

Abstract
To facilitate the wider application of the NADPH-dependent P450BM3, we fused the monooxygenase with a phosphite dehydrogenase (PTDH). The resulting monooxygenase-dehydrogenase fusion enzyme acts as a self-sufficient bifunctional catalyst, accepting phosphite as a cheap electron donor for the regeneration of NADPH.
The well-expressed fusion enzyme was purified and analyzed in comparison to the parent enzymes. Using lauric acid as substrate for P450BM3, it was found that the fusion enzyme had similar substrate affinity and hydroxylation selectivity while it displayed a significantly higher activity than the non-fused monooxygenase. Phosphite-driven conversions of lauric acid at restricted NADPH concentrations confirmed multiple turnovers of the cofactor. Interestingly, both the fusion enzyme and the native P450BM3 displayed enzyme concentration dependent activity and the fused enzyme reached optimal activity at a lower enzyme concentration. This suggests that the fusion enzyme has an improved tendency to form functional oligomers.
To explore the constructed phosphite-driven P450BM3 as a biocatalyst, conversions of the drug compounds omeprazole and rosiglitazone were performed. PTDH-P450BM3 driven by phosphite was found to be more efficient in terms of total turnover when compared with P450BM3 driven by NADPH. The results suggest that PTDH-P450BM3 is an attractive system for use in biocatalytic and drug metabolism studies.
Electronic supplementary material
The online version of this article (doi:10.1007/s00253-016-7993-7) contains supplementary material, which is available to authorized users.
Keywords: Drug metabolites, Enzyme catalysis, Fatty acids, NADPH regeneration, Oxidation, Protein engineering
Introduction
Cytochrome P450 monooxygenases (P450s) are versatile enzymes that catalyze a broad range of hydroxylation, epoxidation, sulfoxidation, deamination, and dehalogenation reactions and are attractive for applied biocatalysis (Sono et al. 1996; Bernhardt 2006). In vivo, the main functions include steroid hydroxylation, fatty acid hydroxylation, and xenobiotic detoxification, which consequently makes P450s of interest to the pharmaceutical industry (Gillam and Hayes 2013; Munro et al. 2013). Similar to other monooxygenases, catalysis requires a suitable substrate, dioxygen, and two electrons to be fed into the catalytic reaction cycle (Meunier et al. 2004). The electrons are transferred to the heme from a reducing cofactor via electron transfer proteins, which may be domains fused to the monooxygenase component or may occur as separate proteins transferring electrons in transient assemblies (Hannemann et al. 2007). The need for auxiliary redox proteins presents an obstacle for the efficient application of P450s because (a) complex cofactors have to be integrated into different proteins or protein domains to obtain a functional catalyst and (b) participating enzymes need to match each other in terms of concentration, stability, affinity, and activity to achieve maximal turnover. In addition, the dependency on NAD(P)H, slow electron transport, and coupling efficiency can limit activity (Lundemo and Woodley 2015).
The highly active bacterial flavocytochrome P450BM3 (CYP102A1 from Bacillus megaterium, EC 1.14.14.1) naturally circumvents some of these limitations. As a single polypeptide P450 (class VIII), it has an FAD- and FMN-containing NADPH-cytochrome P450 reductase (BMR domain) fused to the C-terminus of the P450 domain in a 119 kDa protein, that is active as a dimer (Ruettinger et al. 1989; Neeli et al. 2005). The enzyme can be solubly expressed in good yields in heterologous hosts such as Escherichia coli. It is one of the few P450s with reported activities above 1 s−1, which is partly due to rapid electron transfer through the BMR domain (Munro et al. 1996; Munro et al. 2002). The enzyme efficiently hydroxylates medium- to long-chain fatty acids. Arachidonic acid was reported to be converted with the highest activity (285 s−1). Low uncoupling rates of approx. 3.8% were determined when using laurate as substrate (Noble et al. 1999). After it was shown that the substrate spectrum could be modulated by various mutations, P450BM3 was widely used as a surrogate for class II mammalian P450s. It also is used as a model P450 in mechanistic studies because of easy expression, high activity, and the availability of crystal structures of the separate domains. Whitehouse et al. reviewed recent progress on this enzyme and describe numerous mutants accepting non-natural substrates; many of them are bulky molecules like drugs and related compounds (Whitehouse et al. 2012).
The regeneration of NADPH, which is stoichiometrically consumed in the monooxygenation reaction, remains a major challenge for the application of P450s. Several regeneration approaches have been investigated, of which the enzymatic regeneration of the cofactor is most commonly applied. One possibility is the use of dehydrogenases; popular examples being glucose-6-phosphate dehydrogenase (Ahmed et al. 1999; Falck et al. 2001; Volz et al. 2002; Murataliev et al. 2004; Chen et al. 2008), glucose dehydrogenase (Sulistyaningdyah et al. 2005; Schewe et al. 2008; Schewe et al. 2009), isocitrate dehydrogenase (Capdevila et al. 1996; Schwaneberg et al. 2001; Peters et al. 2003; Landwehr et al. 2006), and formate dehydrogenase (Maurer et al. 2003; Kühnel et al. 2007). Phosphite dehydrogenase (PTDH, EC 1.20.1.1) is one of the most cost efficient options; it regenerates NADPH while oxidizing cheap phosphite to phosphate with the simultaneous reduction of NADP+. The large change in energy of this reaction makes it thermodynamically almost irreversible and only one inhibitor (sulfite) has been described to date (Costas et al. 2001; Woodyer et al. 2003; Relyea and van der Donk 2005).
The recycling of NADPH with phosphite has been reported for fusions of PTDH with several Baeyer-Villiger monooxygenases (BVMOs) (Torres Pazmiño et al. 2009). A codon-optimized gene was used for expression of PTDH-BVMO fusions in E. coli. The employed PTDH contained 18 mutations which led to enhanced activity towards NADP+, soluble expression, and stability (Johannes et al. 2007). Watanabe et al. also reported a fusion of PTDH to a Pdx-PCNA2 construct which functioned as the “PCNA-utilized protein complex of P450cam and its two electron transfer-related proteins” (PUPPET) for the regeneration of NADH (Watanabe et al. 2013).
To facilitate the application of P450BM3 as a practical biocatalyst, we present here a fusion of P450BM3 to the optimized variant of PTDH for cofactor recycling: PTDH-P450BM3. The turnover of NADP+ to NADPH by the fused PTDH domain should enable the use of phosphite as a cheap reductant for performing P450BM3-catalyzed oxygenations with phosphate as a byproduct (Scheme 1). The fusion protein is compared to the regeneration system with the free PTDH and P450BM3 and the P450BM3 without cofactor regeneration system.
Scheme 1.

Schematic model of PTDH coupled to P450BM3 facilitating phosphite-driven cofactor recycling
Materials and methods
Materials
Unless stated otherwise, all chemicals and enzymes were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands), Oriental Yeast Co. (Tokyo, Japan), New England Biolabs (Leiden, The Netherlands), and Agilent (Santa Clara, USA) and used without further purification. Isopropyl-1-thio-β-D-galactopyranoside (IPTG) was ordered at Iris Biotech GmBH (Marktredwitz, Germany). Oligonucleotide primers were obtained from Sigma Genosys. DNA sequencing was performed at GATC Biotech AG (Konstanz, Germany).
Bacterial strains, plasmids, and cloning
E. coli TOP10 (Invitrogen, Carlsbad, CA, USA) was used for genetic manipulations. For expression, a pBAD vector or a modified pBAD vector with an NcoІ restriction site replacing NdeІ were used. The plasmid pPTDH in which the codon-optimized ptxD gene is fused to an N-terminal histidine tag codes the 18-fold mutated PTDH without a fused monooxygenase. The vector pJOE-P450BM3 encoding the wild-type gene cyp102A1 was a kind gift from Prof. B. Hauer (University of Stuttgart) (Scheps et al. 2013). For the creation of the fusion enzyme, the vector pCRE2-PAMO was used as a template. It carries the 18-fold mutant of the phosphite dehydrogenase from Pseudomonas stutzeri WM88 with N-terminal histidine tag and the pamO gene in fusion (Torres Pazmiño et al. 2009). The fusion construct pCre2-P450BM3 was made implementing the In-Fusion technique from Clontech, resulting in the sequence coding for the fusion construct PTDH-P450BM3 (KX768143). Using primers with overhangs matching the expression vector, cyp102A1 encoding P450BM3 was amplified via PCR using pJOE-P450BM3 as template and then hybridized with the respective expression vector, following the recommendations of the manufacturer. For comparison studies with the unfused enzyme, cyp102A1 was subcloned into pBAD. Implementing primers with an overhang matching the target vector, cyp102A1 was amplified via a PCR reaction. The PCR product was purified and utilized as a primer for a second PCR reaction, subcloning the gene encoding P450BM3 into the respective expression vector, yielding pBAD-P450BM3.
Screen for soluble enzyme expression
E. coli TOP10, BL21 (DE3), C43 (DE3) and SHuffle cells harboring pCre2-P450BM3 were pre-grown overnight at 37 °C in ampicillin-supplemented LB medium (5 mL; 50 μg/mL). TB-medium (50 mL, ampicillin-supplemented) in a non-baffled glass shaking flask (250 mL) was inoculated with an OD600 of 0.08 and incubated at 37 °C and 180 rpm. After an OD600 of 0.75–0.85 was reached, expression of PTDH-P450BM3 was induced by the addition of 0.02% arabinose (final concentration). The cells were incubated for 25 h at 30 °C and 135 rpm. Cells were harvested by centrifugation (20 min, 8800×g, 4 °C), washed with ice-cold sodium phosphate buffer (50 mM, pH 7.5) and frozen (−20 °C) until cell lysis by sonication. The cell debris was separated by centrifugation and the content of PTDH-P450BM3 in the cell-free extract was investigated via CO-difference spectroscopy. For the optimization of the arabinose concentration and the temperature for enzyme expression, TB-medium (2.5 mL) in 24-square deep well plates with the sandwich cover system from enzyscreen®, was inoculated with a preculture of E. coli TOP10, harboring pCre2-P450BM3 (5 μL). Cultures were grown at 17 °C (48 h), 24 °C (32 h), 30 °C (16 h), and 37 °C (16 h) and expression was induced directly after inoculation with 0% (as control), 0.002%, 0.02%, and 0.2% of final arabinose. Cells were harvested via centrifugation and cell pellets were frozen (−20 °C, 16 h). Cell lysis was performed enzymatically (200 μL lysis buffer consisting of 2 mg/mL lysozyme, 0.1 mg/mL DNasel, 5 mM MgSO4 in 50 mM Tris/HCl (pH 8)) and with a freeze-and-thaw cycle (plate was shock frozen in liquid nitrogen and incubated in a shaker at 30 °C for 1 h). Samples were taken and the cell debris was separated from the soluble fraction by centrifugation. Both the lyzed cell and the soluble fraction were analyzed for expression of PTDH-P450BM3 via SDS-PAGE.
Enzyme expression
E. coli TOP10 cells harboring pBAD vectors encoding the respective P450BM3 variants were pre-grown overnight (37 °C) in 5 mL ampicillin-supplemented LB medium (50 mg/mL). Four hundred milliliters of TB-medium (ampicillin-supplemented) in non-baffled glass shaking flask (2 L) were inoculated with an OD600 of 0.08 and incubated at 37 °C and 180 rpm. Expression was induced at an OD600 of 0.7–0.8 with arabinose (0.02% w/v) and 5-aminolevulinic acid was added (0.5 mM). After induction, the cells were incubated (17 °C, 180 rpm) over 44 h, harvested via centrifugation, washed once with ice-cold sodium phosphate buffer (50 mM, pH 7.5), and were stored until further use (−20 °C).
The PTDH was expressed as described before by Dudek et al. (Dudek et al. 2013). E. coli TOP10 cells were transformed with the plasmid and grown overnight at 37 °C in 5 mL of LB supplemented with ampicillin (50 μg/mL). The next day cells were diluted (1:100) in TB supplemented with ampicillin (50 μg/mL) and arabinose (0.02% w/v) and incubated for 16 h at 30 °C. Cells were harvested via centrifugation, washed once with ice-cold sodium phosphate buffer (50 mM, pH 7.5), and were stored until further use (−20 °C).
Enzyme purification
For purification, the frozen cells were resuspended in sodium phosphate buffer (50 mM, pH 7.5, optionally supplemented with DNase) at RT and disrupted via sonication on ice. The resulting crude extract was centrifuged and the supernatant/cell-free extract (CFE) diluted to a protein concentration of ∼20 mg/mL. The diluted CFE was then precipitated at an ammonium sulfate (AS) concentration of 55% (w/v) and collected by centrifugation (18,600×g, 15 min, 4 °C). The resulting pellet was resuspended in sodium phosphate buffer (50 mM, 500 mM NaCl pH 7.5) and subjected to an affinity chromatography purification on a 5 mL HF HisTrap column on an Äkta purifier (GE Healthcare). Fractionated elution was carried out with imidazole (250 mM). The buffer was removed by a second AS precipitation with 65% AS. The resulting pellet was resuspended in ice-cold Tris/HCl. After the addition of sucrose (20 mg/mL) for stabilization during storage, the enzyme was shock frozen in liquid nitrogen and stored at −80 °C (van Beek et al. 2015).
The PTDH was purified using Ni Sepharose High Performance (GE Healthcare) as described before (Dudek et al. 2013). Ten percent glycerol (v/v) was added to the purified enzyme, which was shock frozen and stored at −20 °C.
Concentration determination
Concentrations of P450BM3 and PTDH-P450BM3 were determined by measuring CO-difference spectra (Omura and Sato 1964). After the addition of sodium dithionite (spatula tip), the enzyme solution was incubated (5 min) and a baseline spectrum from 400 to 500 nm was recorded. The samples were then bubbled with carbon monoxide (∼60 s) and incubated for 10 min before the UV spectrum from 400 to 500 nm was recorded. After subtraction of the baseline spectrum, the amount of P450 was calculated based on the maximum absorbance at 450 nm minus the absorption at 490 nm (ε450nm = 91 mM−1 cm−1). This method is specific for the native P450-domain, therefore the concentration of native fused PTDH is an estimation that might vary slightly from the actual concentration.
The concentration of unfused PTDH was determined using Waddell’s method (Waddell 1956) and the estimated extinction coefficient at 280 nm (ε280 nm = 26.5 mM−1 cm−1).
Activity determination and kinetic measurements
NADPH oxidation or NADP+ reduction rates were measured spectrophotometrically at 30 °C by following the absorption at 340 nm over 2–5 min (ε340 = 6.2 × 103 M−1 cm−1) in a final volume of 500 μL in a quartz cuvette. For the investigation of the properties of the P450BM3 (fused or WT), typical reaction mixtures contained lauric acid as a substrate (2.5 mM from a 25 mM stock in ethanol/methanol (1:1)) and enzyme (100 nM) in Tris buffer (50 mM, pH 7.5). The reaction was started by the addition of NADPH (250 μM). For the investigation of the PTDH, the reaction mixture contained enzyme (100 nM) and phosphite as the substrate (5 mM). NADP+ was added to start the reaction (250 μM). For the determination of kinetic parameters or concentration-based activity, the concentration of the respective component was varied. All measurements were performed in triplicate. Kinetic parameters were calculated using the Michaelis-Menten equation as a function of the respective substrate concentration in GraphPad Prism 6.
Biotransformations in 96-well plate format
Purified enzyme (0.5 μM), substrate (10 μM), NADPH or NADP+ (0.15 mM), and phosphite (1.5 mM) were mixed in Tris/HCl buffer (50 mM, pH 7.5, 100 μL) in 96-well round-bottomed plates. The reaction mixtures were incubated at 30 °C in a water bath or a plate shaker at 500 rpm. Samples (50 μL) were taken and mixed with 100% acetonitrile (100 μL). After centrifugation (4000 rpm, 20 min, 4 °C), the resulting supernatant (50 μL) was transferred into 40% acetonitrile in MilliQ water (150 μL) and stored at −20 °C prior to analysis by UPLC-MS.
Biotransformations investigating cofactor recycling
Purified enzyme (1 μM), lauric acid (2.25 mM, 10% ethanol/methanol as final concentration), NADPH (50 μM), and phosphite (4 mM) were mixed in Tris/HCl buffer (50 mM, pH 7.5) in plastic falcon tubes (15 mL with 3 mL reaction mixture). For the three different set-ups—PTDH-P450BM3, PTDH + P450BM3, and P450BM3—reaction mixtures without enzyme and without NADPH addition were the controls. The biotransformations were incubated (30 °C, 180 rpm) and samples for analysis in the molybdate assay and by GC-MS were taken in triplicate per time point (20 and 120 μL).
Phosphate quantification via molybdate assay
Samples were taken from in vitro biotransformations to investigate cofactor recycling (20 μL). These were mixed with molybdate reagent (200 μL; 100 mM zinc acetate, 10 mM ammonium molybdate, and 0.1% SDS in MilliQ water, pH 5 (adjusted with HCl; stored in a PE bottle). Before an incubation (30 min, 30 °C), ascorbic acid (50 μL of a 10% w/v stock solution in MilliQ water, pH 5 (adjusted with NaOH)) was added. Measurements were performed in a Powerwave 96-well plate reader (Bio-TEK Instruments) at 850 and 700 nm. Phosphate standards containing potassium dihydroxy phosphate (0.1–5 mM) were used to prepare the calibration curve (Saheki et al. 1985; Dudek et al. 2013).
GC-MS analysis
Samples from biotransformations investigating cofactor recycling (120 μL) were taken, and the conversions stopped in Eppendorf tubes prepared with NaCl (6 mg) and H2SO4 (50%, 10 μL). The reaction mixtures were extracted twice with twice the volume of MTBE/hexane (1:1); the internal standard, decanoic acid (1 mM in the final derivatization mixture) was added during the first extraction with the MTBE/hexane phase. The organic phases were collected and evaporated. Samples were resuspended in MTBE (60 μL), followed by the addition of 1% TMS in BSTFA (60 μL) and incubated at 75 °C for 30 min for derivatization. Tubes were sealed with parafilm during derivatization (Lalman and Bagley 2004; Scheps et al. 2013).
Measurements were performed on a HP-5 column ((5% Phenyl) methylpolysiloxan, 30 m × 0.25 mm × 0.25 μm), with an injection volume of 1 μL at 300 °C (injection and interface temperature) and helium as carrier gas. The method features a temperature gradient from 40 to 300 °C (10 °C/min and 10 min isothermal at 300 °C) with a solvent cut-off of 6 min, a split ratio of 30, and a flow of 1 mL min−1. The detection limit was approximately 4 μM. 12-hydroxylauric acid was used as a model product to estimate the response factor for monohydroxylated lauric acids. Derivatized capric acid, lauric acid, and 12-OH lauric acid elute after 10.6, 12.9, and 16.6 min, respectively. The derivatized products were identified via their fragmentation patterns. 11-hydroxylauric acid (2), 10-hydroxylauric acid (3), and 9-hydroxylauric acid (4) eluted at 18.5, 18.4, and 18.1 min, respectively.
UPLC-MS analysis
Samples were analyzed on a Waters ACQUITY UPLC liquid chromatography system coupled to a Waters Synapt HDMS mass spectrometer equipped with an electrospray (ESI) ionization source. Samples taken during biotransformations were placed in a 96-well Nunc plate (injection volume 5–10 μL). Chromatograph separations were carried out on an ACQUITY UPLC BEH C18 column (130 Å, 1.7 μm × 2.1 mm × 100 mm; Waters, Milford, MA, USA) applying a flow rate of 0.5 mL min−1 at a column temperature of 45 °C. The mobile phase consisted of ultra-pure water supplemented with formic acid (0.1% v/v, phase A) and pure acetonitrile (phase B). The gradient applied for separation was 0.0–6.0 min (10–70% phase B), followed by a return to the initial mobile phase composition over 0.01 min.
The MS analysis was performed under the following conditions using the parameters stated. Positive electrospray ionization (ESI) conditions in V-mode. A generic method with two scan functions was used as follows: m/z 80–1000, cone voltage 20 V and 0.1 s scan time, the collision energy (CE) in function 1 was 20 V and in function 2 an energy ramp of 15–45 V was applied, the transfer cell CE is 12 V. Data was collected in centroid mode. Leucine-enkephalin was used as lock mass (m/z 556.2771) for internal calibration at a concentration of 250 pg mL−1 and a flow rate of 0.04 mL min−1. The MS data was processed in MetaboLynx 4.1 (Waters, Milford, MA, USA) using both the mass defect filter (MDF) and the dealkylation tool. The list of proposed metabolites was reviewed manually. A semiquantitative estimation of substrate conversion and metabolite formation were assessed by calculation of the fraction of the substrate peak area or a product peak area to the total related peak area detected (Kulig et al. 2015).
Results
Enzyme expression and isolation
The plasmid pCre2-PAMO described by Torres Pazmiño et al. was used as a template to create an expression plasmid for the production of P450BM3 fused to the C-terminus of a codon-optimized and stabilized PTDH which carries a His-tag at its N-terminus (Torres Pazmiño et al. 2009). Using this construct, six amino acids (SRSAAG) link the two proteins into one fusion protein of 157 kDa. For producing the P450BM3 as a reference for the non-fused enzyme, the P450BM3-encoding gene was cloned into a regular pBAD vector, resulting in expression of P450BM3 with a C-terminal His-tag (119 kDa) (Fig. S1). The PTDH was expressed from a pBAD vector with an N-terminal His-tag (Dudek et al. 2013).
The expression of PTDH-P450BM3 was optimized in terms of the type of E. coli host strain (Table S1), the expression temperature, and the concentration of arabinose added for induction (Fig. S2). Four E. coli strains with different features for recombinant protein expression were tested for soluble production of PTDH-P450BM3, namely E. coli TOP10, E. coli BL21(DE3), E. coli C43, and E. coli SHuffle. Expression of soluble and functional enzyme was determined by CO-difference spectroscopy. The most efficient strain for production of PTDH-P450BM3 was E. coli TOP10, while almost no expression was detected in E. coli BL21(DE3). To optimize soluble expression in E. coli TOP10, the temperature and concentration of inducer were varied; the best yield was obtained at 17 °C and induction with 0.02–0.2% arabinose. As judged by SDS-PAGE analysis, addition of δ-amino levulinic acid further improved expression. Recombinant production of PTDH-P450BM3 and P450BM3 using E. coli TOP10 in a culture volume of 50 mL yielded 9.5 mg PTDH-P450BM3 and 9.8 mg unfused P450BM3 in the cell-free extract after 42 h cultivation. This shows that the functional expression of both P450BM3 variants is very efficient.
By exploiting the His-tag on both P450BM3 variants, both enzymes could be easily purified. The integrity of the purified P450BM3 and PTDH-P450BM3 was confirmed by the characteristic UV/Vis-spectrum (Fig. 1). The UV/Vis-spectral features of both proteins are nearly identical which confirms the incorporation of the essential heme and flavin cofactors.
Fig. 1.

UV/Vis spectra of purified P450BM3 (solid line, 4.5 μM) and purified PTDH-P450BM3 (dashed line, 2.6 μM)
To verify whether the obtained enzymes were functional, monooxygenase and dehydrogenase activities were measured. For testing P450BM3 activity, conversion of lauric acid (1) by P450BM3 was analyzed. GC analysis revealed formation of monohydroxylation products in positions 11 (2), 10 (3), and 9 (4) in similar amounts (Scheme 2). By monitoring the rate of NADPH consumption using 50 nM enzyme, monooxygenase activities of 6.7 s−1 for P450BM3 and 8.9 s−1 for PTDH-P450BM3 were found, while uncoupling rates (defined as NADPH consumption in the absence of substrate) were determined as 0.22 s−1 for PTDH-P450BM3 and 0.19 s−1 for P450BM3. This shows that the fused enzyme displays a slightly higher monooxygenase activity (Fig. 2a). For PTDH activity, a similar trend was observed with a k obs of 0.71 s−1 for the non-fused PTDH and 2.5 s−1 for the fused PTDH (Fig. 2b). These data show that PTDH-P450BM3 was fully functional, displaying both monooxygenase and dehydrogenase activities.
Scheme 2.

Model reaction catalyzed by P450BM3. NADPH is consumed during the monohydroxylation of lauric acid (1) in position 11, 10 and 9 (2,3,4)
Fig. 2.
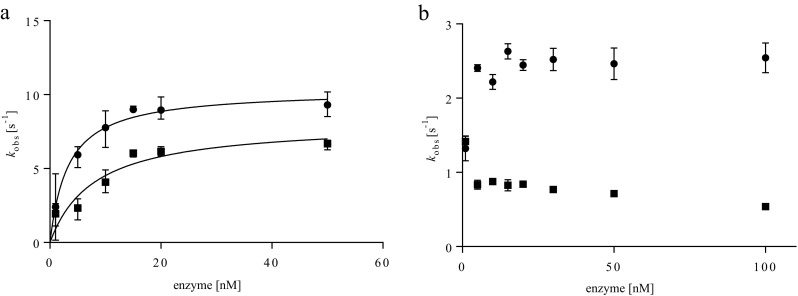
Enzyme concentration dependent activity of unfused P450BM3 (squares), unfused PTDH (squares), and PTDH-P450BM3 (circles). The graphs show the relation between enzyme concentration and the initial activities for a oxidation of 0.20 mM NADPH in the presence of 0.4 mM lauric acid as substrate at varying concentrations of either P450BM3 or PTDH-P450BM3 and b reduction of NADP+ in the presence of 2.0 mM phosphite as substrate at varying concentrations of either PTDH or PTDH-P450BM3. Concentrations of (PTDH)-P450BM3 were determined via CO-difference spectra, PTDH concentrations by Waddell’s method. Experiments were performed in triplicate
Enzyme concentration dependent activity
Neeli et al. demonstrated that inter-monomer electron transfer is required for efficient hydroxylation of lauric acid and therefore the dimeric form of P450BM3 is the catalytically active variant (Neeli et al. 2005). To examine if the oligomerization behavior of the fused enzymes affected the reactivity of the fusion construct, the specific activities of the two non-fused enzymes and PTDH-P450BM3 were determined by following the consumption or generation of NADPH at different enzyme concentrations (1–50 nM for the P450 reaction, 1–100 nM for the PTDH reaction). The observed monooxygenase activity decreased sharply at enzyme concentrations below 10 nM. Hyperbolic fitting of k obs values versus enzyme concentration gave apparent half-saturation concentrations of 8.8 ± 2.1 and 3.3 ± 0.9 nM for P450BM3 and PTDH-P450BM3, respectively (Fig. 2a). A similar enzyme concentration dependence was previously reported for native P450BM3 and is assumed to reflect the dissociation constant K D for the dimerization of P450BM3, being active as a dimer (Neeli et al. 2005). The lower K D for the fusion enzyme suggests that the fusion partner promotes oligomerization.
The rate of phosphite-dependent NADP+ reduction by PTDH (−P450BM3) was determined as a function of the concentration of the fused or non-fused PTDH (Fig. 2b). The k obs for the PTDH reaction was relatively unaffected at enzyme concentrations of 5–100 nM. However, at an enzyme concentration of 1 nM, a decrease in the specific activity was observed for PTDH-P450BM3, while an increase was detected for the non-fused PTDH.
Kinetic parameters of PTDH-P450BM3
The steady state kinetic parameters of PTDH-P450BM3 were determined and compared to those of the non-fused PTDH and P450BM3. For analyzing the P450 kinetic parameters, the lauric acid-dependent oxidation of NADPH was measured in the absence of phosphite for the fusion enzyme and for P450BM3 (Table 1). The phosphite-dependent reduction of NADP+ was measured for PTDH-P450BM3 and PTDH in the absence of lauric acid (Table 2). The kinetic properties of the fused enzyme were found to be in the same range as those of the separate enzymes. While the K m values differed marginally, PTDH-P450BM3 displayed a higher k cat for both enzyme domains (for PTDH activity, 1.9 s−1 for PTDH vs. 2.3 s−1 for PTDH-P450BM3; for P450 activity, 7.3 s−1 for P450BM3 vs. 8.0 s−1 for PTDH-P450BM3). These results confirm that the fused system is fully functional and that both domains are at least as efficient as their non-fused counterparts. When considering lauric acid, the PTDH showed a lower activity than the P450BM3 component. Therefore, the phosphite-dependent generation of NADPH should be the rate-limiting step during bioconversions. Yet, for other P450BM3 substrates, lower rates have been reported which makes PTDH a suitable NADPH regeneration partner.
Table 1.
Kinetics of NADPH dependent lauric acid oxidation. Kinetic parameters of PTDH-P450BM3 and P450BM3 for NADPH and lauric acid were analyzed by following absorption of NADPH in a spectrophotometer at 340 nm. Specific activities were determined with 100 nM of the respective enzyme and varying concentrations of either NADPH or lauric acid. When fixed, NADPH had a concentration of 200 μM and lauric acid of 2.5 mM
| NADPH | Lauric acid | |||||
|---|---|---|---|---|---|---|
| k cat [s−1] | K m [μM] | k cat/K m [s−1 mM−1] | k cat [s−1] | K m [μM] | k cat/K m [s−1 mM−1] | |
| PTDH-P450BM3 | 6.9 ± 0.5 | 15.4 ± 2.3 | 448 | 8.0 ± 0.3 | 688 ± 78 | 11.6 |
| P450BM3 | 6.0 ± 0.3 | 17.8 ± 2.7 | 337 | 7.3 ± 0.2 | 1427 ± 113 | 5.1 |
Table 2.
Kinetics of phosphite-dependent cofactor regeneration. Kinetic parameters of PTDH-P450BM3 and PTDH for NADP+ and phosphite were analyzed by following absorption of NADPH in a spectrophotometer at 340 nm. Specific activities were determined with 100 nM of the respective enzyme and varying concentrations of either NADP+ or phosphite. When fixed, NADP+ had a concentration of 200 μM and phosphite of 2 mM
| NADP+ | Phosphite | |||||
|---|---|---|---|---|---|---|
| k cat [s−1] | K m [μM] | k cat/K m [s−1 mM−1] | k cat [s−1] | K m [μM] | k cat/K m [s−1 mM−1] | |
| PTDH-P450BM3 | 2.10 ± 0.02 | 9.5 ± 0.5 | 221 | 2.30 ± 0.01 | 148 ± 4 | 15.5 |
| PTDH | 1.78 ± 0.02 | 3.9 ± 0.2 | 452 | 1.87 ± 0.02 | 201 ± 10 | 9.3 |
Phosphite-driven drug conversion under screening conditions
The high catalytic activity makes P450BM3 an attractive biocatalyst for drug metabolism studies. Variants of the enzyme can be used for the production of human metabolites (Reinen et al. 2011). The application of PTDH- P450BM3 for exploring drug metabolism under screening conditions was investigated with omeprazole, a treatment for gastroesophageal reflux disease, and rosiglitazone, an antidiabetic drug. Both drugs are metabolized by P450BM3 (Whitehouse et al. 2012).
To investigate the functionality of the PTDH-P450BM3 system, conversion of both drugs was performed with NADPH or with phosphite plus NADP+ as electron source. Furthermore, conversion by the fusion enzyme was compared with conversions catalyzed by P450BM3. For these conversions, 10 μM of either omeprazole or rosiglitazone were incubated at 30 °C with purified P450BM3 or PTDH-P450BM3 in a final reaction volume of 100 μL. Samples were taken after 0 and 4 h and analyzed by UPLC-MS.
Both enzyme variants produced a single metabolite when using omeprazole (5) as substrate. A monohydroxylated metabolite (6) was evident from a mass shift from the parent compound of 15.996 Da. Hydroxylation of one of the methyl groups of the 4-methoxy-3,5-dimethylpyridine moiety in omeprazole was proposed on the basis of fragmentation data (Scheme 3a). As expected, conversion with P450BM3 only occurred when NADPH was added as an electron source and not in reactions with phosphite and NADP+. Around 16% of the hydroxylated metabolite was formed in the NADPH-driven P450BM3 reaction (Fig. 3a). In the NADPH-driven reaction with the fusion enzyme, 25% of the same metabolite was formed. Reactions driven on phosphite as the sole electron donor showed similar behavior in terms of product formation. Under the tested conditions, the bifunctional enzyme was more efficient compared to non-fused P450BM3.
Scheme 3.

Conversions of a omeprazole (5) to its proposed monohydroxylation metabolite (6) and b rosiglitazone (7) and the proposed demethylation (8) and N-oxidation metabolites (9)
Fig. 3.
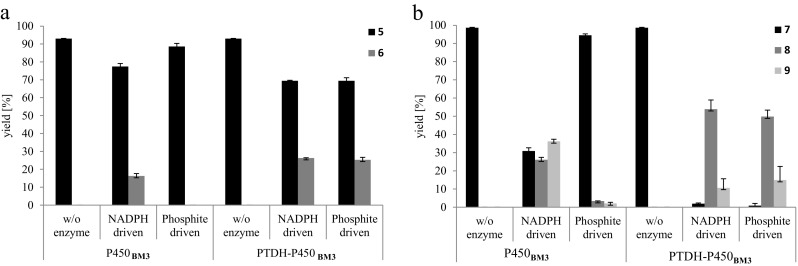
Conversions of a omeprazole and b rosiglitazone by P450BM3 and PTDH-P450BM3. Substrate was used at concentrations of 10 mM in a total volume of 100 mL. Reactions were carried out at 30 °C; reaction progress was measured after 4 h in comparison to 0 h samples by UPLC-MS. Phosphite-driven reactions contained phosphite and NADP+, whereas NADPH-driven reactions contained NADPH and phosphite. The yield of product and remaining substrate was determined as a percentage of the respective peak area to the total related peak area. Reactions were performed in duplicate (5 = omeprazole, 6 = monohydroxylated omeprazole, 7 = rosiglitazone, 8 = demethylated rosiglitazone, 9 = monohydroxylated rosiglitazone)
In reactions with rosiglitazone (MH+ 358.12; 7) two main products were formed: a demethylated metabolite (MH+ 344.11; 8) and an N-oxide (MH+ 374.12; 9) (Scheme 3b) were proposed based on the fragmentation data. The unfused P450BM3 produced these metabolites in reactions with NADPH as electron donor with a yield of 26% for the demethylation and 36% for the N-oxidation product after 4 h. Very little conversion (<5%) occurred when NADP+ and phosphite were used. The ratio of demethylation vs. N-oxidation shifted when using PTDH-P450BM3; around 50% demethylation and 15% N-oxidation product were formed in reactions with the bifunctional enzyme (Fig. 3b). As observed for omeprazole, the total turnover was higher in reactions with the fusion enzyme. Furthermore, conversions with PTDH-P450BM3 did not show a significant difference when changing the electron source (NADPH or phosphite).
Cofactor recycling
To establish the efficiency of the engineered PTDH-P450BM3 fusion enzyme with respect to cofactor recycling, conversions were analyzed in greater detail. Omeprazole and rosiglitazone are poorly soluble in aqueous solution and so lauric acid was used as a model substrate to investigate bioconversions with limited NADPH supply. Reactions were performed with the fused enzyme (PTDH-P450BM3), the non-fused parent proteins (PTDH + P450BM3), and P450BM3 without any cofactor recycling enzyme. Reaction mixtures consisted of 2.25 mM lauric acid, 4 mM phosphite, 50 μM NADPH, and 1.0 μM enzyme. As a consequence, conversions of lauric acid greater than 2.2% relied on cofactor regeneration by the PTDH. Conversions were monitored over time by taking samples for the analysis of phosphate production and the quantification of lauric acid and its metabolites.
Uncoupling was investigated by comparing the amount of phosphate formed, as an indicator of the amount of NADPH regenerated, and the amount of lauric acid oxidized. The concentration of phosphate was measured via a blue reaction product formed with molybdate (Saheki et al. 1985). Formation of phosphate which exceeded the production of hydroxylated lauric acids indicated uncoupling was occurring. Both the conversion of lauric acid and the amount of products formed were quantified after TMS derivatization by GC-MS analysis (Scheps et al. 2013). Three monohydroxylation products of lauric acid were identified via the characteristic elution and fragmentation patterns of their TMS derivatized products (Fig. S3 and Fig. S4) and were formed at varying concentrations. The products were identified as 11-hydroxylauric acid (2), 10-hydroxylauric acid (3), and 9-hydroxylauric acid (4).
Conversions of lauric acid by PTDH-P450BM3 reached completion after less than 2 h with a yield of 2.2 mM of combined monohydroxylation products in a ratio of 32.8% 9-hydroxylauric acid (4), 30.7% 10-hydroxylauric acid (3), and 36.7% 11-hydroxylauric acid (2). Excess phosphate, compared to the total amount of hydroxylated products formed, was produced mainly after conversion of lauric acid was complete. After 4 h, 25% more phosphate than product was formed, but during the first 1.5 h, only 10% phosphate production was uncoupled from substrate hydroxylation and uncoupling was still less earlier in the reaction (<5%). From the amount of monohydroxylated products formed, it was calculated back that phosphite supplied electrons for 44 turnovers of the NADPH cofactor. Even more turnovers of NADPH occurred, if the uncoupling is taken into consideration (Fig. 4a, b).
Fig. 4.
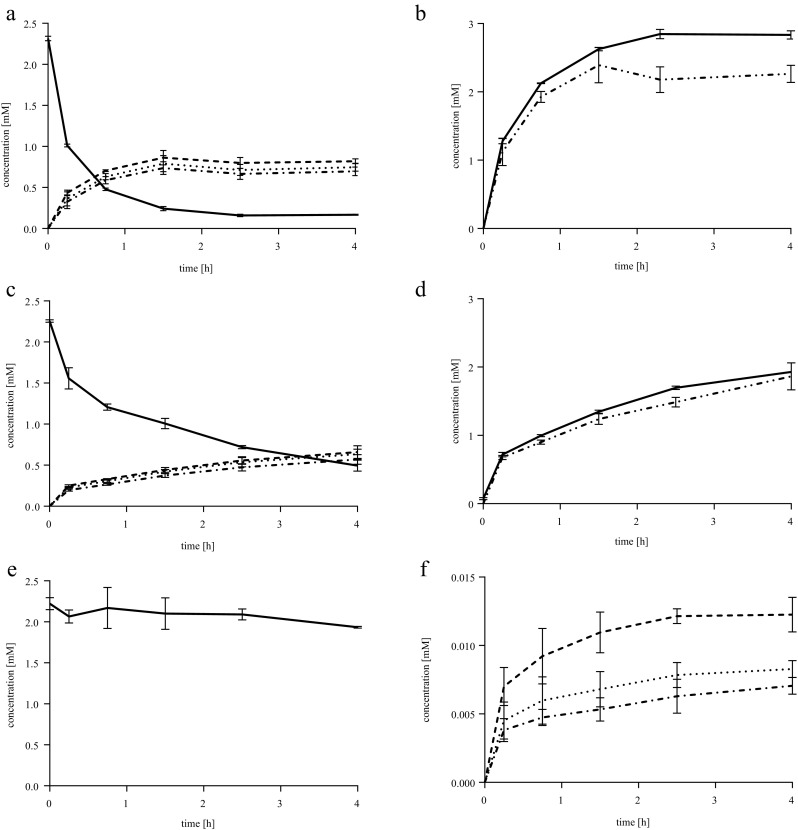
Conversions of lauric acid by different P450BM3 systems. All reactions contained 2.25 mM lauric acid, 4.0 mM phosphite, and 50 μM NADPH, thus making any product formation above 0.05 mM dependent on phosphite oxidation. a, b PTDH-P450BM3. c, d PTDH + P450BM3. e, f P450BM3. Samples were taken and analyzed by GC-MS to determine substrate depletion (a, c, and e; solid line) and product formation (a, c, and f) (9-hydroxylauric acid, dotted line; 10-hydroxylauric acid, dotted dashed line; 11-hydroxylauric acid, dashed line). In b and d, uncoupling was examined by following phosphate production measured with the molybdate assay (solid lines) and comparing it to total product formation determined by GC-MS (double dotted dashed line). For P450BM3 conversions, only a background of phosphate was detected (data not shown). All samples were measured in triplicate. Controls were reaction mixtures without enzyme and reaction mixtures without NADPH
Conversions performed with non-fused PTDH and P450BM3 were less efficient than with the fusion enzyme (production of 1.6 mM monohydroxylated lauric acid after 2.5 h in comparison to 2.2 mM for the fusion enzyme). After 4 h, 1.86 mM of the combined hydroxylation products were formed with a similar ration between 9-, 10-, and 11-hydroxylauric acid as observed for the fused enzyme. The reaction did not reach completion after 4 h. Thus, phosphate production was still coupled to the product formation (approx. 6% uncoupling). In the time frame analyzed, at least 36 turnovers of the NADPH cofactor occurred (Fig. 4c, d).
The P450BM3 without cofactor regeneration system was not able to drive the reaction to a product yield near the concentration of cofactor added (only 27.6 μM hydroxylated product formed, with 1.94 mM of lauric acid remaining, when 50 μM NADPH were supplied). With 30.0% 9-hydroxylauric acid (4), 25.6% 10-hydroxylauric acid (3), and 44.4% 11-hydroxylauric acid (2), the latter was favored more than in the other two reactions (Fig. 4e, f). As expected, no significant phosphate was produced in reactions without PTDH (data not shown).
Interestingly, in controls without NADPH added to the reaction, PTDH-P450BM3 and PTDH + P450BM3 still formed small amounts of product (16.4 and 10.4 μM of total monohydroxylated products), whereas no monohydroxylated products were formed in the controls with P450BM3. Possibly, these conversions are driven by phosphite and minor amounts of NADPH and/or NADP+ co-purified with the enzyme.
Discussion
While the natural fusion of the P450 oxygenase domain to the BMR domain in P450BM3 already makes this P450 a convenient system for oxidative biocatalysis, a NADPH regenerating system is still required for its efficient application. With the design of PTDH-P450BM3, a fully self-sufficient system for phosphite-driven conversion of substrates by P450BM3 was engineered. It utilizes the reducing power of phosphite to generate NADPH from NADP+, which then donates electrons into the P450 reaction cycle for oxidation. This simplifies enzyme handling by providing the benefits of a single biocatalyst, without compromising the catalytic properties of either the PTDH or P450BM3.
Specific activity was fully retained at lower enzyme concentrations for PTDH-P450BM3 compared to P450BM3, indicating a higher affinity for oligomerization. An increase in the specific activity was observed for both enzymes in the fusion. Fogle et al. postulated that product release is the rate-limiting step for the variant stabilized and optimized for the conversion of NADP+ (referred to as 12x-PTDH, lacking mutation A176R compared to the variant fused to P450BM3) (Fogle and van der Donk 2007). The C-terminal modification of the PTDH might lead to structural changes that promote the release of NADPH from the active site. For the P450BM3, the product release and the electron transfer from the BMR to the heme domain are described as rate limiting (Whitehouse et al. 2012). Considering that the PTDH is N-terminally fused to the heme domain of P450BM3, a structural change that influences product release seems likely. Additionally, the assembly of the BMR domain of one P450BM3 with the heme domain of a second P450BM3 may be promoted by the fusion, and therefore the electron transfer may improve.
Phosphite-dependent conversion of omeprazole and rosiglitazone as exemplary drugs by PTDH-P450BM3 showed comparable turnover to NADPH-driven reactions. The total turnover was slightly higher than with conversions containing P450BM3 and NADPH in excess. This indicates that the fused enzyme is more efficient despite the observation that PTDH showed a lower activity than P450BM3 when testing the fused domains separately. This can be explained by the relatively low rate of turnover of these drugs by P450BM3. Butler et al. reported conversion of omeprazole by wild-type P450BM3 implementing glucose-6-phosphate dehydrogenase as a cofactor recycling system. In reaction mixtures with purified enzyme (0.1 μM) and the same concentration of substrate as used here (10 μM), less than 1% product formation was observed within 30 min. Higher conversions are described for mutants in the heme domain of P450BM3 (e.g., F87 V ∼50%) (Butler et al. 2013). In this study, we report a higher turnover for the wild-type P450BM3 without any cofactor recycling system (16%) and a slightly higher turnover for the fusion enzyme (25–26%). The higher yields are most likely caused by the higher enzyme concentrations that we employed and a prolonged reaction time. Clearly, it would be attractive to incorporate known mutations in P450BM3 in order to generate a powerful phosphite-driven PTDH-P450BM3 capable of generating drug metabolites.
The p-hydroxylation of the pyridine ring and N-demethylation of rosiglitazone are both major reactions when considering the metabolism of these drugs in humans while the N-oxidation is not listed as a relevant pathway (Cox et al. 2000). N-demethylation is mainly performed by the human P450 2C8 (Baldwin et al. 1999). Rosiglitazone conversions of up to 99% by mutants of P450BM3 using glucose-6-phosphate dehydrogenase for cofactor recycling have been described by Reinen et al. 2011. In conversions by PTDH-P450BM3, less than 3% of rosiglitazone was detected in reactions after 4 h, suggesting that conversions by the fusion enzyme were comparably efficient. Integrating the mutations described by Reinen et al. might increase the total turnover slightly—ratios for demethylation and N-oxidation products were not described. Interestingly, we observed that the ratio of two formed metabolites from rosiglitazone depended on which P450BM3-based biocatalyst was used; native P450BM3 favored N-oxidation, whereas the fusion enzyme showed a strong preference for formation of one of the major human metabolites, the demethylated product. The N-terminal region of the unfused P450BM3 is described as one of the regions that are involved in the switch of conformation between the open and closed state (Ravichandran et al. 1993). We therefore assume that a fusion to the N-terminus might induce minor changes in the structure of the active site or the substrate access channel that influence regioselectivity.
With lauric acid as a model compound for conversions at higher substrate concentrations (2.25 mM), the fused enzyme proved superior to the non-fused system. Using PTDH-P450BM3, the conversion was complete within 2 h, while using non-fused P450BM3, the reaction did not reach completion within 4 h. In both cases, the reaction was driven far beyond the amount of NADPH added, while significant uncoupling of phosphate production from the formation of the monohydroxylated products only took place after the reaction had reached completion. The ratio of monohydroxylation products of lauric acid was in agreement with what was reported by Miura et al. for the NADPH-driven conversion by wild-type P450BM3 (Miura and Fulco 1975).
Watanabe et al. investigated a fusion of PTDH to the “PCNA-utilized protein complex of P450 and its two electron transfer-related proteins” (PUPPET) for the regeneration of NADH in conversions by P450cam (Watanabe et al. 2013). The PTDH-PUPPET fusion retained, similar to the PTDH-P450BM3 fusion, kinetic parameters comparable to the parent constructs, and the consumption of camphor was driven on phosphite and NAD+. The rate of NAD+ reduction by PTDH was lower than the rate of NADH oxidation by the P450, as we report for the PTDH-P450BM3 system. In both cases, cofactor reduction was assumed to be the rate-limiting step. In biotransformations with 1 mM D-camphor, 20 μM NAD+, and 10 mM phosphite, a turnover of the cofactor of 46 was reached, while here more than 50 turnovers based on phosphate production in conversions of lauric acid are described. Watanabe et al. achieved the same turnover in reactions with the non-fused PTDH after 8 h reaction time, suggesting that unfused PTDH and P450BM3 may reach the same turnover as the fused system over an extended incubation. While Watanabe et al. implement a NADH-dependent system, here the recycling of the more expensive NADPH is investigated. This demonstrates the capability of the PTDH to cooperate with different electron donor systems of P450s. Coupling to various P450s enhances the scope of substrate conversions that can be driven on phosphite.
A limiting factor in the conversions reported by Watanabe et al. was a loss of activity of the PTDH during incubations with phosphite. The use of a stabilized version of the PTDH for our studies might have prevented this effect for conversions by PTDH-P450BM3. The main limiting factor was the substrate solubility for rosiglitazone and omeprazole and substrate and/or product inhibition in case of lauric acid. It is expected that these bottlenecks can be overcome by enzyme immobilization, as described for the PTDH-PUPPET fusion and non-fused P450BM3 (Maurer et al. 2003; Weber et al. 2010; Lee et al. 2014; Tan et al. 2015).
Overall, these findings suggest that the designed fusion enzyme PTDH-P450BM3 is an efficient bifunctional biocatalyst with potential application in drug metabolism studies and for the production of fine chemicals.
Electronic supplementary material
(PDF 282 kb)
Acknowledgements
This project was funded by the European Union FP7 People Initiative as the Marie-Curie International Training Network P4FIFTY 289217 with an Early Researcher position (to N. Beyer) and an Experienced Researcher position (to J. K. Kulig). We thank Prof. B. Hauer (University of Stuttgart) for supplying the vector pJOE-P450BM3 encoding the wild-type gene.
Compliance with ethical standards
Funding
The Early Researcher position of N. Beyer and the Experienced Researcher position of J. K. Kulig was funded by the European Union FP7 People Initiative as the Marie-Curie International Training Network P4FIFTY (289217).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals by any of the authors.
References
- Ahmed F, Al-Mutairi EH, Avery KL, Cullis PM, Primrose WU, Roberts GCK, Willis CL. An unusual matrix of stereocomplementarity in the hydroxylation of monohydroxy fatty acids catalysed by cytochrome P-450 from Bacillus megaterium with potential application in biotransformations. Chem Commun. 1999;20:2049–2050. doi: 10.1039/a905974a. [DOI] [Google Scholar]
- Baldwin SJ, Clarke SE, Chenery RJ. Characterisation of the cytochrome P450 enzymes involved in the in vitro metabolism of granisetron. J Clin Pharmacol. 1999;48:424–432. doi: 10.1046/j.1365-2125.1999.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt R. Cytochromes P450 as versatile biocatalysts. J Biotechnol. 2006;124:128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Butler CF, Peet C, Mason AE, Voice MW, Leys D, Munro AW. Key mutations alter the cytochrome P450 BM3 conformational landscape and remove inherent substrate bias. J Biol Chem. 2013;288:25387–25399. doi: 10.1074/jbc.M113.479717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila JH, Wei S, Helvig C, Falck JR, Belosludtsev Y, Truan G, Graham-Lorence SE, Peterson JA. The highly stereoselective oxidation of polyunsaturated fatty acids by cytochrome P450BM-3. J Biol Chem. 1996;271:22663–22671. doi: 10.1074/jbc.271.37.22663. [DOI] [PubMed] [Google Scholar]
- Chen C-KJ, Shokhireva TK, Berry RE, Zhang H, Walker FA. The effect of mutation of F87 on the properties of CYP102A1-CYP4C7 chimeras: altered regiospecificity and substrate selectivity. J Biol Inorg Chem. 2008;13:813–824. doi: 10.1007/s00775-008-0368-5. [DOI] [PubMed] [Google Scholar]
- Costas AMG, White AK, Metcalf WW. Purification and characterization of a novel phosphorus-oxidizing enzyme from Pseudomonas stutzeri WM88. J Biol Chem. 2001;276:17429–17436. doi: 10.1074/jbc.M011764200. [DOI] [PubMed] [Google Scholar]
- Cox PJ, Ryan DA, Hollis FJ, Harris A-M, Miller AK, Vousden M, Cowley H. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–780. [PubMed] [Google Scholar]
- Dudek HM, Popken P, van Bloois E, Duetz WA, Fraaije MW. A generic, whole-cell-based screening method for Baeyer-Villiger monooxygenases. J Biomol Screen. 2013;18:678–687. doi: 10.1177/1087057113480390. [DOI] [PubMed] [Google Scholar]
- Falck J, Reddy YK, Haines DC, Reddy KM, Krishna UM, Graham S, Murry B, Peterson JA. Practical, enantiospecific syntheses of 14,15-EET and leukotoxin B (vernolic acid) Tetrahedron Lett. 2001;42:4131–4133. doi: 10.1016/S0040-4039(01)00694-3. [DOI] [Google Scholar]
- Fogle EJ, van der Donk WA (2007) Pre-steady-state studies of phosphite dehydrogenase demonstrate that hydride transfer is fully rate limiting. Biochemistry 46:13101–13108 [DOI] [PMC free article] [PubMed]
- Gillam EMJ, Hayes MA. The evolution of cytochrome P450 enzymes as biocatalysts in drug discovery and development. Curr Top Med Chem. 2013;13:2254–2280. doi: 10.2174/15680266113136660158. [DOI] [PubMed] [Google Scholar]
- Hannemann F, Bichet A, Ewen KM, Bernhardt R. Cytochrome P450 systems—biological variations of electron transport chains. Biochim Biophys Acta. 2007;1770:330–344. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Johannes TW, Woodyer RD, Zhao H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase. Biotechnol Bioeng. 2007;96:18–26. doi: 10.1002/bit.21168. [DOI] [PubMed] [Google Scholar]
- Kühnel K, Maurer SC, Galeyeva Y, Frey W, Laschat S, Urlacher VB. Hydroxylation of dodecanoic acid and (2R,4R,6R,8R)-tetramethyldecanol on a preparative scale using an NADH-dependent CYP102A1 mutant. Adv Synth Catal. 2007;349:1451–1461. doi: 10.1002/adsc.200700054. [DOI] [Google Scholar]
- Kulig JK, Spandolf C, Hyde R, Ruzzini AC, Eltis LD, Grönberg G, Hayes MA, Grogan G. A P450 fusion library of heme domains from Rhodococcus jostii RHA1 and its evaluation for the biotransformation of drug molecules. Bioorg Med Chem. 2015;23:5603–5609. doi: 10.1016/j.bmc.2015.07.025. [DOI] [PubMed] [Google Scholar]
- Lalman JA, Bagley DM. Extracting long-chain fatty acids from a fermentation medium. J Am Oil Chem Soc. 2004;81:105–110. doi: 10.1007/s11746-004-0866-y. [DOI] [Google Scholar]
- Landwehr M, Hochrein L, Otey CR, Kasrayan A, Bäckvall JE, Arnold FH. Enantioselective α-hydroxylation of 2-arylacetic acid derivatives and buspirone catalyzed by engineered cytochrome P450 BM-3. J Am Chem Soc. 2006;128:6058–6059. doi: 10.1021/ja061261x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Nam DH, Lee SH, Park JH, Park SJ, Lee SH, Park CB, Jeong KJ. New platform for cytochrome P450 reaction combining in situ immobilization on biopolymer. Bioconjug Chem. 2014;25:2101–2104. doi: 10.1021/bc500404j. [DOI] [PubMed] [Google Scholar]
- Lundemo MT, Woodley JM. Guidelines for development and implementation of biocatalytic P450 processes. Appl Microbiol Biotechnol. 2015;99:2465–2483. doi: 10.1007/s00253-015-6403-x. [DOI] [PubMed] [Google Scholar]
- Maurer SC, Schulze H, Schmid RD, Urlacher V. Immobilisation of P450BM-3 and an NADP(+) cofactor recycling system: towards a technical application of heme-containing monooxygenases in fine chemical synthesis. Adv Synth Catal. 2003;345:802–810. doi: 10.1002/adsc.200303021. [DOI] [Google Scholar]
- Meunier B, De Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- Miura Y, Fulco AJ (1975) W-1, w-2 and w-3 hydroxylation of long-chain fatty acids, amides and alcohols by a soluble enzyme system from Bacillus megaterium. Biochim Biophys Acta:305–317 [DOI] [PubMed]
- Munro AW, Daff S, Coggins JR, Lindsay JG, Chapman SK. Probing electron transfer in flavocytochrome P-450 BM3 and its component domains. Eur J Biochem. 1996;239:403–409. doi: 10.1111/j.1432-1033.1996.0403u.x. [DOI] [PubMed] [Google Scholar]
- Munro AW, Leys DG, McLean KJ, Marshall KR, Ost TWB, Daff S, Miles CS, Chapman SK, Lysek DA, Moser CC, Page CC, Dutton PL. P450 BM3: the very model of a modern flavocytochrome. Trends Biochem Sci. 2002;27:250–257. doi: 10.1016/S0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- Munro AW, Girvan HM, Mason AE, Dunford AJ, McLean KJ. What makes a P450 tick? Trends Biochem Sci. 2013;38:140–150. doi: 10.1016/j.tibs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Murataliev MB, Trinh LN, Moser LV, Bates RB, Walker FA. Chimeragenesis of the fatty acid binding site of cytochrome P450BM3. Replacement of residues 73–84 with the homologous residues from the insect cytochrome P450 CYP4C7. Biochemistry. 2004;43:1771–1780. doi: 10.1021/bi035674b. [DOI] [PubMed] [Google Scholar]
- Neeli R, Girvan HM, Lawrence A, Warren MJ, Leys D, Scrutton NS, Munro AW. The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett. 2005;579:5582–5588. doi: 10.1016/j.febslet.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Noble MA, Miles CS, Chapman SK, Lysek DA, MacKay AC, Reid GA, Hanzlik RP, Munro AW. Roles of key active-site residues in flavocytochrome P450 BM3. Biochem J. 1999;339:371–379. doi: 10.1042/bj3390371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- Peters MW, Meinhold P, Glieder A, Arnold FH. Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J Am Chem Soc. 2003;125:13442–13450. doi: 10.1021/ja0303790. [DOI] [PubMed] [Google Scholar]
- Ravichandran KG, Boddupalli SS, Hasermann CA, Peterson JA, Deisenhofer J. Crystal structure of hemoprotein domain of P450BM-3, a prototype for microsomal P450’s. Science. 1993;261:731–736. doi: 10.1126/science.8342039. [DOI] [PubMed] [Google Scholar]
- Reinen J, Van Leeuwen JS, Li Y, Sun L, Grootenhuis PDJ, Decker CJ, Saunders J, Vermeulen NPE, Commandeur JNM. Efficient screening of cytochrome P450 BM3 mutants for their metabolic activity and diversity toward a wide set of drug-like molecules in chemical space. Drug Metab Dispos. 2011;39:1568–1576. doi: 10.1124/dmd.111.039461. [DOI] [PubMed] [Google Scholar]
- Relyea HA, van der Donk WA (2005) Mechanism and applications of phosphite dehydrogenase. Bioorg Chem 33:171–189 [DOI] [PubMed]
- Ruettinger RT, Wen LP, Fulco J. Coding nucleotide, 5’ regulatory, and deduced amino acid sequences of P-450(BM-3), a single peptide cytochrome P-450:NADPH-P-450 reductase from Bacillus megaterium. J Biol Chem. 1989;264:10987–10995. [PubMed] [Google Scholar]
- Saheki S, Takeda A, Shimazu T. Assay of inorganic phosphate in the mild pH range, suitable for measurement of glycogen phosphorylase activity. Anal Biochem. 1985;148:277–281. doi: 10.1016/0003-2697(85)90229-5. [DOI] [PubMed] [Google Scholar]
- Scheps D, Honda Malca S, Richter SM, Marisch K, Nestl BM, Hauer B. Synthesis of ω-hydroxy dodecanoic acid based on an engineered CYP153A fusion construct. Microb Biotechnol. 2013;6:694–707. doi: 10.1111/1751-7915.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schewe H, Kaup B-A, Schrader J. Improvement of P450BM-3 whole-cell biocatalysis by integrating heterologous cofactor regeneration combining glucose facilitator and dehydrogenase in E. coli. Appl Microbiol Biotechnol. 2008;78:55–65. doi: 10.1007/s00253-007-1277-1. [DOI] [PubMed] [Google Scholar]
- Schewe H, Holtmann D, Schrader J. P450(BM-3)-catalyzed whole-cell biotransformation of a-pinene with recombinant Escherichia coli in an aqueous-organic two-phase system. Appl Microbiol Biotechnol. 2009;83:849–857. doi: 10.1007/s00253-009-1917-8. [DOI] [PubMed] [Google Scholar]
- Schwaneberg U, Otey C, Cirino PC, Farinas E, Arnold FA. Cost-effective whole-cell assay for labratory evolution of hydroxylases in Escherichia coli. J Biomol Screen. 2001;6:111–117. doi: 10.1177/108705710100600207. [DOI] [PubMed] [Google Scholar]
- Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Sulistyaningdyah WT, Ogawa J, Li QS, Maeda C, Yano Y, Schmid RD, Shimizu S. Hydroxylation activity of P450BM-3 mutant F87V towards aromatic compounds and its application to the synthesis of hydroquinone derivatives from phenolic compounds. Appl Microbiol Biotechnol. 2005;67:556–562. doi: 10.1007/s00253-004-1761-9. [DOI] [PubMed] [Google Scholar]
- Tan CY, Hirakawa H, Nagamune T. Supramolecular protein assembly supports immobilization of a cytochrome P450 monooxygenase system as water-insoluble gel. Sci Rep. 2015;5:1–8. doi: 10.1038/srep08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres Pazmiño DE, Riebel A, de Lange J, Rudroff F, Mihovilovic MD, Fraaije MW. Efficient biooxidations catalyzed by a new generation of self-sufficient Baeyer-Villiger monooxygenases. Chembiochem. 2009;10:2595–2598. doi: 10.1002/cbic.200900480. [DOI] [PubMed] [Google Scholar]
- van Beek HL, Beyer N, Janssen DB, Fraaije MW. Lyophilization conditions for the storage of monooxygenases. J Biotechnol. 2015;203:41–44. doi: 10.1016/j.jbiotec.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Volz TJ, Rock DA, Jones JP. Evidence for two different active oxygen species in cytochrome P450 BM3 mediated sulfoxidation and N-dealkylation reactions. J Am Chem Soc. 2002;124:9724–9725. doi: 10.1021/ja026699l. [DOI] [PubMed] [Google Scholar]
- Waddell WJ. A simple ultraviolet spectrophotometric method for the determination of protein. J Lab Clin Med. 1956;48:311–314. [PubMed] [Google Scholar]
- Watanabe H, Hirakawa H, Nagamune T. Phosphite-driven self-sufficient cytochrome P450. ChemCatChem. 2013;5:3835–3840. doi: 10.1002/cctc.201300445. [DOI] [Google Scholar]
- Weber E, Sirim D, Schreiber T, Thomas B, Pleiss J, Hunger M, Gläser R, Urlacher VB. Immobilization of P450 BM-3 monooxygenase on mesoporous molecular sieves with different pore diameters. J Mol Catal B Enzym. 2010;64:29–37. doi: 10.1016/j.molcatb.2010.01.020. [DOI] [Google Scholar]
- Whitehouse CJC, Bell SG, Wong L-L. P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev. 2012;41:1218–1260. doi: 10.1039/C1CS15192D. [DOI] [PubMed] [Google Scholar]
- Woodyer R, van der Donk WA, Zhao H (2003) Relaxing the nicotinamide cofactor specificity of phosphite dehydrogenase by rational design. Biochemistry 42:11604–11614 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 282 kb)