

Abstract
Background
This study aimed to assess the significance of serum levels of vascular endothelial growth factor (VEGF) in non-alcoholic fatty liver disease (NAFLD).
Methods
Sixty-seven consecutive NAFLD patients and 47 healthy controls who visited our liver clinics between May 2008 and December 2010 were included. The NAFLD diagnosis required elevated alanine aminotransferase and/or gamma-glutamyl transpeptidase levels, evidence of hepatic steatosis on ultrasound and/or liver histology, and exclusion of other causes of liver injury. Serum VEGF levels were determined by an enzyme immunoassay. Liver biopsy was obtained in 34 NAFLD patients. Histological lesions were scored by a liver histopathologist.
Results
Serum VEGF levels tended to be lower in matched NAFLD patients than in healthy controls (296±146 vs. 365±186 pg/mL, P=0.092); levels in patients with non-alcoholic steatohepatitis (NASH) also tended to be lower than in those with simple fatty liver (FL) (279±149 vs. 359±190 pg/mL, P=0.095); while VEGF levels were significantly lower in NASH patients than in healthy controls (279±149 vs. 365±186 pg/mL, P=0.041). VEGF levels offered poor predictability for the differentiation between NAFLD patients and controls or between NASH and FL patients. However, patients with high VEGF levels (≥300 pg/mL) were significantly more likely to have FL, either in the total NAFLD population (67% vs. 35%, P=0.019) or in the 34 NAFLD patients with liver biopsy (57% vs. 15%, P=0.023), while those with high VEGF levels also had a significantly lower mean fibrosis score (0.7±0.9 vs. 1.6±1.0, P=0.017).
Conclusion
Our data suggest that serum VEGF levels are equally high in healthy controls and in patients with simple fatty liver, but tend to decrease when NASH develops.
Keywords: Nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, vascular endothelial growth factor, vascular endothelial growth factor receptors, angiogenesis markers
Introduction
Non-alcoholic fatty liver disease (NAFLD) represents a major public health problem with a prevalence as high as 30% in many populations [1,2]. Liver biopsy is considered to be the gold standard for the assessment of the presence and severity of hepatic injury in NAFLD, but it is an invasive procedure that has also been associated with sampling errors [3,4]. Therefore, there is growing scientific interest in imaging studies and serum-based assays that aim to detect the presence of NAFLD, and to distinguish simple fatty liver (FL) from non-alcoholic steatohepatitis (NASH), the more severe and progressive type of NAFLD [5].
Considering that NASH is characterized by marked activation of inflammatory cells [6-9] and upregulation of several soluble inflammatory mediators, the role of different cytokines and chemokines in NAFLD has been evaluated by several groups [10]. Liver inflammation, which is a key element of NASH, has been shown to be triggered by activated cytokines induced by lipotoxicity. Although the pathogenesis of NASH is still not fully understood, some studies suggested that angiogenesis might play a role in its progression [11,12]. Inflammatory cells are able to initiate angiogenesis through different pathways, thereby contributing to the formation of new vasculature in the liver [13,14]. Although angiogenesis has been well documented in patients with chronic viral hepatitis [15,16], information regarding angiogenesis in NAFLD is very limited.
Vascular endothelial growth factor (VEGF) is a proangiogenic factor implicated in the angiogenetic process. The aim of this study was to evaluate the possible significance of serum VEGF levels in patients with NAFLD.
Patients and methods
Sixty-seven consecutive patients with NAFLD who visited our outpatient liver clinics between May 2008 and December 2010 were included. The diagnosis of NAFLD was based on the following criteria: elevated alanine aminotransferase (ALT) and/or gamma-glutamyl transpeptidase (GGT) levels; evidence of hepatic steatosis on ultrasonography and/or liver histology; and exclusion of other causes of liver injury. In particular, all patients had negative serological markers for hepatitis B (HBsAg), hepatitis C (anti-HCV) and human immunodeficiency virus (anti-HIV), weekly alcohol consumption less than 210 g for men or 140 g for women, no use of potentially hepatotoxic agents, no evidence of metabolic or autoimmune liver disease, and absence of any known systemic disease with potential liver involvement. The history of alcohol use was taken from the patients and was confirmed by the patients’ relatives or friends.
Demographic characteristics and medical history were recorded for all patients, together with any history of known arterial hypertension or the presence of diabetes mellitus, diagnosed on the basis of antidiabetic treatment and/or fasting glucose >126 mg/dL on more than one occasion.
Forty-seven healthy controls matched for age, sex and body mass index (BMI) with 47 of the aforementioned 67 patients were also enrolled. Controls were either subjects who visited the outpatient clinics of our hospital for routine examinations during the same period, or hospital staff members. Controls had normal glucose metabolism and liver biochemistry and no evidence of hepatic steatosis on abdominal ultrasound.
The study was approved by the Hippokratio Hospital Institutional Review Board.
Anthropometric assessments
Participants’ body weight was measured with a digital scale (Seca Robusta 813, Hamburg, Germany) to the nearest 100 g and height was measured to the nearest 0.5 cm. Waist circumference (WC) was tape-measured to the nearest 0.1 cm. Increased WC was defined as >102 cm for men and >88 cm for women.
Laboratory markers
The laboratory data recorded included complete blood count, prothrombin time, urea, creatinine, urate, liver enzymes (ALT, aspartate aminotransferase, alkaline phosphatase, and GGT), total protein, albumin, as well as detection of HBsAg, anti-HBc, anti-HBs, anti-HCV, anti-HIV, and liver autoantibodies.
Serum VEGF levels were measured using a commercially available sensitive enzyme-linked immunosorbent assay (ELISA, Quantikine/immunoassay kit, R&D Systems, Minneapolis, MN, USA). The intra- and inter-assay coefficients of variation were <7% and <10%, respectively. Serum levels of caspase-generated fragments of keratin-18 (K18) as well as of and soluble Fas (sFas) were also measured by ELISA based immunoassays (M30-Apoptosense ELISA assay, PEVIVA, Alexis, Grünwald, Germany and Human Fas/TNFRSF6 Quantikine ELISA Kit, R&D Systems, Minneapolis, MN, USA, respectively).
Definitions
Patients were considered obese if they had a BMI ≥30 kg/m2. Metabolic syndrome was defined according to the National Cholesterol Education Program: Adult Treatment Panel III criteria [17].
Patients with NAFLD were classified into those with FL and those with NASH, according to the widely accepted histological criteria described below and/or to a recently published formula based on serum K18 and sFas levels, shown to correctly classify 88% of NAFLD patients [18].
Transient elastography (TE)
Liver stiffness was measured (in kPa) using the standard probe of TE (Fibroscan, Echosens, France) in 48 of the 67 patients, but the results were reliable in only 46. The result was considered reliable if 10 successful measurements were obtained, with a success ratio >60% and a ratio of interquartile range to mean stiffness <30%. For patients who underwent both TE and liver biopsy, liver stiffness measurement was performed a few hours before liver biopsy in most or within 4 weeks before or after liver biopsy in some patients.
Liver histology
Adequate liver biopsies were obtained in 34 of the 67 NAFLD patients. Histological lesions were classified according to the Brunt classification by one blinded liver histopathologist (DT). A liver biopsy was considered to be adequate if at least 6 portal tracts were identified and the specimen length was ≥1.5 cm. The diagnosis of NASH was made according to the Brunt classification criteria [19], as modified by Kleiner et al [20]. Global grading of necroinflammatory activity and staging of fibrosis were assessed according to Brunt et al [19]. Severity of steatosis and NAFLD activity score were evaluated according to Kleiner et al [20].
Statistical analysis
Quantitative variables with normal distribution were expressed as mean values ± standard deviation (SD) and those with abnormal distribution as median values (range). Statistical analysis was performed using the Mann-Whitney test for comparisons of quantitative variables between groups, Spearman’s coefficient for correlations of quantitative variables, and a two-tailed Fisher’s exact test for qualitative data. The accuracy of VEGF levels for predicting early (stage: 0-1) or advanced (stage: 2-4) liver disease was assessed by the area under the receiver operating characteristic curve (AUROC). A two-tailed P-value of <0.05 was considered to be statistically significant.
Results
Of the 67 NAFLD patients, 21 (31%) were diagnosed with FL and 46 (69%) with NASH. FL and NASH were diagnosed in 11 (32%) and 23 (68%) of the 34 patients who underwent liver biopsy, and in 10 (30%) and 23 (70%) of the remaining 33 patients without a liver biopsy according to the K-18/sFas formula. In the 34 patients with a liver biopsy, the accuracy of the K-18/sFas formula in differentiating between the histological presence of FL and NASH was 91% (31/34).
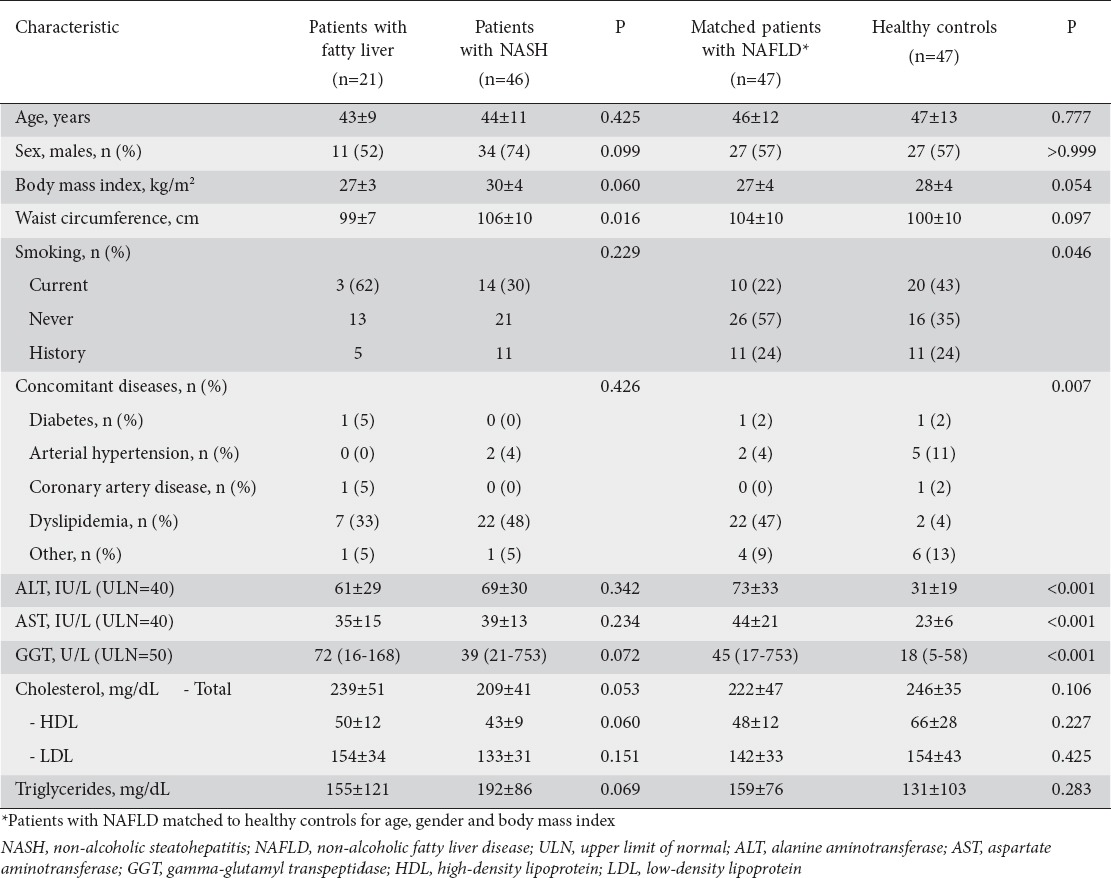
Baseline patient characteristics are shown in Table 1. Patients with NASH had higher WC (107±10 vs. 103±13 cm, P=0.016) compared to patients with FL. Though the differences were not statistically significant, NASH patients also tended to have higher BMI (30±4 vs. 29±5 kg/m2, P=0.06), higher triglyceride levels (175±75 vs. 155±104 mg/dL, P=0.069) and lower HDL levels (44±10 vs. 51±13 mg/dL, P=0.060) compared to those with FL.
Table 1.
Demographic, anthropometric, clinical and laboratory characteristics of the study population
In the 47 NAFLD patients who were matched to healthy controls, VEGF levels were lower in patients than in controls (296±146 vs. 365±186 pg/mL) but the difference did not reach statistical significance (P=0.092). Given that no difference in the VEGF levels was found between patients with FL and controls (336±136 vs. 365±186 pg/mL, P=0.532), the previous finding was attributed to patients with NASH. In particular, patients with NASH had significantly lower VEGF levels compared to healthy subjects (279±149 vs. 365±186 pg/mL, P=0.041). In addition, serum VEGF levels tended to be lower in the 46 patients with NASH compared to the 21 patients with FL (289±147 vs. 359±190 pg/mL, P=0.095) (Fig. 1). A similar trend in serum VEGF levels was also observed in the 34 patients who underwent liver biopsy (FL: 326±130 vs. NASH: 253±149 pg/mL, P=0.098). These data suggest that serum VEGF levels tend to decrease with the progression from simple steatosis to steatohepatitis.
Figure 1.
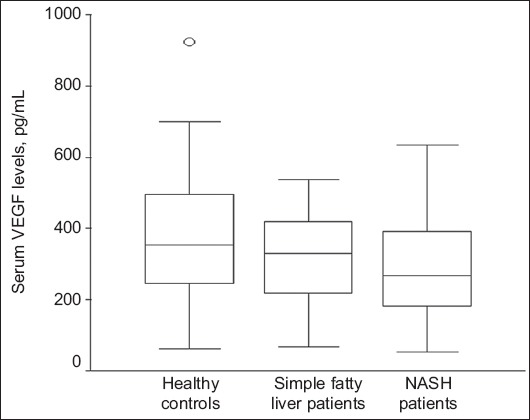
Serum levels of vascular endothelial growth factor (VEGF) in healthy controls and in patients with simple fatty liver or non-alcoholic steatohepatitis (NASH)
Controls vs. NASH: P=0.041, fatty liver vs. NASH: P=0.095. Box and whisker plots express medians, and interquartile and overall ranges. The outlying values are plotted individually
Serum VEGF levels were not able to differentiate effectively between NAFLD patients and controls (AUROC: 0.601, 95% CI 0.486-0.715; P=0.092) (Fig. 2A). In the 67 NAFLD patients, serum VEGF levels also could not differentiate between FL and NASH patients (AUROC: 0.628, 95% CI 0.485-0.770; P=0.095) (Fig. 2B). In the 34 NAFLD patients with liver biopsy, VEGF levels were numerically but not statistically higher in patients with an early stage of fibrosis (0-1) than in those with an advanced stage (2-4) (302±141 vs. 239±149 pg/mL, P=0.129), while there was no significant correlation between VEGF levels and fibrosis stage (r=-0.268, P=0.125).
Figure 2.
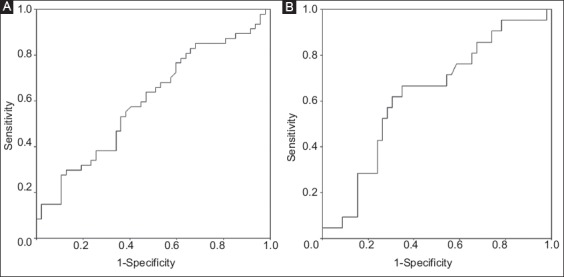
(A) Area under the receiving operating characteristic curve (AUROC) of serum levels of vascular endothelial growth factor for the discrimination between patients with non-alcoholic fatty liver disease and healthy controls (AUROC: 0.601, 95% CI 0.486-0.715; P=0.092) (B) or between patients with simple fatty liver and those with non-alcoholic steatohepatitis (AUROC: 0.628, 95% CI 0.485-0.770; P=0.095)
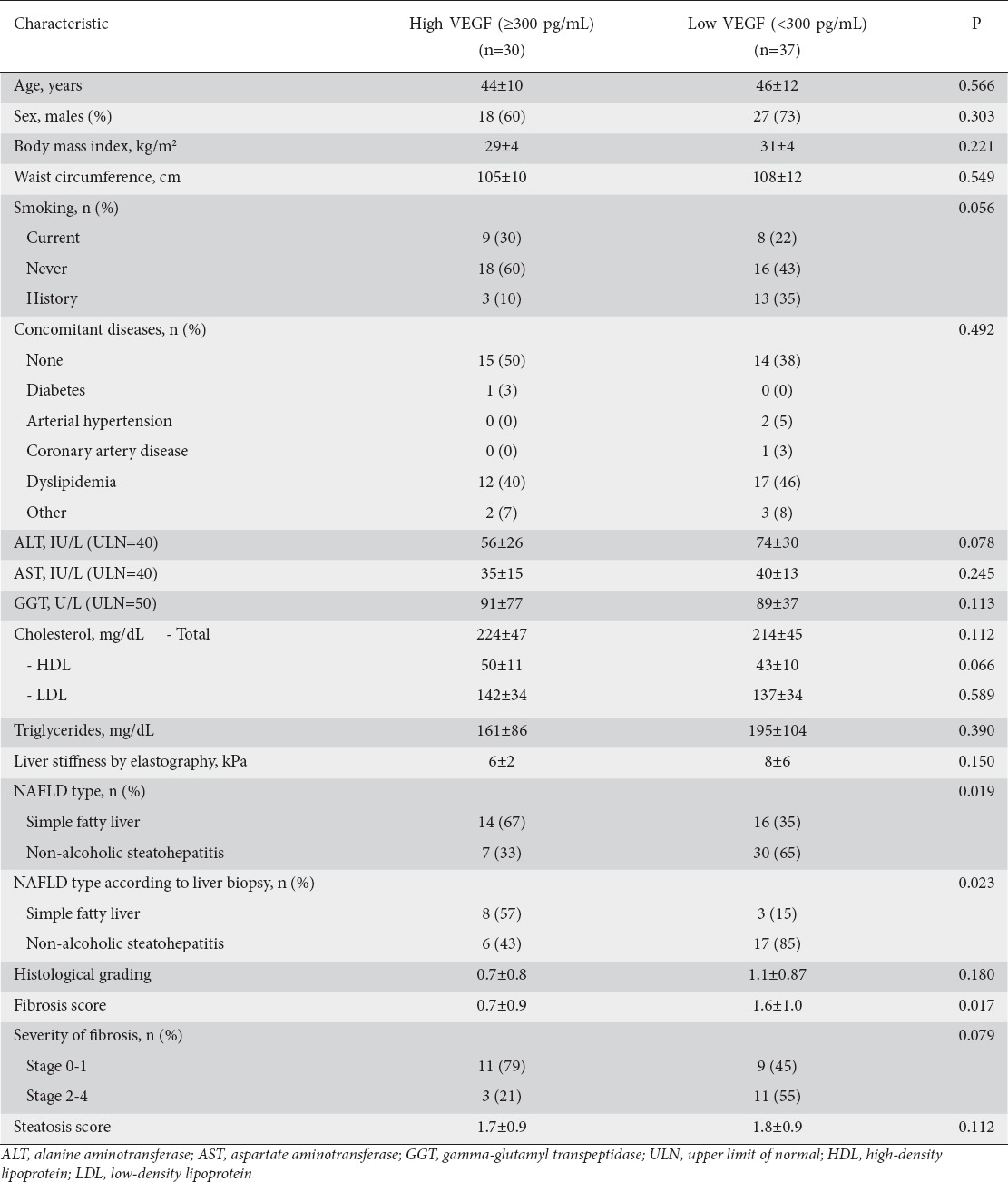
When we split our NAFLD patients according to their median VEGF value, patients with high (≥300 pg/mL) VEGF levels were found to have FL significantly more frequently, both in the total NAFLD population (67% vs. 35%, P=0.019) and in the 34 NAFLD patients with liver biopsy (57% vs. 15%, P=0.023). Patients with high VEGF levels also had a lower mean fibrosis score (0.7±0.9 vs. 1.6±1.0, P=0.017) (Table 2).
Table 2.
Vascular endothelial growth factor (VEGF) serum levels in patients with non-alcoholic fatty liver disease (NAFLD) in relation to their characteristics
No significant correlation was observed between serum VEGF levels and age (r=0.104, P=0.271), BMI (r=-0.109, P=0.247), sex (r=0.090, P=0.342), liver inflammation expressed by histological grading (r=-0,116, P=0.513), hepatic steatosis (r=0.182, P=0.319) or liver stiffness at elastography (r=-0.117, P=0.430). Additionally, no significant correlation was found between serum VEGF levels and cholesterol (r=0.176, P=0.526), triglyceride (r=0.064, P=0.616) or GGT values (r=0.064, P=0.526). Interestingly, serum VEGF levels had a significant inverse correlation with aspartate aminotransferase (r=-0.262, P=0.007) and ALT values (r=-0.275, P=0.005).
Discussion
Like all chronic liver diseases, NAFLD is characterized by a wide spectrum of inflammation and fibrosis. NASH, the most severe form of NAFLD, can progress to cirrhosis and may eventually lead to hepatocellular carcinoma [21-23]. The two-hit hypothesis, based on the initial intrahepatic accumulation of triglycerides followed at a second stage by inflammatory mediators, gut endotoxin, mitochondrial dysfunction and endoplasmic reticulum stress causing liver injury, has provided some explanation for the pathogenesis of NASH, but has recently been challenged. Currently, the pathophysiology of NAFLD is considered to be multifactorial, including a relation between changes in microvasculature and the progression of fibrosis [24-27]. Tissue damage from both fat accumulation and lipotoxicity results in reduced sinusoidal perfusion and changes in sinusoidal architecture. Additionally, several cytokines activated by lipotoxicity further induce the migration of inflammatory cells, which contribute to angiogenesis in the inflammatory foci [28]. During disease progression, fatty hepatocytes, tissue inflammation and perisinusoidal fibrosis constrict the sinusoidal lumens and impair the sinusoidal perfusion, leading to hypoxia [28,29]. All these events provoking liver damage can initiate angiogenesis [26]. As a consequence, angiogenesis leading to new vasculature formation may have prognostic value in disease progression. The idea that interfering with angiogenesis might be a potential target to avoid progression of liver disease has stimulated research into several markers for angiogenesis in chronic liver diseases.
Angiogenesis has been documented in cases of viral hepatitis, but information regarding angiogenesis in NAFLD is very limited. Angiogenesis in chronic hepatitis C has been reported to be induced more frequently than in patients with chronic hepatitis B or healthy controls [30]. Salcedo et al concluded that serum VEGF, angiopoietin 2 and tyrosine kinase 2 levels could be useful as noninvasive markers of response to therapy and disease progression in patients with chronic hepatitis C [31]. Moreover, enhanced angiogenesis has been described in association with hepatocellular carcinoma in patients with chronic hepatitis and cirrhosis [32,33]. In our study, VEGF serum levels did not differ between patients with FL and healthy controls, but were found to be lower in NASH patients compared to healthy controls (P=0.041) and tended to be lower in patients with NASH compared to those with FL (P=0.098). These findings suggest that serum VEGF levels tend to decrease during the progression from healthy liver or simple steatosis to steatohepatitis.
Most of the data on angiogenesis in NAFLD come from animal models, while the few reports on serum VEGF levels in NAFLD patients have been controversial. Kitade et al showed that angiogenesis is involved in the development of NASH-related liver fibrosis and carcinogenesis in leptin-deficient rats [34]. In a rat model, it was also shown that renin inhibition may have favorable effects on liver fibrogenesis in NASH, through inhibition of angiotensin-II, tumor growth factor β and VEGF [35]. More recently, Yang et al suggested that anti-VEGF receptor agents ameliorate hepatic venous dysregulation, microcirculatory dysfunction, splanchnic venous pooling and ascites in NASH cirrhotic rats [36]. In humans, Amarapurkar et al reported that immunohistochemical VEGF hepatic expression was seen in 29% of NASH patients or 46% of patients with chronic liver disease of various etiologies, being more common in the early stages of fibrosis [37]. Yilmaz et al reported no significant difference in serum VEGF levels, but significantly lower serum levels of soluble VEGF receptor 1 in NAFLD patients compared to healthy controls, with lower soluble VEGF receptor 1 levels being associated with increased fibrosis [38]. On the other hand, Coulon et al found that serum VEGF serum levels were significantly higher in patients with FL compared to healthy controls, but only relatively higher in patients with NASH compared to healthy controls. In the same study, the concentration of soluble VEGF receptor 1 was significantly higher in the serum of FL and NASH patients compared to controls [39]. Moreover, Tarantino et al showed that serum VEGF levels were higher in NASH patients compared to patients with FL or healthy controls, but serum VEGF level was not a useful marker for differentiation between FL and NASH patients [40]. More recently, Ciupinska-Kajor et al reported that the immunohistochemical hepatic expression of VEGF was higher in simple steatosis and borderline NASH in severely obese patients and in NASH in non-obese patients with NAFLD [41]. In NASH, centrilobular arteries and increased microvessel density are more commonly detected in advanced fibrotic stages, suggesting a possible association between neoangiogenesis and NASH progression to cirrhosis [42].
In addition to the existing controversial data, our results also suggest that serum VEGF levels cannot accurately differentiate healthy controls from NAFLD patients, or patients with simple fatty liver from those with NASH. However, our finding of higher serum VEGF levels in healthy controls, or even perhaps in patients with FL, compared to those with NASH may seem strange, since increased angiogenesis is usually thought to be present in more advanced liver disease, as mentioned above. One explanation for such discrepant findings may be the type of assay used to determine serum VEGF levels. Only free VEGF was measured in our study, which means that the detected VEGF levels may, at least in some cases, underestimate the circulating VEGF levels in serum. In addition, we neither assessed the hepatic expression of VEGF nor measured VEGF receptors, which may offer important information about the role of VEGF in the pathogenesis of NAFLD. Another reason for these conflicting results could be the heterogeneity of patients and controls among the different studies. Finally, angiogenesis can be useful early in the course of NAFLD, reflecting the autohealing ability of the liver. However, beyond a critical point, angiogenesis may become injurious itself and VEGF inhibitors could predominate, thus resulting in a reduction in circulating VEGF levels. Alternatively, our results could reflect a possible dichotomous effect of VEGF on NAFLD progression, similar to that reported for adipose tissue dysfunction [43].
In conclusion, serum VEGF levels offer poor predictability in differentiating healthy controls from NAFLD patients or patients with FL from those with NASH. However, our data suggest that serum VEGF levels are similarly high in healthy controls and patients with FL and tend to decrease when NASH develops. This is a challenging finding, because it may indicate a dichotomous effect of VEGF levels in NAFLD; this question needs to be further evaluated in larger studies in order to clarify the role of VEGF in the pathogenesis of NAFLD.
Summary Box.
What is already known:
Non-alcoholic steatohepatitis (NASH) is characterized by marked elevation of inflammatory cells
Angiogenesis might play a role in the progression of NASH
Vascular endothelial growth factor (VEGF) is a proangiogenic factor implicated in the angiogenetic process
In the human setting, the few reports on serum VEGF levels in patients with non-alcoholic fatty liver disease (NAFLD) are controversial
What the new findings are:
VEGF levels cannot reliably differentiate between patients with NAFLD and healthy controls or between patients with simple fatty liver (FL) and NASH
Serum levels of VEGF are lower in patients with NASH than in healthy controls and relatively lower in NASH than in patients with FL
NAFLD patients with high VEGF levels (≥300 pg/mL) have a lower incidence of NASH and a lower mean fibrosis score
Biography
Laiko General Hospital, Athens, Greece; Hippokration General Hospital, Athens, Greece; Medical School, National and Kapodistrian University of Athens, Greece
Footnotes
Conflict of interest: None
References
- 1.Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol. 2003;98:2042–2047. doi: 10.1111/j.1572-0241.2003.07659.x. [DOI] [PubMed] [Google Scholar]
- 2.Satapathy SK, Sanyal AJ. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35:221–235. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 3.Nalbantoglu IL, Brunt EM. Role of liver biopsy in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:9026–9037. doi: 10.3748/wjg.v20.i27.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ratziu V, Charlotte F, Heurtier A, et al. LIDO Study Group. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128:1898–1906. doi: 10.1053/j.gastro.2005.03.084. [DOI] [PubMed] [Google Scholar]
- 5.Hernaez R, Lazo M, Bonekamp S, et al. Diagnostic accuracy and reliability of ultrasonography for the detection of fatty liver: a meta-analysis. Hepatology. 2011;54:1082–1090. doi: 10.1002/hep.24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hübscher SG. Histological assessment of non-alcoholic fatty liver disease. Histopathology. 2006;49:450–465. doi: 10.1111/j.1365-2559.2006.02416.x. [DOI] [PubMed] [Google Scholar]
- 7.Tiniakos DG. Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis: histological diagnostic criteria and scoring systems. Eur J Gastroenterol Hepatol. 2010;22:643–650. doi: 10.1097/MEG.0b013e32832ca0cb. [DOI] [PubMed] [Google Scholar]
- 8.Brunt EM. Nonalcoholic steatohepatitis: pathologic features and differential diagnosis. Semin Diagn Pathol. 2005;22:330–338. doi: 10.1053/j.semdp.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Brunt EM, Kleiner DE, Wilson LA, et al. NASH Clinical Research Network. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology. 2009;49:809–820. doi: 10.1002/hep.22724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18:727–735. doi: 10.3748/wjg.v18.i8.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cayón A, Crespo J, Guerra AR, Pons-Romero F. [Gene expression in obese patients with non-alcoholic steatohepatitis] Rev Esp Enferm Dig. 2008;100:212–218. doi: 10.4321/s1130-01082008000400004. [DOI] [PubMed] [Google Scholar]
- 12.Kitade M, Yoshiji H, Noguchi R, et al. Crosstalk between angiogenesis, cytokeratin-18, and insulin resistance in the progression of non-alcoholic steatohepatitis. World J Gastroenterol. 2009;15:5193–5199. doi: 10.3748/wjg.15.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salcedo X, Medina J, Sanz-Cameno P, García-Buey L, Martín-Vilchez S, Moreno-Otero R. Review article: angiogenesis soluble factors as liver disease markers. Aliment Pharmacol Ther. 2005;22:23–30. doi: 10.1111/j.1365-2036.2005.02532.x. [DOI] [PubMed] [Google Scholar]
- 14.Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2011;31:146–162. doi: 10.1111/j.1478-3231.2010.02369.x. [DOI] [PubMed] [Google Scholar]
- 15.Medina J, Arroyo AG, Sánchez-Madrid F, Moreno-Otero R. Angiogenesis in chronic inflammatory liver disease. Hepatology. 2004;39:1185–1195. doi: 10.1002/hep.20193. [DOI] [PubMed] [Google Scholar]
- 16.Lai WK, Adams DH. Angiogenesis and chronic inflammation;the potential for novel therapeutic approaches in chronic liver disease. J Hepatol. 2005;42:7–11. doi: 10.1016/j.jhep.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 18.Tamimi TI, Elgouhari HM, Alkhouri N, et al. An apoptosis panel for nonalcoholic steatohepatitis diagnosis. J Hepatol. 2011;54:1224–1229. doi: 10.1016/j.jhep.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 20.Kleiner DE, Brunt EM, Van Natta M, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 21.McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40(Suppl 1):S17–S29. doi: 10.1097/01.mcg.0000168645.86658.22. [DOI] [PubMed] [Google Scholar]
- 22.Edmison J, McCullough AJ. Pathogenesis of non-alcoholic steatohepatitis: human data. Clin Liver Dis. 2007;11:75–104. doi: 10.1016/j.cld.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1769–1777. doi: 10.1053/j.gastro.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellentani S, Marino M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD) Ann Hepatol. 2009;8(Suppl 1):S4–S8. [PubMed] [Google Scholar]
- 25.Sanal MG. The blind men ‘see’ the elephant-the many faces of fatty liver disease. World J Gastroenterol. 2008;14:831–844. doi: 10.3748/wjg.14.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elpek G. Angiogenesis and liver fibrosis. World J Hepatol. 2015;7:377–391. doi: 10.4254/wjh.v7.i3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451–496. doi: 10.1146/annurev-pathol-012615-044224. [DOI] [PubMed] [Google Scholar]
- 28.McCuskey RS, Ito Y, Robertson GR, McCuskey MK, Perry M, Farrell GC. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology. 2004;40:386–393. doi: 10.1002/hep.20302. [DOI] [PubMed] [Google Scholar]
- 29.Kukla M. Angiogenesis: a phenomenon which aggravates chronic liver disease progression. Hepatol Int. 2013;7:4–12. doi: 10.1007/s12072-012-9391-2. [DOI] [PubMed] [Google Scholar]
- 30.Mazzanti R, Messerini L, Monsacchi L, et al. Chronic viral hepatitis induced by hepatitis C but not hepatitis B virus infection correlates with increased liver angiogenesis. Hepatology. 1997;25:229–234. doi: 10.1002/hep.510250142. [DOI] [PubMed] [Google Scholar]
- 31.Salcedo X, Medina J, Sanz-Cameno P, et al. The potential of angiogenesis soluble markers in chronic hepatitis C. Hepatology. 2005;42:696–701. doi: 10.1002/hep.20828. [DOI] [PubMed] [Google Scholar]
- 32.Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41:864–880. doi: 10.1016/j.jhep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Deli G, Jin CH, Mu R, et al. Immunohistochemical assessment of angiogenesis in hepatocellular carcinoma and surrounding cirrhotic liver tissues. World J Gastroenterol. 2005;11:960–963. doi: 10.3748/wjg.v11.i7.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitade M, Yoshiji H, Kojima H, et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology. 2006;44:983–991. doi: 10.1002/hep.21338. [DOI] [PubMed] [Google Scholar]
- 35.Aihara Y, Yoshiji H, Noguchi R, et al. Direct renin inhibitor, aliskiren, attenuates the progression of non-alcoholic steatohepatitis in the rat model. Hepatol Res. 2013;43:1241–1250. doi: 10.1111/hepr.12081. [DOI] [PubMed] [Google Scholar]
- 36.Yang YY, Liu RS, Lee PC, et al. Anti-VEGFR agents ameliorate hepatic venous dysregulation/microcirculatory dysfunction, splanchnic venous pooling and ascites of NASH-cirrhotic rat. Liver Int. 2014;34:521–534. doi: 10.1111/liv.12299. [DOI] [PubMed] [Google Scholar]
- 37.Amarapurkar AD, Amarapurkar DN, Vibhav S, Patel ND. Angiogenesis in chronic liver disease. Ann Hepatol. 2007;6:170–173. [PubMed] [Google Scholar]
- 38.Yilmaz Y, Yonal O, Kurt R, et al. Circulating levels of vascular endothelial growth factor A and its soluble receptor in patients with biopsy-proven nonalcoholic fatty liver disease. Arch Med Res. 2011;42:38–43. doi: 10.1016/j.arcmed.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Coulon S, Francque S, Colle I, et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine. 2012;59:442–449. doi: 10.1016/j.cyto.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Tarantino G, Conca P, Pasanisi F, et al. Could inflammatory markers help diagnose nonalcoholic steatohepatitis? Eur J Gastroenterol Hepatol. 2009;21:504–511. doi: 10.1097/MEG.0b013e3283229b40. [DOI] [PubMed] [Google Scholar]
- 41.Ciupińska-Kajor M, Hartleb M, Kajor M, et al. Hepatic angiogenesis and fibrosis are common features in morbidly obese patients. Hepatol Int. 2013;7:233–240. doi: 10.1007/s12072-011-9320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gill RM, Belt P, Wilson L, Bass NM, Ferrell LD. Centrizonal arteries and microvessels in nonalcoholic steatohepatitis. Am J Surg Pathol. 2011;35:1400–1404. doi: 10.1097/PAS.0b013e3182254283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun K, Wernstedt Asterholm I, Kusminski CM, et al. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A. 2012;109:5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]