binding sites extend this experimental site in two different directions, one encompassing another frequently mutated residue, E545K, and the other the activation loop. These binding sites hold significant promise for the design of novel, allosteric, selective inhibitors of the E542K or E545K oncogenic mutant enzymes.
Supplementary Material
Acknowledgments
We thank Jesse Yoder for helpful discussions and critical reading of the manuscript. This work was supported by NIH Grants CA062924 and CA043460 and by The Virginia and D.K. Ludwig Fund for Cancer Research. SBG is an Alexander and Margaret Stewart Trust Cancer fellow. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. This research used resources from the Berkeley Center for Structural Biology supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We acknowledge the use and service of the JHU SOM Mass Spectrometry and Proteomics Core supported by the Sidney Kimmel Comprehensive Cancer Center (NCI center grant 2P30 CA006973) and the use of the JHU SOM Eukaryotic Tissue Culture Facility.
Protein structures have been deposited in the Protein Data Bank with the following accession codes: 5SW8, 5SWG, 5SWO, 5SWP, 5SWR, 5SWT, 5SX8, 5SX9, 5SXA, 5SXB, 5SXC, 5SXD, 5SXE, 5SXF, 5SXI, 5SXJ, 5SXK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller BW, Przepiorka D, de Claro RA, Lee K, Nie L, Simpson N, Gudi R, Saber H, Shord S, Bullock J, Marathe D, Mehrotra N, Hsieh LS, Ghosh D, Brown J, Kane RC, Justice R, Kaminskas E, Farrell AT, Pazdur R. Clin Cancer Res. 2015;21:1525–1529. doi: 10.1158/1078-0432.CCR-14-2522. [DOI] [PubMed] [Google Scholar]
- 2.Castillo JJ, Furman M, Winer ES. Expert Opin Invest Drugs. 2012;21:15–22. doi: 10.1517/13543784.2012.640318. [DOI] [PubMed] [Google Scholar]
- 3.Herman SEM, Johnson AJ. Clin Cancer Res. 2012;18:4013–4018. doi: 10.1158/1078-0432.CCR-11-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei M, Wang X, Song Z, Jiao M, Ding J, Meng LH, Zhang A. Med Res Rev. 2015;35:720–752. doi: 10.1002/med.21341. [DOI] [PubMed] [Google Scholar]
- 5.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O’Brien SM. N Engl J Med. 2014;370:997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dirican E, Akkiprik M, Özer A. Tumour Biol. 2016;37:7033–7045. doi: 10.1007/s13277-016-4924-2. [DOI] [PubMed] [Google Scholar]
- 7.Du L, Chen X, Cao Y, Lu L, Zhang F, Bornstein S, Li Y, Owens P, Malkoski S, Said S, Jin F, Kulesz-Martin M, Gross N, Wang XJ, Lu SL. Oncogene. 2016;35:4641–4652. doi: 10.1038/onc.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D, Wang M, Jiang N, Zhang Y, Bian X, Wang X, Roberts TM, Zhao JJ, Liu P, Cheng H. Oncotarget. 2016;7:13153–13166. doi: 10.18632/oncotarget.7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao Y, Samuels Y, Li Q, Krokowski D, Guan BJ, Wang C, Jin Z, Dong B, Cao B, Feng X, Xiang M, Xu C, Fink S, Meropol NJ, Xu Y, Conlon RA, Markowitz S, Kinzler KW, Velculescu VE, Brunengraber H, Willis JE, LaFramboise T, Hatzoglou M, Zhang GF, Vogelstein B, Wang Z. Nat Commun. 2016;7:11971. doi: 10.1038/ncomms11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 11.Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, Dhanak D, Copeland RA, Gontarek RR, Tummino PJ, Luo L. Biochem J. 2008;409:519–524. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- 12.Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM. Proc Natl Acad Sci USA. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hon WC, Berndt A, Williams RL. Oncogene. 2012;31:3655–3666. doi: 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, Frazier K, Ng S, Smith A, Verhagen J, Haznedar J, Huh K, Iwanowicz E, Xin X, Menezes D, Merritt H, Lee I, Wiesmann M, Kaufman S, Crawford K, Chin M, Bussiere D, Shoemaker K, Zaror I, Maira SM, Voliva CF. ACS Med Chem Lett. 2011;2:774–779. doi: 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chène P, Shoemaker K, Pover AD, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, García-Echeverría C, Sellers WR, Voliva CF. Mol Cancer Ther. 2012;11:317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 16.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, Hamon J, Fabbro D, Caravatti G. Bioorg Med Chem Lett. 2013;23:3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Baselga J, Im S-A, Iwata H, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng L-M, Hurvitz S, Masuda N, Cortes J, De Laurentiis M, Arteaga C, Jiang Z, Jonat W, Hachemi S, Le Mouhaer S, Di Tomaso E, Urban P, Massacesi C, Campone M. SABCS Abstracts. 2015:S6–1. [Google Scholar]
- 18.Mayer IA, Abramson V, Balko J, Sanders M, Juric D, Solit D, Li Y, Cantley L, Winer E, Arteaga C. Cancer Res. 2015;75:CT232. [Google Scholar]
- 19.Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM. Proc Natl Acad Sci USA. 2006;103:16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJH, Withers DJ, Vanhaesebroeck B. Nature. 2006;441:366–370. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 21.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 23.Miller MS, Schmidt-Kittler O, Bolduc DM, Brower ET, Chaves-Moreira D, Allaire M, Kinzler KW, Jennings IG, Thompson PE, Amzel LM, Vogelstein B, Gabelli SB. Oncotarget. 2014;5:5198–5208. doi: 10.18632/oncotarget.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindsley CW. Curr Top Med Chem. 2010;10:458–477. doi: 10.2174/156802610790980602. [DOI] [PubMed] [Google Scholar]
- 25.Yan L. Cancer Res. 2009;69:DDT01-1. [Google Scholar]
- 26.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 27.Tripathy D, Chien AJ, Hylton N, Buxton MB, Ewing CA, Wallace AM, Forero A, Kaplan HG, Nanda R, Albain KS, Moulder SL, Haley BB, DeMichele A, Symmans WF, Veer Lvan’t; Paoloni M, Esserman L, Berry DA, Yee D. J Clin Oncol. 2015;33 (suppl; abstr 524) [Google Scholar]
- 28.Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, Fenga C, Spandidos DA, Libra M, Tsatsakis AM. Int J Oncol. 2016;48:869–885. doi: 10.3892/ijo.2015.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowan-Jacob SW, Jahnke W, Knapp S. Future Med Chem. 2014;6:541–561. doi: 10.4155/fmc.13.216. [DOI] [PubMed] [Google Scholar]
- 30.Wu P, Clausen MH, Nielsen TE. Pharmacol Ther. 2015;156:59–68. doi: 10.1016/j.pharmthera.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H. Proc Natl Acad Sci USA. 2015;112:15910–15915. doi: 10.1073/pnas.1518946112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noble CG, Lim SP, Arora R, Yokokawa F, Nilar S, Seh CC, Wright SK, Benson TE, Smith PW, Shi PY. J Biol Chem. 2016;291:8541–8548. doi: 10.1074/jbc.M115.710731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caliandro R, Belviso DB, Aresta BM, de Candia M, Altomare CD. Future Med Chem. 2013;5:1121–1140. doi: 10.4155/fmc.13.84. [DOI] [PubMed] [Google Scholar]
- 34.Chilingaryan Z, Yin Z, Oakley AJ. Int J Mol Sci. 2012;13:12857–12879. doi: 10.3390/ijms131012857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 36.Halgren TA. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 37.Halgren T. Chem Biol Drug Des. 2007;69:146–148. doi: 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 41.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 42.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Mol Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manallack DT, Hughes RA, Thompson PE. J Med Chem. 2005;48:3449–3462. doi: 10.1021/jm040217u. [DOI] [PubMed] [Google Scholar]
- 45.Drees BE, Weipert A, Hudson H, Ferguson CG, Chakravarty L, Prestwich GD. Comb Chem High Throughput Screen. 2003;6:321–330. doi: 10.2174/138620703106298572. [DOI] [PubMed] [Google Scholar]
- 46.La J, Latham CF, Tinetti RN, Johnson A, Tyssen D, Huber KD, Sluis-Cremer N, Simpson JS, Headey SJ, Chalmers DK, Tachedjian G. Proc Natl Acad Sci USA. 2015;112:6979–6984. doi: 10.1073/pnas.1423900112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baell JB, Holloway GA. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.